

Abstract
Seipinopathy is an autosomal dominant neurodegenerative disease caused by mutations of the Berardinelli-Seip Congenital Lipodystrophy 2 (BSCL2) gene. We report the first Japanese case of seipinopathy with a heterozygous mutation of p.N88S in the BSCL2 gene. The patient showed bilateral hyperreflexia of the biceps, triceps, brachioradialis, and knee, as well as the pes cavus and distal dominant weakness and atrophy of both arms and legs, suggesting the involvement of both upper and lower motor neurons. Mutations of the BSCL2 gene have been known to cause motor neuron degeneration through endoplasmic reticulum stress. Seipinopathy should be considered in patients with symptoms mimicking amyotrophic lateral sclerosis.
Keywords: BSCL2, seipinopathy, Japanese patient, upper motor neuron involvement, pyramidal signs
Introduction
Seipinopathy is an autosomal dominant, neurodegenerative disease caused by mutations of Berardinelli-Seip Congenital Lipodystrophy 2 (BSCL2) gene (1). While loss-of-function mutations in the BSCL2 gene causes lipodystrophy, heterozygous missense mutations, such as p.N88S, p.S90L, p.S90W and p.R96H, have been identified as the cause of seipinopathy through the gain of a toxic function (2-4). Seipinopathy has several phenotypes that are described as follows (5). The Silver syndrome phenotype is characterized by amyotrophy of the small hand muscles and spasticity of the lower limbs. The distal hereditary motor neuropathy type V (dHMN-V) phenotype is characterized by weakness or amyotrophy, primarily in the small hand muscles and late-onset gait disturbance. The hereditary spastic paraparesis phenotype is characterized by spastic paraparesis in the lower limbs. The Charcot-Marie-Tooth disease (CMT) phenotype is characterized by distal muscle weakness and amyotrophy of the lower limbs (rather than the upper limbs). Pyramidal signs are occasionally seen, and depending on the presence or absence of sensory disturbance, the CMT phenotype is further divided into spinal CMT and hereditary motor and sensory neuropathy type Ⅱ (HMSN-Ⅱ). Thus, seipinopathy can lead to symptoms that mimic amyotrophic lateral sclerosis. In fact, mutations of the BSCL2 gene have been known to cause motor neuron degeneration through endoplasmic reticulum stress (1). Seipinopathy should be considered in patients with both upper and lower motor neuron involvement.
Case Report
We herein report the case of a 63-year-old Japanese woman who was not good at physical activities like running when she was an elementary school student. After 30 years of age, she gradually developed walking difficulty. She noticed weakness and atrophy in both hands at 60 years of age and both legs at 63 years of age, without body weight loss. Her paternal grandfather and father also had walking difficulty, but they lived until 88 and 91 years of age, respectively. Her elder son had pes cavus. A physical examination revealed bilateral hyperreflexia of the biceps, triceps, brachioradialis, and knee, as well as pes cavus and distal dominant weakness and atrophy of both hands and legs (Fig. 1, 2). However, a careful examination revealed no fasciculation. The patient was negative for Babinski and Chaddock signs, and showed an indifferent planter response; however, a left Trömner sign was noted. A cranial nerve and sensory examination (both superficial and deep sensation), blood and cerebrospinal fluid tests, magnetic resonance imaging of the head and cervical vertebrae, and sensory nerve conduction studies all revealed normal findings. Motor nerve conduction studies showed normal conduction velocities with decreased compound muscle action potentials (CMAPs) bilaterally in the tibial nerves and the right median nerve (Table). Needle electromyography revealed chronic neurogenic changes in the bilateral first dorsal interossei and the tibialis anterior. The central conduction time was not evaluated. A genetic analysis revealed a heterozygous mutation of the p.N88S in BSCL2 gene (Fig. 3).
Figure 1.
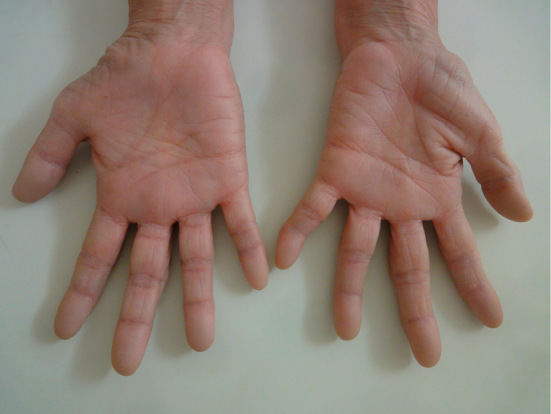
The patient showed characteristic atrophy of both hands, which was more severe in thenar eminence than in hypothenar eminence.
Figure 2.
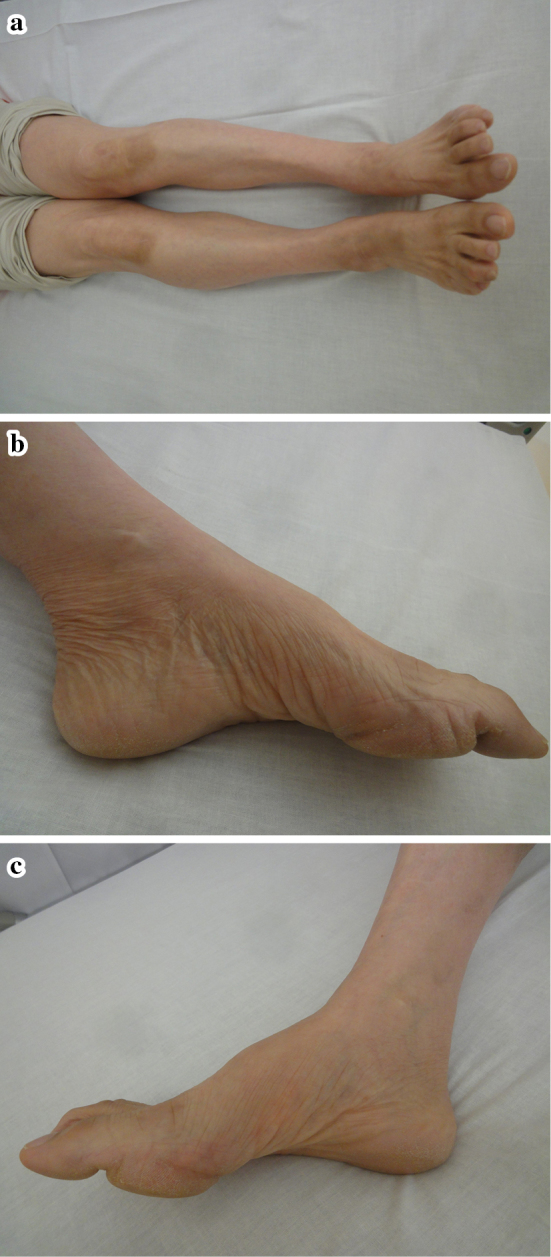
a-c: The patient showed distal dominant atrophy of both legs, including pes cavus. These features are often seen in Charcot-Marie-Tooth disease.
Table.
Nerve Conduction Studies.
| Motor nerve study | Lat (ms) | CMAP (mV) | CV (m/s) |
|---|---|---|---|
| Right median nerve | |||
| Wrist | 2.76 | 4.73 | 59.7 |
| Elbow | 5.94 | 3.46 | |
| Right ulnar nerve | |||
| Wrist | 2.9 | 12.02 | 68.5 |
| Elbow | 6.98 | 12.26 | 61.3 |
| Right tibial nerve | |||
| Ankle | 2.85 | 0.33 | 39.3 |
| Knee | 12 | 0.25 | |
| Left tibial nerve | |||
| Ankle | 3.65 | 0.22 | 40.9 |
| Knee | 12.2 | 0.24 | |
| Sensory nerve study | SNAP (μV) | CV (m/s) | |
| Right median nerve | 16.2 | 61.9 | |
| Right ulnar nerve | 11.3 | 57.3 | |
| Left sural nerve | 5.2 | 54.7 |
Lat: latency, ms: milliseconds, CMAP: compound muscle action potential, mV: millivolt, CV: conduction velocity, m/s: meter/second, SNAP: sensory nerve action potential
Figure 3.

The chromatogram indicated that the patient was heterozygous for a p.N88S mutation in the BSCL2 gene (arrow).
Discussion
Numerous cases of seipinopathy have been reported in Europe; thus, it was considered to only occur in Europe. However, four cases of seipinopathy have been reported (two cases each) in China and Korea (3,6-8). Taiwanese cases of seipinopathy with a heterozygous mutation of p.S90L and p.R96H in the BSCL2 gene were also reported in 2016 (4). To the best of our knowledge, this is the first report of seipinopathy in a Japanese patient (with the exception of meeting abstracts) with both upper and lower motor symptoms. As seipinopathy is a broad-spectrum disease, it may often be misdiagnosed; thus, the incidence around the world may be higher than reported. In our case, the presence of pes cavus, distal dominant weakness, and atrophy of both arms and legs suggested peripheral neuropathy, and at first we assumed that these features were suitable for typical CMT. However, sensory disturbance was not seen and the co-existing pyramidal signs [i.e., Trömner sign (only left-side) and bilateral hyperreflexia] suggested the involvement of upper motor neurons, irrespective of the absence of Babinski and Chaddock signs. Thus, the clinical diagnosis was dHMN with spasticity (9) or HMSN-V (CMT with spasticity). A genetic analysis confirmed seipinopathy mimicking spinal CMT with a heterozygous mutation of p.N88S in the BSCL2 gene. No specific signs or symptoms suggestive of endoplasmic reticulum stress, which plays a causative role in neurodegeneration, were noted. Thus, there may not be an effective treatment at present.
In conclusion, seipinopathy should be considered in patients with both upper and lower motor neuron impairment. Several cases of seipinopathy were recently reported in Asia, and there may be more patients in Japan. Since seipinopathy sometimes progresses slowly, a family history of seipinopathy may not be present. However, a genetic analysis is essential for making the correct diagnosis.
The authors state that they have no Conflict of Interest (COI).
Financial Support
This research is supported by funding from the following sources.
Dr. Takashima received funding through grants from the Nervous and Mental Disorders and Research Committee for Applying Health and Technology of the Ministry of Health, Welfare and Labour, Japan. This research is also supported by the research program for conquering intractable disease (15ek0109010h0002) from the Japan Agency for Medical Research and Development, AMED.
References
- 1. Ito D, Suzuki N. Seipinopathy: a novel endoplasmic reticulum stress-associated disease. Brain 132: 8-15, 2009. [DOI] [PubMed] [Google Scholar]
- 2. Windpassinger C, Auer-Grumbach M, Irobi J, et al. . Heterozygous missense mutations in BSCL2 are associated with distal hereditary motor neuropathy and Silver syndrome. Nature Genetics 36: 271-276, 2004. [DOI] [PubMed] [Google Scholar]
- 3. Choi BO, Park MH, Chung KW, et al. . Clinical and histopathological study of Charcot-Marie-Tooth neuropathy with a novel S90W mutation in BSCL2. Neurogenetics 14: 35-42, 2013. [DOI] [PubMed] [Google Scholar]
- 4. Hsiao CT, Tsai PC, Lin CC, et al. . Clinical and molecular characterization of BSCL2 mutations in a Taiwanese cohort with hereditary neuropathy. PloS One 11: e0147677, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Auer-Grumbach M, Schlotter-Weigel B, Lochmuller H, et al. . Phenotypes of the N88S Berardinelli-Seip congenital lipodystrophy 2 mutation. Ann Neurol 57: 415-424, 2005. [DOI] [PubMed] [Google Scholar]
- 6. Cho HJ, Sung DH, Ki CS. Identification of de novo BSCL2 Ser90Leu mutation in a Korean family with Silver syndrome and distal hereditary motor neuropathy. Muscle Nerve 36: 384-386, 2007. [DOI] [PubMed] [Google Scholar]
- 7. Chen B, Zheng R, Luan X, Zhang W, Wang Z, Yuan Y. Clincial and pathological study of distal motor neuropathy with N88S mutation in BSCL2. Neuropathology 29: 543-547, 2009. [DOI] [PubMed] [Google Scholar]
- 8. Cen Z, Lu X, Wang Z, Ouyang Z, Xie F, Luo W. BSCL2 S90L mutation in a Chinese family with Silver syndrome with a review of the literature. J Clin Neurosci 22: 429-430, 2015. [DOI] [PubMed] [Google Scholar]
- 9. Rossor AM, Kalmar B, Greensmith L, Reilly MM. The distal hereditary motor neuropathies. J Neurol Neurosurg Psychiatry 83: 6-14, 2012. [DOI] [PubMed] [Google Scholar]