

Abstract
A 63-year-old Japanese woman with advanced lung adenocarcinoma developed isolated adrenocorticotropin deficiency caused by immune checkpoint inhibitor (ICI)-related hypophysitis following 8 months of nivolumab therapy. Prompt corticosteroid replacement therapy effectively relieved her secondary adrenal insufficiency symptoms and allowed her to pursue nivolumab therapy, which had been effective for the control of lung adenocarcinoma. Human leukocyte antigen (HLA) typing revealed the presence of the DRB1*04:05-DQA1*03:03-DQB1*04:01 haplotype, which is associated with susceptibility to autoimmune polyglandular syndrome with pituitary disorder in the Japanese population. This case suggests that genetic factors, such as HLA, contribute to the development of endocrinopathies induced by ICIs.
Keywords: hypophysitis, nivolumab, lung adenocarcinoma, hydrocortisone, human leukocyte antigen
Introduction
Isolated adrenocorticotropin deficiency (IAD) is a rare pituitary disorder characterized by secondary adrenal insufficiency (AI) with low or absent cortisol production, but normal secretion of pituitary hormones other than adrenocorticotropic hormone (ACTH), typically in the absence of structural pituitary defects, and an uncertain etiology (1). Corticosteroid replacement therapy is usually effective for the management of IAD.
Immune checkpoint inhibitors (ICIs) comprise a class of drugs that can be used effectively for cancer therapy (2), because they cause reactivation of cytotoxic T cells that destroy tumor cells. By creating an imbalance in the immune system, ICIs generate dysimmune toxicities (autoimmunity), which are referred to as immune-related adverse events (IRAEs), that involve a variety of organs, including the lung, gut, skin, muscle, nerves, and endocrine system (3,4). Common endocrine IRAEs include hypophysitis and thyroid dysfunction, and uncommon IRAEs include primary AI and type 1 diabetes mellitus (T1D). The factors that predict IRAEs remain unclear.
Hypophysitis, or inflammatory processes in the pituitary gland, can cause hypopituitarism characterized by one or multiple deficits in several anterior pituitary hormones, including thyroid-stimulating hormone (TSH), ACTH, and gonadotropins (5,6). ICI-related hypophysitis is frequently (up to 17% of cases) associated with ipilibmab, an anti-cytotoxic T lymphocyte antigen-4 antibody, and patients with ipilibmab-induced hypophysitis typically experience headache, multiple anterior pituitary hormone defects, and reversible enlargement of the pituitary gland (7-12). In contrast, hypophysitis is an extremely rare event (<1%) in patients treated with other ICIs, such as nivolumab/pembrolizmab, an anti-programmed cell death protein 1 (PD-1) monoclonal antibody (13-15). However, few studies have so far investigated the detailed clinical characteristics of hypophysitis induced by anti-PD-1 agents.
Several case reports are available about IAD during nivolumab therapy for metastatic melanoma (16-20). We herein report on a patient with advanced lung adenocarcinoma (LAC) who developed IAD during nivolumab therapy. In addition, previously reported cases of IAD in association with nivolumab treatment are reviewed.
Case Report
A 63-year-old Japanese woman was admitted to our hospital in December 2016 because of 1 week of anorexia, fatigue, and general weakness. She had a paternal family history of cerebral infarction. The patient had given birth three times in her 20s and had no history of head trauma or endocrinological disorder. The patient drank 1 L beer per day and had smoked 40 cigarettes per day (60 pack-years smoking) from 26 to 56 years of age (May 2008) when she was diagnosed with advanced squamous cell carcinoma (SCC) of the esophagus involving the surrounding lymph nodes and trachea (cT4N2M1, stage IVb) (21). She had received definitive chemoradiotherapy with 4 courses of intravenous (IV) cisplatin and 5-fluorouracil (totals of 350 mg and 15,600 mg, respectively) and neck external radiation therapy (total of 60 Gy) for esophageal SCC. The therapy had been effective for two years, but the patient eventually developed local recurrence of esophageal SCC and underwent salvage surgery by transthoracic excision of the esophagus in January 2010.
The patient developed primary hypothyroidism due to the previous neck external irradiation and began thyroid hormone replacement therapy with oral levothyroxine (75 μg/day) in 2012.
A 1.2-cm tumor was detected in the upper lobe of the right lung by follow-up computed tomography (CT) in March 2014 (Fig. 1A). The patient underwent wedge resection to treat the right lung tumor in June of the same year. The histopathological features were consistent with LAC (Fig. 2), and the margin was negative (pT1aN0M0, stage IA) (22). A genetic analysis detected no epidermal growth factor receptor mutations or anaplastic lymphoma kinase rearrangement.
Figure 1.

Chest computed tomography (CT) scans. (A) Chest CT performed in March 2014 showing a 1.2-cm tumor at the apex of the right lung (white arrow). (B) Chest CT performed in April 2016 showing 1.6- and a 2.3-cm tumors on the right pleura (long white arrows) and a 1.0-cm tumor in the lower lobe of the left lung (short white arrow). (C, D) Chest CT performed in July 2016 (C) and November 2016 (D) showing marked reduction in the diameters of the lung and pleural tumors.
Figure 2.
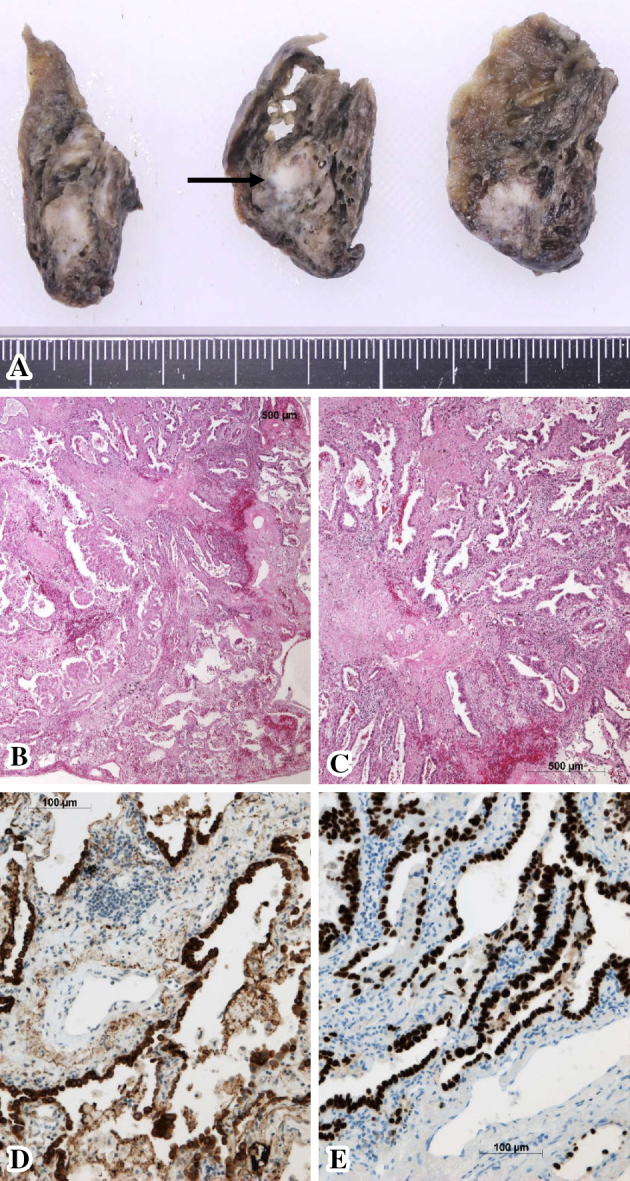
Histopathological findings of the resected lung tissue (July 2014). (A) The gross appearance of the cut surface of the apex of the right upper lung shows a 1.5-cm tumor (black arrow). (B-E) A microscopic examination of the right lung tumor. Proliferation of moderately and poorly differentiated atypical glands was observed (B, C: Hematoxylin and Eosin staining), indicating lung adenocarcinoma. The cytoplasm of the tumor cells was immunohistochemically positive for surfactant protein A (D), and the tumor cell nuclei were positive for thyroid transcription factor-1 (E).
Follow-up CT performed in October 2015 detected multiple tumors in both lungs and on the right pleura, and a transesophageal needle biopsy revealed metastases from LAC. Fluorodeoxyglucose-positron emission tomography showed no abnormal area of accumulation except for the lung and pleural tumors. The patient received 4 courses of chemotherapy with IV cisplatin and gemcitabine (totals, 300 mg and 2,000 mg, respectively) from January to March 2016, but a chest CT scan showed enlargement of the lung and pleural tumors (Fig. 1B), with a Response Evaluation Criteria in Solid Tumors (RECIST) (23) classification of progressive disease. The patient started nivolumab treatment [140 mg (3 mg/kg IV), every 2 weeks] in April 2016 as second-line chemotherapy for the lung and pleural metastases from LAC (24).
Nivolumab therapy effectively controlled the metastatic LAC, and a CT scan performed in July 2016 showed a marked reduction in the tumor size in both lungs (Fig. 1C), with a RECIST classification of partial response.
In November 2016, the patient's body length, body weight, blood pressure, and pulse rate were 161 cm, 47 kg, 121/67 mmHg, and 76 beats per minute, respectively. Her serum TSH (2.31 μIU/mL) and free thyroxine (1.22 ng/dL) levels were normal under replacement therapy with oral levothyroxine (75 μg/day), and her serum electrolyte levels (sodium, 143 mEq/L; potassium, 4.2 mEq/L; and chloride, 108 mEq/L) were also normal. Because the nivolumab therapy remained effective (Fig. 1D), the patient received a 17th round of nivolumab therapy but subsequently developed acute anorexia, fatigue, and myalgia and became unable to walk because of general weakness.
According to a physical examination at admission, the patient's consciousness was clear, and her body weight, body temperature, blood pressure, and pulse rate were 43 kg, 36.9°C, 77/57 mmHg, and 111 beats per minute, respectively. She presented with weakness and pain upon pressure in the muscles of the extremities, but had no headache, struma, abdominal pain, diarrhea, rash, vitiligo, skin pigmentation, or peripheral edema. No heart murmur or chest rale was detected. Laboratory data showed hyponatremia (serum sodium, 117 mEq/L), low levels of plasma ACTH (3.1 pg/mL), serum cortisol (1.6 μg/dL), and dehydroepiandrosterone sulfate (39 ng/mL), and high levels of serum creatine phosphokinase (180 IU/L), C-reactive protein (1.86 mg/dL), aldosterone (26.1 ng/dL), and plasma renin activity (11.1 ng/mL/h) (Table 1). The patient's serum free thyroxine level (1.49 ng/dL) was normal under continued replacement therapy with oral levothyroxine (75 μg/day), whereas the serum TSH level (11.01 μIU/mL) was high. Because the patient was suspected of having AI, she commenced corticosteroid replacement therapy with oral hydrocortisone (15 mg/day) on day 2 of admission, just after undergoing a rapid ACTH stimulation test. On day 4, the patient's test results indicated secondary AI (Table 2A).
Table 1.
Laboratory Findings on Admission (December 2016).
| Hematology | ||
| Red blood cells | 447×104/μL | (386-492) |
| Hemoglobin | 13.4 g/dL | (11.6-14.8) |
| Hematocrit | 37.8 % | (35.1-44.4) |
| White blood cells | 4,100 /μL | (3,300-8,600) |
| Platelets | 29.3×104/μL | (15.8-34.8) |
| Blood chemistry | ||
| Casual plasma glucose | 83 mg/dL | (70-109) |
| Glycated hemoglobin | 5.9 % | (4.6-6.2) |
| Total protein | 6.4 g/dL | (6.6-8.1) |
| Albumin | 4.0 g/dL | (4.1-5.1) |
| Aspartate aminotransferase | 26 IU/L | (13-30) |
| Alanine aminotransferase | 13 IU/L | (7-23) |
| Creatine phosphokinase | 180 IU/L | (41-153) |
| Urea nitrogen | 9.2 mg/dL | (8.0-18.4) |
| Creatinine | 0.59 mg/dL | (0.46-0.79) |
| Sodium | 117 mEq/L | (137-147) |
| Potassium | 3.9 mEq/L | (3.5-4.7) |
| Chloride | 85 mEq/L | (98-108) |
| Calcium | 9.2 mg/dL | (8.8-10.1) |
| C-reactive protein | 1.86 mg/dL | (0-0.14) |
| Immunoglobulin G4 | 15 mg/dL | (0-70) |
| Carcinoembryonic antigen | 2.30 ng/mL | (0-4.76) |
| Sialyl Lewis x | 23.8 U/mL | (0-37.9) |
| Cytokeratin-19 fragments | 1.5 ng/mL | (0-2.1) |
| Squamous cell carcinoma antigen | 0.9 ng/mL | (0-1.5) |
| Plasma osmolality | 241 mOsm/L | (275-290) |
| Arginine vasopressin | 1.8 pg/mL | |
| Thyroid-stimulating hormone | 11.01 μIU/mL | (0.50-5.00) |
| Free thyroxine | 1.49 ng/dL | (0.90-1.70) |
| Free triiodothyronine | 2.28 pg/mL | (2.30-4.00) |
| Adrenocorticotropic hormone | 3.1 pg/mL | (7.2-63.3) |
| Cortisol | 1.6 μg/dL | (4.5-21.1) |
| Dehydroepiandrosterone sulfate | 39 ng/mL | (70-1,770) |
| Aldosterone | 26.1 ng/dL | (3.0-15.9) |
| Plasma renin activity | 11.1 ng/mL/h | (0.2-2.3) |
| Noradrenaline | 0.98 ng/mL | (0.1-0.5) |
| Adrenaline | 0.02 ng/mL | (0-0.1) |
| Dopamine | 0.03 ng/mL | (0-0.03) |
Blood samples were taken in the morning (10 AM) with the patient in the supine position. The patient was receiving thyroid hormone replacement therapy with oral levothyroxine (75 μg/day) for primary hypothyroidism due to previous neck external irradiation.
The reference range for each parameter is shown in parentheses.
Table 2.
Endocrinological Investigation.
A. Rapid ACTH stimulation test in December 2016 (day 2 after admission)
| Time (min) | |||
|---|---|---|---|
| 0 | 30 | 60 | |
| Serum cortisol (μg/dL) | 1.7 | 9.1 | 11.8 |
| Plasma ACTH (pg/mL) | 2.3 | N.M. | N.M. |
Synthetic ACTH 1–24 (cosyntropin hydroxide 0.25 mg) was administered intravenously in the morning (9 AM).
B. CRH/GRF/TRH/LHRH stimulation test in December 2016 (day 6 after admission)
| Time (min) | ||||||
|---|---|---|---|---|---|---|
| 0 | 15 | 30 | 60 | 90 | 120 | |
| Plasma ACTH (pg/mL) | 4.3 | 4.9 | 4.6 | 5.7 | 5.6 | 5.5 |
| Serum cortisol (μg/dL) | 0.6 | 0.7 | 0.6 | 0.8 | 0.6 | 0.4 |
| Serum TSH (μIU/mL) | 0.98 | 12.01 | 17.08 | 13.28 | 9.17 | 7.23 |
| Serum GH (ng/mL) | 0.86 | 8.49 | 11.00 | 7.26 | 3.45 | 1.96 |
| Serum prolactin (ng/mL) | 17.6 | 95.4 | 110.2 | 77.1 | 50.1 | 37.7 |
| Serum LH (mIU/mL) | 15.2 | 23.0 | 29.9 | 33.2 | 36.9 | 37.4 |
| Serum FSH (mIU/mL) | 43.6 | 44.0 | 46.5 | 47.9 | 50.1 | 55.3 |
The following were administered intravenously in the morning (9 AM): human corticotropin-releasing hormone (CRH; 100 μg), growth hormone-releasing factor (GRF; 100 μg), thyrotropin-releasing hormone (TRH; 500 μg), and luteinizing hormone-releasing hormone (LHRH; 100 μg). The test was conducted after stopping oral hydrocortisone (15 mg/day) replacement therapy for 1 day. The patient had low serum levels of insulin-like growth factor 1 (48 ng/mL; reference range, 66-194 ng/mL) and estradiol (5.0 pg/mL; reference range, <39.0 pg/mL) together with normal serum free thyroxine levels (1.46 ng/dL; reference range, 0.90-1.70 ng/dL) under replacement therapy with oral levothyroxine (75 μg/day) for primary hypothyroidism due to previous neck external irradiation.
C. GH-releasing peptide-2 stimulation test performed in December 2016 (day 8 after admission)
| Time (min) | |||||
|---|---|---|---|---|---|
| 0 | 15 | 30 | 45 | 60 | |
| Plasma ACTH (pg/mL) | 4.0 | 4.8 | 4.8 | 4.7 | 4.7 |
| Serum cortisol (μg/dL) | 0.6 | 0.6 | 0.4 | 0.5 | 0.3 |
| Serum GH (ng/mL) | 1.22 | 37.0 | 25.72 | 17.18 | 11.87 |
GH-releasing peptide-2 (100 μg) was administered intravenously in the morning (9 AM). The test was conducted after stopping replacement therapy with oral hydrocortisone (15 mg/day) for 1 day.
ACTH: adrenocorticotropic hormone, FSH: follicle-stimulating hormone, GH: growth hormone, LH: luteinizing hormone, N.M.: not measured, TSH: thyroid-stimulating hormone
Dynamic tests for secretion of pituitary hormones showed normal release of growth hormone (GH), TSH, and prolactin and age-appropriate release of luteinizing hormone and follicle-stimulating hormone in the absence of ACTH release after a corticotropin-releasing hormone load (Table 2B). A GH-releasing peptide-2 loading test also showed no ACTH release, while the GH release was normal (Table 2C). Brain magnetic resonance imaging (MRI) scans revealed a symmetric round-shaped pituitary gland with a slightly thickened hypophyseal stalk and a pituitary height of 5.6 mm (Fig. 3A-C). These findings indicated a diagnosis of IAD caused by nivolumab-induced hypophysitis (1,25). The patient tested negative for pituitary autoantibody (Bio Medical Laboratories, Tokyo, Japan). She was also negative for other organ-specific autoantibodies, such as thyroid peroxidase antibody (<16 IU/mL), thyroglobulin antibody (<28 IU/mL), TSH-binding inhibitory immunoglobulin (<1.0 IU/L), glutamic acid decarboxylase antibody (<5.0 U/mL), insulin antibody (<125.0 nU/mL), insulinoma-associated antigen-2 antibody (<0.4 U/mL), gastric parietal cell antibody (titer <1:10), intrinsic factor antibody, adrenocortical antibody (titer <1:10), anti-nuclear antibody (titer <1:40), Sjögren syndrome A and B antibodies (<0.5 U/mL), Jo-1 antibody (<0.5 IU/mL), mitochondrial antibody (<6.9 U/mL), anti-neutrophil cytoplasmic antibody, and rheumatoid factor. Human leukocyte antigen (HLA) typing revealed the presence of A*24:02/26:01, B*15:01/59:01, and C*01:02/03:03 class I genes and DRB1*04:05/12:02, DQB1*03:01/04:01, DQA1*03:03/06:01, and DPB1*04:02/05:01 class II genes.
Figure 3.

Magnetic resonance imaging (MRI) scans. (A-C) Brain MRI scans obtained at the diagnosis of isolated adrenocorticotropin deficiency (December 2016). Sagittal T1-weighted plain MRI scans (A) showing a symmetric round-shaped pituitary gland and a slightly thickened hypophyseal stalk. A normal high-intensity signal was observed in the posterior lobe of the pituitary. Gadolinium-enhanced MRI scans (B, sagittal plane; C, coronal plane) showing homogeneous enhancement of the hypophyseal stalk and pituitary gland with a pituitary intermediate lobe cyst. The width, length, and height of the pituitary gland are 14.2, 12.4, and 5.6 mm, respectively. (D-F) Brain MRI scans obtained after corticosteroid replacement therapy (April 2017). Sagittal T1-weighted plain MRI scans (D) showing no abnormalities in the hypothalamus, hypophyseal stalk, or pituitary gland. Gadolinium-enhanced MRI scans (E, sagittal plane; F, coronal plane) showing homogeneous enhancement of the hypophysial stalk and pituitary gland, with a small pituitary intermediate lobe cyst. The width, length, and height of the pituitary gland are 11.4, 12.1, and 4.4 mm, respectively.
Thyroid ultrasonography showed normally smooth echogenicity in a mildly atrophic thyroid gland, with no abnormal color-flow Doppler finding, consistent with primary hypothyroidism due to the previous neck external radiation therapy.
The patient experienced improvements in anorexia, fatigue, myalgia, and weakness and became ambulatory. In addition, her hyponatremia was corrected within a week (Table 3). Her body weight, temperature, blood pressure, and pulse rate were 45.4 kg, 36.3°C, 106/71 mmHg, and 80 beats per minute, respectively on day 14. Her serum levels of sodium (141 mEq/L), potassium (4.2 mEq/L), chloride (106 mEq/L), creatine phosphokinase (38 IU/L), C-reactive protein (0.01 mg/dL), TSH (2.63 μIU/mL), free thyroxine (1.35 ng/dL), and aldosterone (3.3 ng/dL), the plasma level of arginine vasopressin (1.2 pg/mL), and plasma renin activity (1.6 ng/mL/h) were measured. Because pituitary dysfunction induced by ICIs is usually permanent (10), cancer treatment with nivolumab was planned, and the patient continued hormone replacement therapy with oral hydrocortisone (15 mg/day) for IAD. The patient received an 18th round of nivolumab therapy for metastatic LAC on day 22 of admission without complication and was discharged on day 24 of admission.
Table 3.
Serial Changes in Serum Sodium Levels before and after Corticosteroid Replacement Therapy.
| Time (day after admission) | ||||||
|---|---|---|---|---|---|---|
| 1 | 3 | 6 | 10 | 14 | 21 | |
| Serum sodium (mEq/L) | 117 | 124 | 139 | 141 | 144 | 141 |
The patient began therapy with oral hydrocortisone (15 mg/day) for her adrenal insufficiency secondary to isolated adrenocorticotropin deficiency on Day 2 after admission.
Brain MRI performed in April 2017 revealed a normal-shaped pituitary gland with a pituitary height of 4.4 mm (Fig. 3D-F), which was indicative of improvement in the hypophysitis. An endocrinological examination showed persistent secondary AI in the absence of other pituitary hormone deficits. The clinical course of the patient was uneventful during nivolumab therapy for metastatic LAC and hormone replacement therapies with oral levothyroxine and hydrocortisone for primary hypothyroidism as a result of the previous neck irradiation and AI secondary to IAD, respectively.
Discussion
An elderly Japanese patient with advanced LAC developed IAD that was caused by ICI-related hypophysitis and manifested as acute anorexia, general weakness, and severe hyponatremia (Table 3) following eight months of nivolumab therapy. Prompt corticosteroid replacement therapy had an immediate effect on the secondary AI and allowed her to pursue nivolumab therapy, which had been effective for the control of advanced LAC.
Table 4 summarizes the characteristics of the patients who exhibited IAD related to cancer treatment with nivolumab; all patients were Japanese. These cases included both men and women adults who developed IAD that manifested predominantly as acute anorexia after three to nine months of nivolumab therapy in a dose-independent manner. None of the patients presented with headache at onset, but two had MRI findings showing reversible, mild, or slight enlargement of the pituitary gland. Pituitary and thyroid autoantibody tests in these two cases revealed negative results.
Table 4.
Summary of Reported Patients who Exhibited Isolated Adrenocorticotropin Deficiency (IAD) during Cancer Treatment with Nivolumab.
| Case | Age/sex (ethnicity) | Target cancer | Regimen | Time to onset of IAD (months) | Major symptoms at IAD onset | Reversible pituitary gland enlargement on MRI* | Pituitary autoantibodies** | Thyroid autoantibodies*** | Major complicating disorders | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 68/M (Japanese) | Melanoma (in-transit metastasis) |
2 mg/kg IV, every 3 weeks | 7 | N.A. | Undetermined | N.A. | N.A. | None | (16) |
| 2 | 76/F (Japanese) | Melanoma | 2 mg/kg IV, every 3 weeks | 5 | Anorexia, bradykinesia | N.A. | N.A. | N.A. | None | (17) |
| 3 | 50/M (Japanese) | Melanoma (mediastinal lymph node metastases) |
2 mg/kg IV, every 3 weeks | 3.5 | Anorexia, fatigue, weakness | Mild | Negative | Negative | None | (18) |
| 4 | 55/M (Japanese) | Melanoma (right adrenal gland metastasis) |
2 mg/kg IV, every 3 weeks | 3 | Anorexia, malaise, myalgia | Undetermined | N.A. | N.A. | Interstitial pneumonia, hypothyroidism | (19) |
| 5 | 39/M (Japanese) | Melanoma (lungs, bones, and liver metastases) |
2 mg/kg IV, every 3 weeks | 9 | General malaise | Undetermined | N.A. | N.A. | None | (20) |
| 6 | 50/F (Japanese) | Melanoma (lung metastases) |
2 mg/kg IV, every 3 weeks | 9 | Fever, fatigue, dizziness, walking difficulty | Undetermined | N.A. | N.A. | None | (20) |
| 7 | 63/F (Japanese) | Lung adenocarcinoma (pleural metastases) |
3 mg/kg IV, every 2 weeks | 8 | Anorexia, fatigue, myalgia, weakness | Slight | Negative | Negative | Primary hypothyroidism due to previous neck external irradiation for primary esophageal cancer (SCC) | Present case |
*In cases 1, 4, 5, and 6, the brain MRI findings obtained at the diagnosis of IAD are described, but those prior to IAD onset or after corticosteroid treatment were unavailable for comparison. Therefore, the determination of reversible enlargement of the pituitary gland was not possible.
**Cases 3 and 7 tested negative for anti-pituitary cell antibody using a commercially available kit that employs a substrate from rat pituitary sections.
***Thyroid autoantibodies include thyroglobulin antibody, thyroid peroxidase antibody, and thyroid-stimulating hormone-binding inhibitory immunoglobulin.
F: female, IV: intravenous, M: male, MRI: magnetic resonance imaging, N.A.: data not available, SCC: squamous cell carcinoma
Although a definitive diagnosis of hypophysitis can be achieved only via histology from a trans-sphenoidal biopsy specimen, a non-invasive diagnosis can be putatively made by evaluating MRI scans and patterns of endocrinological deficits in pituitary hormones (5). The distinctive MRI characteristics of hypophysitis include pre-contrast symmetric enlargement of the pituitary gland, which is occasionally accompanied by stalk thickening, and post-contrast anterohypophyseal enhancement. However, the pituitary enlargement in ICI-related hypophysitis can be mild and only becomes evident in some patients after comparison with imaging data obtained prior to ICI therapy or after the resolution of pituitary inflammation (8). The patterns of pituitary hormone deficits in patients with hypophysitis may be associated with the degree of pituitary inflammation, and ACTH secretion may be disturbed initially and solely by mild pituitary inflammation (5,6). In the present case, MRI scans performed at the IAD diagnosis and four months after corticosteroid replacement therapy revealed reversible and slight enlargement of the pituitary gland (Fig. 3). In addition, the patient did not present with headache, which would be expected in the presence of some degree of pituitary inflammation. Thus, the IAD of the present patient was deemed to have been caused by nivolumab-induced hypophysitis, probably with a slight degree of pituitary inflammation.
The time to onset of endocrine IRAEs induced by ipilibmab, including hypophysitis, is typically nine weeks after the initiation of therapy (9,10). This latency period is roughly the same as that of nivolumab-induced endocrine IRAEs (median 4-18 weeks) (13). In contrast, the latency periods for nivolumab-induced IAD (3-9 months) in previously reported patients and our own are typically much longer (Table 4). The reason for the longer drug exposure period prior to nivolumab-induced IAD onset remains unclear. The present case suggests that nivolumab-induced hypophysitis may occur even six months after administration, and therefore, long-term monitoring should be considered.
The diagnosis of autoimmunity can be made by detecting circulating organ-specific autoantibodies. However, the actual roles that pituitary autoantibody tests play are controversial because of methodological difficulties, and because no specific pituitary antigens have been associated with hypophysitis (5,6,26). Studies have shown substantial differences in the sensitivity of pituitary autoantibody detection among pituitary substrates from different species; animal pituitary substrates are associated with lower sensitivity than human pituitary substrates (27). No study has reported positivity on a pituitary autoantibody test using animal pituitary substrates in patients with hypophysitis induced by nivolumab, including the present patient evaluated with rat pituitary substrates (Table 4). However, one report described positive results for a human substrate pituitary autoantibody test conducted in patients with hypophysitis induced by ipilibmab (28). These findings suggest that our patient exhibited nivolumab-induced autoimmune hypophysitis with a false-negative result on the pituitary autoantibody test.
Although the factors predicting endocrine IRAEs remain unclear, several case studies of Japanese patients with ICI-related T1D have revealed the presence of genes that induce susceptibility to T1D, such as the HLA-DRB1*04:05-DQB1*04:01 and HLA-B*40:02 haplotypes (29,30). Few studies have investigated this type of genetic association for other endocrine IRAEs, but a recent case study of two Japanese patients with nivolumab-induced hypophysitis showed the presence of HLAs that have been associated with lymphocytic hypophysitis, such as HLA-A2, A24, B7, DR1, and DR4 (20). In the present case, the patient had the HLA class II DRB1*04:05-DQA1*03:03-DQB1*04:01 haplotype, which is associated with susceptibility to pituitary dysfunction associated with polyglandular autoimmune syndrome (PGAS) type 3 in the Japanese population (31). PGAS is a group of endocrine and non-endocrine organ-specific autoimmune disorders (32). Our patient did not exhibit a clinically evident organ-specific autoimmune disorder until the development of nivolumab-induced hypophysitis. However, because ICIs can induce multiple endocrine gland injuries via autoimmune mechanisms, nivolumab-induced hypophysitis probably occurred in our patient because of a genetic predisposition to polyglandular autoimmunity, and the possibility of subsequent development of other endocrine IRAEs was considered.
Patients with untreated IAD often exhibit mild elevations in serum TSH levels because of the absence of the physiological inhibitory effects of cortisol on pituitary TSH secretion (1), disturbed thyroid hormone synthesis and/or secretion induced by cortisol deficiency (33), or a concomitant autoimmune thyroid disorder (34). In the present case, the patient had a high serum TSH level that normalized rapidly after corticosteroid replacement for IAD, and she had normal serum free thyroxine levels while under a fixed dose of oral levothyroxine replacement for primary hypothyroidism due to previous neck external irradiation. In addition, she had no serological or imaging evidence of an autoimmune thyroid disorder. These findings suggest that the elevated TSH level when our patient developed IAD was caused by the absence of a physiological inhibitory effect of cortisol on TSH secretion.
In conclusion, we herein described a case of IAD caused by nivolumab-induced hypophysitis in a patient with advanced LAC. The present case suggests that endocrine IRAEs induced by ICIs occur in patients with certain genetic backgrounds, such as those with HLA genes associated with susceptibility to polyglandular autoimmunity. The findings from the present case suggest that autoimmune hypophysitis accompanied by a mild degree of pituitary inflammation is a cause of IAD.
The authors state that they have no Conflict of Interest (COI).
Acknowledgement
The authors thank the clinical laboratory technicians of Uonuma Institute of Community Medicine, Niigata University Medical and Dental Hospital, for their technical support.
References
- 1. Andrioli M, Pecori Giraldi F, Cavagnini F. Isolated corticotrophin deficiency. Pituitary 9: 289-295, 2006. [DOI] [PubMed] [Google Scholar]
- 2. Pennock GK, Chow LQ. The evolving role of immune checkpoint inhibitors in cancer treatment. Oncologist 20: 812-822, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Michot JM, Bigenwald C, Champiat S, et al. Immune-related adverse events with immune checkpoint blockade: a comprehensive review. Eur J Cancer 54: 139-148, 2016. [DOI] [PubMed] [Google Scholar]
- 4. Torino F, Corsello SM, Salvatori R. Endocrinological side-effects of immune checkpoint inhibitors. Curr Opin Oncol 28: 278-287, 2016. [DOI] [PubMed] [Google Scholar]
- 5. Falorni A, Minarelli V, Bartoloni E, Alunno A, Gerli R. Diagnosis and classification of autoimmune hypophysitis. Autoimmun Rev 13: 412-416, 2014. [DOI] [PubMed] [Google Scholar]
- 6. Bellastella G, Maiorino MI, Bizzarro A, et al. Revisitation of autoimmune hypophysitis: knowledge and uncertainties on pathophysiological and clinical aspects. Pituitary 19: 625-642, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Spain L, Diem S, Larkin J. Management of toxicities of immune checkpoint inhibitors. Cancer Treat Rev 44: 51-60, 2016. [DOI] [PubMed] [Google Scholar]
- 8. Faje A. Immunotherapy and hypophysitis: clinical presentation, treatment, and biologic insights. Pituitary 19: 82-92, 2016. [DOI] [PubMed] [Google Scholar]
- 9. Joshi MN, Whitelaw BC, Palomar MT, Wu Y, Carroll PV. Immune checkpoint inhibitor-related hypophysitis and endocrine dysfunction: clinical review. Clin Endocrinol (Oxf) 85: 331-339, 2016. [DOI] [PubMed] [Google Scholar]
- 10. Torino F, Barnabei A, Paragliola RM, Marchetti P, Salvatori R, Corsello SM. Endocrine side-effects of anti-cancer drugs: mAbs and pituitary dysfunction: clinical evidence and pathogenic hypotheses. Eur J Endocrinol 169: R153-R164, 2013. [DOI] [PubMed] [Google Scholar]
- 11. Albarel F, Gaudy C, Castinetti F, et al. Long-term follow-up of ipilimumab-induced hypophysitis, a common adverse event of the anti-CTLA-4 antibody in melanoma. Eur J Endocrinol 172: 195-204, 2015. [DOI] [PubMed] [Google Scholar]
- 12. Araujo PB, Coelho MC, Arruda M, Gadelha MR, Neto LV. Ipilimumab-induced hypophysitis: review of the literature. J Endocrinol Invest 38: 1159-1166, 2015. [DOI] [PubMed] [Google Scholar]
- 13. Eigentler TK, Hassel JC, Berking C, et al. Diagnosis, monitoring and management of immune-related adverse drug reactions of anti-PD-1 antibody therapy. Cancer Treat Rev 45: 7-18, 2016. [DOI] [PubMed] [Google Scholar]
- 14. Rossi E, Sgambato A, De Chiara G, et al. Endocrinopathies induced by immune-checkpoint inhibitors in advanced non-small cell lung cancer. Expert Rev Clin Pharmacol 9: 419-428, 2016. [DOI] [PubMed] [Google Scholar]
- 15. Costa R, Carneiro BA, Agulnik M, et al. Toxicity profile of approved anti-PD-1 monoclonal antibodies in solid tumors: a systematic review and meta-analysis of randomized clinical trials. Oncotarget 8: 8910-8920, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Fujimura T, Kambayashi Y, Furudate S, et al. Isolated adrenocorticotropic hormone deficiency possibly caused by nivolumab in a metastatic melanoma patient. J Dermatol 44: e13-e14, 2017. [DOI] [PubMed] [Google Scholar]
- 17. Narahira A, Yanagi T, Cho KY, et al. Isolated adrenocorticotropic hormone deficiency associated with nivolumab therapy. J Dermatol 44: e70, 2017. [DOI] [PubMed] [Google Scholar]
- 18. Okano Y, Satoh T, Horiguchi K, et al. Nivolumab-induced hypophysitis in a patient with advanced malignant melanoma. Endocr J 63: 905-912, 2016. [DOI] [PubMed] [Google Scholar]
- 19. Ishikawa M, Oashi K. Case of hypophysitis caused by nivolumab. J Dermatol 44: 109-110, 2017. [DOI] [PubMed] [Google Scholar]
- 20. Kitajima K, Ashida K, Wada N, et al. Isolated ACTH deficiency probably induced by autoimmune-related mechanism evoked with nivolumab. Jpn J Clin Oncol 18: 1-4, 2017. [DOI] [PubMed] [Google Scholar]
- 21. Rice TW, Ishwaran H, Blackstone EH, Hofstetter WL, Kelsen DP, Apperson-Hansen C; Worldwide Esophageal Cancer Collaboration Investigators Recommendations for clinical staging (cTNM) of cancer of the esophagus and esophagogastric junction for the 8th edition AJCC/UICC staging manuals. Dis Esophagus 29: 913-919, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Goldstraw P, Ball D, Jett JR, et al. Non-small-cell lung cancer. Lancet 378: 1727-1740, 2011. [DOI] [PubMed] [Google Scholar]
- 23. Eisenhauer EA, Therasse P, Bogaerts J, et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer 45: 228-247, 2009. [DOI] [PubMed] [Google Scholar]
- 24. Kazandjian D, Suzman DL, Blumenthal G, et al. FDA approval summary: Nivolumab for the treatment of metastatic non-small cell lung cancer with progression on or after platinum-based chemotherapy. Oncologist 21: 634-642, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kageyama K, Nigawara T, Sakihara S, et al. Diagnostic usefulness of the growth hormone-releasing peptide-2 test as a substitute for the insulin tolerance test in hypopituitarism. Endocr J 55: 777-783, 2008. [DOI] [PubMed] [Google Scholar]
- 26. Glezer A, Bronstein MD. Pituitary autoimmune disease: nuances in clinical presentation. Endocrine 42: 74-79, 2012. [DOI] [PubMed] [Google Scholar]
- 27. Ricciuti A, De Remigis A, Landek-Salgado MA, et al. Detection of pituitary antibodies by immunofluorescence: approach and results in patients with pituitary diseases. J Clin Endocrinol Metab 99: 1758-1766, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Iwama S, De Remigis A, Callahan MK, Slovin SF, Wolchok JD, Caturegli P. Pituitary expression of CTLA-4 mediates hypophysitis secondary to administration of CTLA-4 blocking antibody. Sci Transl Med 6: 230ra45, 2014. [DOI] [PubMed] [Google Scholar]
- 29. Okamoto M, Okamoto M, Gotoh K, et al. Fulminant type 1 diabetes mellitus with anti-programmed cell death-1 therapy. J Diabetes Investig 7: 915-918, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Munakata W, Ohashi K, Yamauchi N, Tobinai K. Fulminant type I diabetes mellitus associated with nivolumab in a patient with relapsed classical Hodgkin lymphoma. Int J Hematol 105: 383-386, 2017. [DOI] [PubMed] [Google Scholar]
- 31. Hashimoto K, Maruyama H, Nishiyama M, et al. Susceptibility alleles and haplotypes of human leukocyte antigen DRB1, DQA1, and DQB1 in autoimmune polyglandular syndrome type III in Japanese population. Horm Res 64: 253-260, 2005. [DOI] [PubMed] [Google Scholar]
- 32. Cutolo M. Autoimmune polyendocrine syndromes. Autoimmun Rev 13: 85-89, 2014. [DOI] [PubMed] [Google Scholar]
- 33. Tamura M, Yokoyama N, Nishikawa T, et al. Improvement of hypothyroidism after glucocorticoid replacement in isolated adrenocorticotropin deficiency. Intern Med 34: 559-563, 1995. [DOI] [PubMed] [Google Scholar]
- 34. Murakami T, Wada S, Katayama Y, Nemoto Y, Kugai N, Nagata N. Thyroid dysfunction in isolated adrenocorticotropic hormone (ACTH) deficiency: case report and literature review. Endocr J 40: 473-478, 1993. [DOI] [PubMed] [Google Scholar]