

Abstract
Idiopathic pulmonary fibrosis (IPF) is a progressive, fatal disease that can only be cured by lung transplantation. Pharmacological agents play a role in preserving lung function and prolonging survival until a suitable donor organ becomes available. However, data on the effects of newer antifibrotic therapies on lung transplantation outcomes in IPF patients are lacking. The nine patients included in this case series were treated with nintedanib 150 mg twice daily for 3–30 (mean 13 ± 9) months before lung transplant surgery. Lung function was relatively preserved during nintedanib therapy, and no acute IPF exacerbations occurred. Transplant surgery was generally uneventful; eight of the nine patients are still alive. There were no extraordinary bleeding complications or issues with postoperative thoracic wound healing or dehiscence. Only one patient experienced bronchial anastomotic stenosis a few months later. In conclusion, nintedanib preserved lung function up to transplantation, was well tolerated, and had no detrimental effects on the short‐term outcome of lung transplant.
Keywords: Idiopathic pulmonary fibrosis, lung transplant, nintedanib
Introduction
Idiopathic pulmonary fibrosis (IPF) is a progressive, fatal disease characterized by chronic, fibrosing interstitial pneumonia of unknown cause, associated with a histopathological and/or radiological pattern of usual interstitial pneumonia (UIP) 1. Median survival time after diagnosis is 2.5–3.5 years, although the course of IPF is unpredictable and heterogeneous 2. While disease progression is slow in the majority of patients, others experience rapid decline in lung function and have shorter survival 2. One possible explanation for this is differences in gene expression between patients with the rapid progressor versus slow progressor phenotype. Patients whose disease progresses quickly have shown upregulation of the molecular pathways involved in fibroblast differentiation and inflammation 3, 4. Other factors contributing to inter‐patient variability in the clinical behaviour of IPF include the degree of inflammation and differences in innate and adaptive immunity 5.
The only curative therapy for IPF is lung transplantation. However, the availability of donor organs significantly limits the number of transplants that can be performed. Therefore, pharmacological therapies that prolong patient survival until suitable organs become available are important 6, 7. Newer antifibrotic agents (e.g. pirfenidone, nintedanib) are potential therapeutic options in this setting. Pirfenidone has both antifibrotic and anti‐inflammatory effects, mediated in part by the inhibition of the production and activity of transforming growth factor‐beta 8. Nintedanib is a tyrosine kinase inhibitor that blocks receptors for platelet‐derived growth factor (PDGF), vascular endothelial growth factor (VEGF), and fibroblast growth factor (FGF) 9.
Randomized clinical trial data show that treatment with pirfenidone and nintedanib has a number of beneficial effects in patients with IPF, including slowing the decline in lung function and exercise tolerance, prolonging the time to first acute exacerbation, and improving progression‐free survival compared with placebo 10, 11, 12, 13, 14. The use of both agents has increased after their approval by the US Food and Drug Administration (FDA) in October 2014 15. However, there is a lack of data on the safety of new antifibrotic agents in IPF patients undergoing lung transplantation. The PDGF, VEGF, and FGF blockade induced by nintedanib 9 have the theoretical potential to increase perioperative bleeding risk, impair postoperative wound healing, and/or cause bronchial anastomotic complications after lung transplantation. There is currently no information on the use of nintedanib in patients with IPF awaiting a lung transplant, but there is a need to better understand the risks and benefits of nintedanib use in this setting.
Data from a series of nine IPF patients treated with nintedanib until lung transplantation are presented to highlight the efficacy and safety of nintedanib as a bridge to transplant.
Case Series
Methods and Results
Patients were treated at three centres from northern Italy. All patients had a definitive diagnosis of IPF and were treated with nintedanib prior to lung transplantation. Pre‐transplant characteristics of individual patients are given in Table 1. Data about the perioperative and postoperative course were collected.
Table 1.
Pre‐transplant characteristics of the study population.
| i.d., M/F | Age at Tx (years) | BMI (kg/m2) | FVC, % pred. (start therapy) | DLCO, % pred. | 6MWD (meters) | GAP score, n | mPAP (mmHg) | LAS score, n | Comorbidities |
|---|---|---|---|---|---|---|---|---|---|
| 1, M | 55 | 24 | 43 | 24 | 360 | 5 | 20 | 39 | Arterial hypertension |
| 2, F | 63 | 25 | 55 | 40 | 450 | 5 | 18 | 34 | Reflux; hiatal hernia; previous HCV infection |
| 3, M | 57 | 28 | 40 | 33 | 210 | 5 | 20 | 35 | Osteoporosis |
| 4, M | 48 | 23 | 43 | 36 | 450 | 4 | 17 | 35 | None |
| 5, M | 56 | 28 | 48 | 25 | 270 | 5 | 25 | 39 | Dyslipidaemia; OSA |
| 6, M | 60 | 25 | 53 | 22 | 454 | 4 | 26 | 40.7 | Coronary artery disease post‐stenting |
| 7, M | 65 | 25 | 81 | 19 | 144 | 4 | 25 | 32.2 | None |
| 8, M | 66 | 22 | 43 | 30 | 505 | 7 | 22 | 30 | None |
| 9, F | 60 | 29 | 59 | 24 | 390 | 4 | 10 | 33.7 | Carotid artery disease; hiatal hernia |
| Mean ± SD | 59 ± 5.6 | 25 ± 3.5 | 51.6 ± 12.7 | 28 ± 7 | 359 ± 124 | 4.7 ± 0.9 | 20.3 ± 5.0 | 35.6 ± 3.7 |
6MWD, 6‐min walk distance; BMI, body mass index; DLCO, diffusing capacity of the lung for carbon monoxide; F, female; FVC, forced vital capacity; GAP, gender‐age‐physiology; HCV, hepatitis C virus; i.d., patient identification number; LAS, lung allocation score; M, male; mPAP, mean pulmonary artery pressure; OSA, obstructive sleep apnoea; pred., predicted; SD, standard deviation; Tx, treatment.
Nine IPF patients (7 male, 2 female; mean age 59 ± 5.6 years) were treated with nintedanib and subsequently underwent lung transplantation (bilateral in six patients). All patients were receiving continuous oxygen therapy prior to transplantation. In seven patients, IPF diagnosis was based on histological samples consistent with a UIP pattern; of these, only one had undergone cryobiopsy. Time on the transplant waiting list was 10.3 ± 5.7 months, and mean lung allocation score (LAS) at the time of being added to the transplant list was 35.6 ± 3.7.
Before Lung Transplant
Nintedanib, 150 mg twice daily, was initiated 3–30 months prior to lung transplant surgery and continued until the day of transplantation (mean therapy duration 13 ± 9 months). Concomitant medication included aspirin (n = 2) and aspirin + clopidogrel (n = 1). No acute exacerbations of IPF occurred during treatment with nintedanib. Diarrhoea was reported by three patients; this was of mild intensity and did not require nintedanib dosage adjustment. No hepatic dysfunction or weight loss occurred during nintedanib therapy, and no patients discontinued treatment due to severe side effects or adverse events.
In seven patients with available functional data 12 months before starting treatment, we observed a median 7.8% predicted (range 5–12) decline/year in forced vital capacity (FVC) (Fig. 1). After 12 weeks of nintedanib treatment, in eight patients with available data, there was a median 3.2% predicted (range 1–7) decline in FVC (decline was <5% predicted and >5% predicted in six and two patients, respectively) (Fig. 1), whereas after 6 months of follow up, only one patient had a ≥10% predicted decreased in FVC from baseline. The predicted median FVC decline per year (%pred.) was 10 (range 2–18). Only one patient had mild secondary pulmonary hypertension on right heart catheterization (mean pulmonary artery pressure (mPAP) > 25 mmHg).
Figure 1.
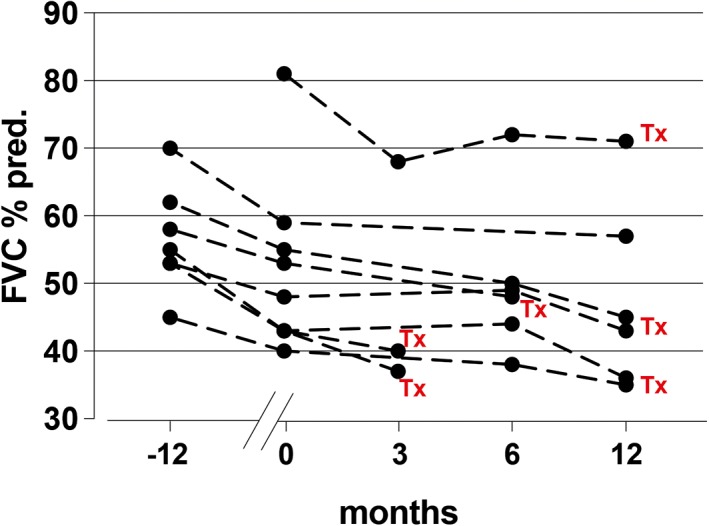
Change in force vital capacity (FVC, % predicted) 12 months before starting nintedanib until lung transplantation.
During and After Lung Transplant
Overall, the transplantation procedures went smoothly for all patients; four patients required intraoperative support with veno‐arterial or veno‐venous extracorporeal membrane oxygenation (ECMO), and one patient needed support after surgery in the intensive care unit (ICU). No patient experienced extraordinary bleeding problems during surgery. The patient in the ICU who was under intraoperative veno‐venous ECMO had blood loss after surgery due to mild thoracic wound bleeding, which was closed by stitches without any haemodynamic consequence. Median time to extubation was 25 h (range 22–120). Median duration of stay in the ICU was 10 days (range 4–33), and median hospital stay was 32 days (range 21–51). There were no problems with postoperative thoracic wound dehiscence in any patient. Postoperative immunosuppression was cyclosporine + azathioprine + corticosteroids in three patients, cyclosporine + mycophenolate mofetil + corticosteroids in four patients, azathioprine + corticosteroids and tacrolimus in one patient, and tacrolimus + mycophenolate mofetil + corticosteroids in one patient. No major side effects due to possible drug interactions with previous antifibrotic therapy were observed in the first month after transplantation. Primary graft dysfunction (PGD) occurred in five patients (in three patients only PGD0; one patient PGD 48 and one patient PGD0,PGD24, PGD48,PGD72). Two patients experienced early postoperative wound infection, and two had acute cellular rejection during the first 6 months after transplantation. Anastomotic airway complications such as dehiscence were not reported after hospital discharge (postoperative day 30). In one patient, airway narrowing was observed 4 months after transplant and required bronchoscopic balloon dilation, which was successful. At the most recent follow up (median 12 months, range 8–12), eight of nine patients were alive and had stable pulmonary function. One patient died from septic shock 7 months after transplant.
Discussion
These data document, for the first time, the usefulness and safety of nintedanib administered prior to lung transplant surgery in patients with IPF. There was no increase in the risk of bleeding and/or anastomotic complications and impaired wound healing during and after lung transplantation despite treatment with nintedanib for up to 30 (mean 13) months prior to surgery. In addition, our findings confirm the ability of nintedanib to slow lung function decline in patients with advanced IPF. Until now, data on the use of antifibrotic therapy in patients undergoing lung transplantation were limited, particularly for nintedanib, which was only used by two of nine patients in a recent case series describing preoperative course and post‐transplant outcomes in IPF patients 16. Leuschner et al. recently reported the outcomes after transplant of 30 IPF patients treated with antifibrotic therapy (7 of 30 patients were treated with nintedanib) compared with 32 untreated IPF patients 17. This single‐centre study showed no increased risk of either bleeding or wound healing in patients treated with antifibrotic therapy compared with untreated IPF patients. Furthermore, postoperative outcomes were observed with equal frequency between treated and untreated IPF patients 17. Differently with previous studies, here, we report the experience of three major transplant centres in Italy focusing on the newer antifibrotic therapy, nintedanib, to allow for more precise observations, which are strictly related to one type of drug.
The attenuation of functional decline in patients with IPF seen in our series is extremely important because this extends the time that patients can be on the transplant waiting list. Waitlist mortality for IPF patients is higher than that of those with other diagnoses, probably due to the clinical behaviour and rapid progression of IPF compared with other diseases 18. In addition, the lack of available donor lungs highlights the need for effective medical therapy in end‐stage IPF patients waiting for a transplant. The average LAS of 35 at the time of transplantation in our cohort may seem fairly low but accurately reflects the general population of IPF patients transplanted over the last 5–10 years.
No serious side effects were noted during nintedanib therapy, confirming the good safety profile of the drug. In particular, it is highly relevant that we found no problem with bleeding or thoracic wound healing in the postoperative period. The PDGF, VEGF, and FGF blockade induced by nintedanib 9 is associated with a theoretical possibility of increased perioperative bleeding risk, impaired postoperative wound healing, and/or bronchial anastomotic complications, and this remains a topic of debate. The potential for such postoperative complications during nintedanib therapy could result in some clinicians stopping treatment before transplant surgery or deferring transplantation completely. Based on our experience, nintedanib appears to be safe in IPF patients undergoing lung transplantation. We did not observe any of the above complications or any deleterious effects of nintedanib on perioperative or medium‐term outcomes.
The short half‐life of nintedanib is relevant in the transplantation setting. In addition, drug interactions with calcineurin inhibitors (secondary to altered hepatic (CYP3A4‐mediated) metabolism resulting in changes in tacrolimus/cyclosporine trough levels) are of concern after lung transplantation. However, nintedanib is mainly a substrate of P‐glycoprotein (P‐gp) and only weakly interferes with CYP3A4. This is probably another explanation for why no major drug interaction‐related side effects were noted in our cohort.
This case series is limited by its retrospective design and small patient numbers, and therefore, inferences about the efficacy and safety of nintedanib in larger groups of IPF patients must be made with caution. Nevertheless, this is the first case series that has considered patients treated with the same antifibrotic, allowing us to draw conclusions regarding the drug–surgical procedure interaction with only one type of drug. Furthermore, IPF is a rare disease, and lung transplantation is infrequent due to chronic donor organ shortages; combining patients from three centres allowed the inclusion of as many patients as possible. Our findings are only generalizable to IPF patients with a disease severity and characteristics similar to those in this case series. In addition, our findings need to be confirmed in a larger patient population.
Overall, the outcomes highlighted in this case series indicate that nintedanib is a safe therapeutic option that acts as a bridge to lung transplantation in patients with IPF.
Disclosure Statements
This study was approved by the Institutional Review Board and was designed in accordance with the Helsinki declaration. Patients gave informed consent for their data to be used for research purposes prior to the surgical procedure.
Acknowledgments
Medical writing assistance was provided by Nicola Ryan and Melanie Gatt (PhD) on behalf of Springer Healthcare Italy, funded by Boehringer Ingelheim, Italy.
Balestro, E. , Solidoro, P. , Parigi, P. , Boffini, M. , Lucianetti, A. , Rea, F. Safety of nintedanib before lung transplant: an Italian case series. Respirology Case Reports. 2018;e00312. https://doi.org/10.1002/rcr2.312
Associate Editor: Trevor Williams
References
- 1. Raghu G, Rochwerg B, Zhang Y, et al. 2015. American Thoracic Society, European Respiratory Society, Japanese Respiratory Society, Latin American Thoracic Association An official ATS/ERS/JRS/ALAT clinical practice guideline: treatment of idiopathic pulmonary fibrosis. An update of the 2011 clinical practice guideline. Am. J. Respir. Crit. Care Med. 192:e3–19. [DOI] [PubMed] [Google Scholar]
- 2. Ley B, Collard HR, and King TE. 2011. Clinical course and prediction of survival in idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 183:431–440. [DOI] [PubMed] [Google Scholar]
- 3. Selman M, Carrillo G, Estrada A, et al. 2007. Accelerated variant of idiopathic pulmonary fibrosis: clinical behaviour and gene expression pattern. PLoS One 2:e482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Boon K, Bailey NW, Yang J, et al. 2009. Molecular phenotypes distinguish patients with relatively stable from progressive idiopathic pulmonary fibrosis (IPF). PLoS One 4:e5134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Balestro E, Calabrese F, Turato G, et al. 2016. Immune inflammation and disease progression in idiopathic pulmonary fibrosis. PLoS One 11:e0154516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Glanville AR, and Estenne M. 2003. Indications, patient selection and timing of referral for lung transplantation. Eur. Respir. J. 22:845–852. [DOI] [PubMed] [Google Scholar]
- 7. Organ Procurement and Transplantation Network (OPTN) , and Scientific Registry of Transplant Recipients (SRTR) . 2012. in Annual data report (OPTN/SRTR 2011). Rockville, MD, Department of Health and Human Services, Health Resources and Services Administration, Healthcare Systems Bureau, Division of Transplantation. [Google Scholar]
- 8. Iyer SN, Gurujeyalakshmi G, and Giri SN. 1999. Effects of pirfenidone on transforming growth factor‐beta gene expression at the transcriptional level in bleomycin hamster model of lung fibrosis. J. Pharmacol. Exp. Ther. 291:367–373. [PubMed] [Google Scholar]
- 9. Wollin L, Wex E, Pautsch A, et al. 2015. Mode of action of nintedanib in the treatment of idiopathic pulmonary fibrosis. Eur. Respir. J. 45:1434–1445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. King TE, Bradford WZ, and Castro‐Bernardini S. 2014. A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N. Engl. J. Med. 370:2083–2092. [DOI] [PubMed] [Google Scholar]
- 11. Taniguchi H, Ebina M, Kondoh Y, et al. 2010. Pirfenidone in idiopathic pulmonary fibrosis. Eur. Respir. J. 35:821–829. [DOI] [PubMed] [Google Scholar]
- 12. Noble PW, Albera C, Bradford WZ, et al. 2011. Pirfenidone in patients with idiopathic pulmonary fibrosis (CAPACITY): two randomised trials. Lancet 377:1760–1769. [DOI] [PubMed] [Google Scholar]
- 13. Richeldi L, du Bois RM, Raghu G, et al. 2014. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N. Engl. J. Med. 370:2071–2082. [DOI] [PubMed] [Google Scholar]
- 14. Richeldi L, Costabel U, Selman M, et al. 2011. Efficacy of a tyrosine kinase inhibitor in idiopathic pulmonary fibrosis. N. Engl. J. Med. 365:1079–1087. [DOI] [PubMed] [Google Scholar]
- 15.FDA approves pirfenidone (Esbriet) and nintedanib (Ofev) for IPF. Media release, 17 Oct 2017. Available at: http://pulmccm.org/main/2014/interstitial‐lung‐disease‐review/fda‐approves‐pirfenidone‐esbriet‐nintedanib‐ofev‐ipf/ (accessed 18 July 2017).
- 16. Delanote I, Wuyts WA, Yserbyt J, et al. 2016. Safety and efficacy of bridging to lung transplantation with antifibrotic drugs in idiopathic pulmonary fibrosis: a case series. BMC Pulm. Med. 16:156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Leuschner G, Stocker F, Veit T, et al. 2018. Outcome of lung transplantation in idiopathic pulmonary fibrosis with previous anti‐fibrotic therapy. J. Heart Lung Transplant. 37:268–274. [DOI] [PubMed] [Google Scholar]
- 18. Kistler KD, Nalysnyk L, Rotella P, et al. 2014. Lung transplantation in idiopathic pulmonary fibrosis: a systematic review of the literature. BMC Pulm. Med. 14:139. [DOI] [PMC free article] [PubMed] [Google Scholar]