

Summary
Nasu-Hakola disease (NHD) is a rare autosomal recessive disorder, characterized by progressive presenile dementia and formation of multifocal bone cysts, caused by genetic mutations of either triggering receptor expressed on myeloid cells 2 (TREM2) or TYRO protein tyrosine kinase binding protein (TYROBP), alternatively named DNAX-activation protein 12 (DAP12), both of which are expressed on microglia in the brain and form the receptor-adaptor complex that chiefly recognizes anionic lipids. TREM2 transmits the signals involved in microglial survival, proliferation, chemotaxis, and phagocytosis. A recent study indicated that a loss of TREM2 function causes greater amounts of amyloid-β (Aβ) deposition in the hippocampus of a mouse model of Alzheimer's disease (AD) owing to a dysfunctional response of microglia to amyloid plaques, suggesting that TREM2 facilitates Aβ clearance by microglia. TREM2/DAP12-mediated microglial response limits diffusion and toxicity of amyloid plaques by forming a protective barrier. However, the levels of Aβ deposition in postmortem brains of NHD, where the biological function of the TREM2/DAP12 signaling pathway is completely lost, remain to be investigated. By immunohistochemistry, we studied the expression of Aβ and phosphorylated tau (p-tau) in the frontal cortex and the hippocampus of five NHD cases. Although we identified several small Aβ-immunoreactive spheroids, amyloid plaques were almost undetectable in NHD brains. We found a small number of p-tau-immunoreactive neurofibrillary tangle (NFT)-bearing neurons in NHD brains. Because AD pathology is less evident in NHD than the full-brown AD, it does not play an active role in the development of NHD.
Keywords: Alzheimer's disease, amyloid-β, Nasu-Hakola disease, phosphorylated tau
1. Introduction
Nasu-Hakola disease (NHD), also designated polycystic lipomembranous osteodysplasia with sclerosing leukoencephalopathy (PLOSL; OMIM 221770), is a rare autosomal recessive disorder, characterized by progressive presenile dementia and formation of multifocal bone cysts, caused by genetic mutations of either triggering receptor expressed on myeloid cells 2 (TREM2) or TYRO protein tyrosine kinase binding protein (TYROBP), alternatively named DNAX-activation protein 12 (DAP12), both of which are expressed on microglia in the brain (1). Clinically, the patients with NHD show recurrent bone fractures during the third decade of life, and a frontal lobe syndrome during the fourth decade of life, and progressive dementia and death until the fifth decade of life (2). Pathologically, the brains of NHD patients exhibit extensive demyelination designated leukoencephalopathy, astrogliosis, accumulation of axonal spheroids, and remarkable activation of microglia predominantly in the white matter of frontal and temporal lobes and the basal ganglia (3). TREM2, expressed exclusively on microglia in the brain, serves as a receptor for anionic lipids, lipoproteins and apolipoproteins (4,5). TREM2 forms a complex with DAP12, a signaling adaptor having an immunoreceptor tyrosine-based activation motif (ITAM) capable of recruiting the protein tyrosine kinase Syk that transduces a variety of downstream signals (6). TREM2 transmits microglial signals involved in survival, proliferation, chemotaxis, and phagocytosis (7).
Alzheimer's disease (AD) is characterized by the hallmark pathology comprised of widespread amyloid-β (Aβ) deposition, formation of neurofibrillary tangles (NFTs) composed of abnormally phosphorylated tau (p-tau), extensive neurodegeneration, and profound activation of microglia in the brain (8). Recent studies identified rare genetic variants of TREM2, such as R47H and R62H, closely associated with an increased risk for development of AD (9).
A recent study showed that TREM2 deficiency generates greater amounts of Aβ deposition in the hippocampus of 5XFAD mice, a mouse model of AD, due to a dysfunctional response of microglia to amyloid plaques, suggesting that TREM2 facilitates Aβ clearance by microglia (4). TREM2 interacts with fibrillar Aβ decorated with anionic and zwitterionic lipids (4). Aβ-lipoprotein complexes are efficiently taken up by microglia, depending on TREM2 (5). TREM2 deficiency induces apoptosis of microglia and reduces recruitment of microglia around Aβ plaques in the brains of mouse models of AD (4). Furthermore, microglial processes enriched in TREM2 tightly surround early amyloid fibrils and plaques, and promote their compaction and insulation (10). In TREM2- or DAP12-deficient mouse models of AD, microglia showed a markedly reduced ability to envelope amyloid deposits, leading to an increase in less compact and more diffuse plaques associated with greater neuritic damage (10). These observations suggest that TREM2/DAP12-mediated microglial response limits diffusion and toxicity of amyloid plaques by forming a protective barrier (10,11). However, at present, the levels of Aβ deposition in postmortem NHD brains, where the biological function of TREM2/DAP12 signaling pathway is completely lost, remain unknown. A previous study reported a 48-year-old man of NHD with numerous senile plaques and neurofibrillary tangles throughout the cerebral cortex (12), suggesting that an impaired TREM2/DAP12 signaling function facilitates Aβ accumulation in the human brain. In the present study by immunohistochemistry, we investigate the expression of Aβ and p-tau in NHD brains to clarify whether the Alzheimer's disease pathology is augmented in NHD.
2. Materials and Methods
2.1. Human brain tissues
The brain autopsies were performed at the National Center Hospital, National Center of Neurology and Psychiatry (NCNP), Japan and affiliated hospitals of Research Resource Network (RRN), Japan. The comprehensive examination by an established neuropathologist (YS) validated the pathological diagnosis. In all cases, written informed consent was obtained. The Ethics Committee of the NCNP for the Human Brain Research and the Human Research Ethics Committee of the Meiji Pharmaceutical University (MPU) approved the present study.
For immunohistochemical studies, serial sections of the frontal cortex and the hippocampus were prepared from five AD patients, composed of a 68-year-old woman (AD1), a 68-year-old woman (AD3), a 56-year-old man (AD4), a 59-year-old man (AD5), and an 80-year-old man (AD8) and five NHD patients, composed of a 42-year-old man (NHD1), a 48-year-old woman (NHD2), a 44-year-old man (NHD3), a 32-year-old woman (NHD4), and a 38-year-old man (NHD5). The homozygous mutation of a single base deletion of 141G (c.141delG) in exon 3 of DAP12 was identified in NHD1, NHD2, and NHD5, while the genetic analysis was not performed in NHD3 or NHD4. All AD cases were satisfied with the Consortium to Establish a Registry for Alzheimer's Disease (CERAD) criteria for diagnosis of definite AD (13). They were categorized into the stage C of amyloid deposition and the stage VI of neurofibrillary degeneration, following the Braak's staging (14).
2.2. Immunohistochemistry
After deparaffination, tissue sections were heated in 10 mM citrate sodium buffer, pH 6.0 by autoclave at 110°C for 15 min in a temperature-controlled pressure chamber (Biocare Medical, Pacheco, CA, USA). Then, they were treated at room temperature (RT) for 5 min with formic acid before labeling with anti-Aβ antibody. They were incubated with phosphate-buffered saline (PBS) containing 10% normal goat serum at RT for 15 min to block non-specific staining, followed by incubation in a moist chamber at 4°C overnight with mouse monoclonal anti-Aβ peptide antibody at a concentration of 1 µg/mL (12B2; Immunobiological Laboratories, Gunma, Japan) that reacts with Aβ40, Aβ42, and Aβ43 or mouse monoclonal anti-phosphorylated tau (Ser202, Thr205) antibody at 0.025 µg/mL (AT8; ThermoFisher Scientific, Waltham, MA, USA). After washing with PBS, tissue sections were incubated at RT for 30 min with alkaline phosphatase (AP)-conjugated secondary antibody (Nichirei, Tokyo, Japan), followed by exposure to Warp Red chromogen (Biocare Medical). For double immunolabeling, following heat treatment, tissue sections were treated with 3% hydrogen peroxide-containing water to block the endogenous peroxidase activity, and immunolabeled at 4°C overnight with rabbit polyclonal anti-Iba1 antibody at 0.5 µg/mL (Wako Pure Chemical, Tokyo, Japan) for a marker specific for microglia. They were incubated at RT for 30 min with horseradish peroxidase (HRP)-conjugated secondary antibody (Nichirei), followed by exposure to diaminobenzidine tetrahydrochloride (DAB) substrate (Vector, Burlingame, CA, USA). The tissue sections were processed for a counterstain with hematoxylin. Negative controls underwent all the steps except for exposure to the primary antibody.
3. Results and Discussion
In mouse models of AD, the loss of function of TREM2 increases Aβ plaque burden possibly through decreased phagocytic clearance of Aβ by microglia (4). Loss of TREM2 function reduces the ability of microglia to engulf Aβ (15). AD patients with the TREM2 variant of R47H showed fewer microglia surrounding plaques, increased numbers of filamentous non-compacted plaques, and more p-tau-positive neurites around plaques (10). In all NHD cases, we found several small Aβ-immunoreactive deposits, and some of them might represent axonal spheroids located chiefly in the white matter of the frontal cortex and the hippocampus (Figure 1a–c). They were spherical in shape with smooth margins devoid of the core. Iba1+ microglia occasionally contacted these spheroids (Figure 1c). In support of these observations, a previous study showed the persistent accumulation of Aβ42 in axonal spheroids in a rat model of traumatic injury (16). In contrast, amyloid plaques, compact or diffuse, and amyloid angiopathy were almost undetectable in any cases. Aβ-immunoreactive spheroids consisted of 4–22 spots/20 fields in the frontal cortex and 2–16 spots/20 fields in the hippocampus of NHD brains under microscopic examination at a magnification of 200 ×. In contrast, the deposition of Aβ was much extensive in AD brains (Figure 1d). The omission of the primary antibody did not show any positive reactions. In NHD brains except for NHD5, we identified a small cluster of NFT-bearing neurons labeled by anti-p-tau antibody located predominantly in the hippocampus (Figure 2a–c). NFT-bearing neurons consisted of 0–11 neurons/20 fields in the frontal cortex and 0–34 neurons/20 fields in the hippocampus of NHD brains under microscopic examination at a magnification of 200 ×. We found a trend for the age-dependent increase in p-tau-immunoreactive NFT-bearing neurons. In contrast, numerous p-tau-immunoreactive neuronal deposits, composed of NFTs and dystrophic neurites, were observed in AD brains (Figure 2d). These results indicated that the loss of function of TREM2/DAP12 signaling pathway does not accelerate AD pathology in NHD brains.
Figure 1.
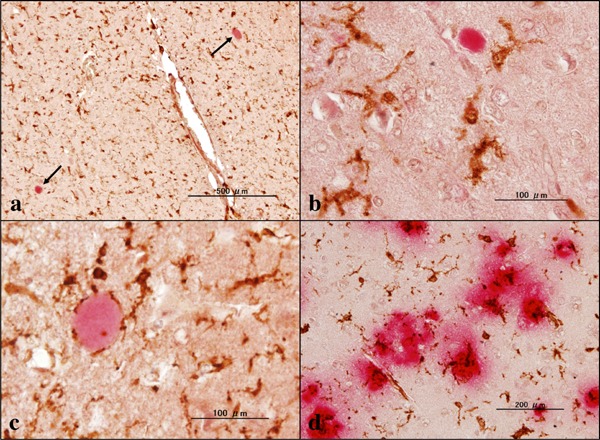
Aβ immunoreactivity in NHD and AD brains. (a) the hippocampus white matter of NHD (NHD4), Aβ (red) and Iba1 (brown), spheroids (arrows), (b) the frontal lobe white matter of NHD (NHD5), Aβ (red) and Iba1 (brown), spheroid, (c) the hippocampus white matter of NHD (NHD2), Aβ (red) and Iba1 (brown), spheroid, and (d) the frontal cortex of AD (AD8), Aβ (red) and Iba1 (brown), amyloid plaques.
Figure 2.
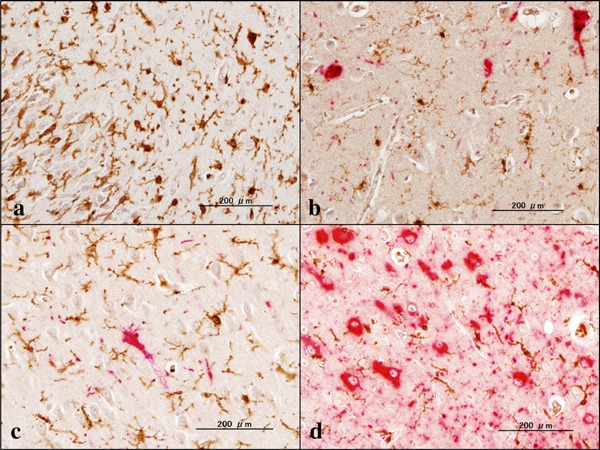
P-tau immunoreactivity in NHD and AD brains. (a) the hippocampus of NHD (NHD5), p-tau (red) and Iba1 (brown), no NFT, (b) the hippocampus of NHD (NHD1), p-tau (red) and Iba1 (brown), several NFTs, (c) the hippocampus of NHD (NHD2), p-tau (red) and Iba1 (brown), one NFT, and (d) the hippocampus of AD (AD5), p-tau (red) and Iba1 (brown), numerous NFTs.
The discrepancy between TREM2-deficient AD mice showing greater amounts of Aβ deposition in the hippocampus (4) and NHD patients not exhibiting the acceleration of Aβ deposition in the frontal cortex and the hippocampus is attributable to a difference in species, ages, and genetic backgrounds. Plaque accumulation is exacerbated at later ages in a TREM2 knockout AD mouse model (17). In AD patients, Aβ plaques appear a decade or two before clinical symptoms of AD (18). In the present study, NHD patients are 32- to 48-year-old. Therefore, they could be affected by the early AD pathology according to their ages. Importantly, by using florbetapir-amyloid-positron emission tomography (PET), a recent study demonstrated extensive Aβ deposition in the grey matter of the inferior frontal and occipital lobes of a 39-year-old Italian NHD woman with a homozygous Q33X mutation in TREM2, suggesting the existence of overlapping pathogenic mechanisms between NHD and AD (19). In the present study, three cases of NHD showed the homozygous mutation of c.141delG in exon 3 of DAP12. A recent study showed that deficiency of DAP12 does not modify the number and size of Aβ plaque deposition in the prefrontal cortex and the hippocampus of APP/PSEN1 mice, although DAP12 deficiency reduces plaque compaction, microglial clustering, and phagocytosis (20). Furthermore, phosphorylation of tau is attenuated in female APP/ PSEN1 mice with loss of DAP12. In contrast, silencing of brain TREM2 exacerbates tau pathology in P301S tau transgenic mice, associated with neuroinflammation-induced overactivation of tau kinases, such as cyclin dependent kinase 5 (CDK5) and glycogen synthase kinase 3 beta (GSK3B) (21). Thus, the absence of either DAP12 or TREM2 produces apparently opposing effects on progression of AD pathology in mouse models of AD.
In summary, we identified no obvious Aβ plaques and a small number of p-tau-immunoreactive NFT-bearing neurons in NHD brains. Because AD pathology is less evident in NHD than the full-brown AD, it does not play an active role in the development of NHD.
Acknowledgements
The authors thank Drs. Kenji Jinnai, Nobutaka Arai, Kiyotaka Nakamagoe, Nobutaka Motohashi, and Saburo Yagishita for providing us brain samples. This work was supported by grants from the Research on Intractable Diseases, entitled “Clinicopathological and genetic studies of Nasu-Hakola disease” (H21-Nanchi-Ippan-201; H22-Nanchi-Ippan-136), the Ministry of Health, Labour and Welfare of Japan, and grants from the JSPS KAKENHI (C25430054 and 16K07043) and the Dementia Drug Resource Development Center (DRC) project (S1511016), the Ministry of Education, Culture, Sports, Science and Technology (MEXT), Japan.
References
- 1. Klünemann HH, Ridha BH, Magy L, Wherrett JR, Hemelsoet DM, Keen RW, De Bleecker JL, Rossor MN, Marienhagen J, Klein HE, Peltonen L, Paloneva J. The genetic causes of basal ganglia calcification, dementia, and bone cysts: DAP12 and TREM2. Neurology. 2005; 64:1502-1507. [DOI] [PubMed] [Google Scholar]
- 2. Bianchin MM, Capella HM, Chaves DL, Steindel M, Grisard EC, Ganev GG, da Silva JP, Júnior, Neto Evaldo S, Poffo MA, Walz R, Carlotti CG, Júnior, Sakamoto AC. Nasu-Hakola disease (polycystic lipomembranous osteodysplasia with sclerosing leukoencephalopathy - PLOSL): A dementia associated with bone cystic lesions. From clinical to genetic and molecular aspects. Cell Mol Neurobiol. 2004; 24:1-24. [DOI] [PubMed] [Google Scholar]
- 3. Satoh J, Tabunoki H, Ishida T, Yagishita S, Jinnai K, Futamura N, Kobayashi M, Toyoshima I, Yoshioka T, Enomoto K, Arai N, Arima K. Immunohistochemical characterization of microglia in Nasu-Hakola disease brains. Neuropathology. 2011; 31:363-375. [DOI] [PubMed] [Google Scholar]
- 4. Wang Y, Cella M, Mallinson K, Ulrich JD, Young KL, Robinette ML, Gilfillan S, Krishnan GM, Sudhakar S, Zinselmeyer BH, Holtzman DM, Cirrito JR, Colonna M. TREM2 lipid sensing sustains the microglial response in an Alzheimer's disease model. Cell. 2015; 160:1061-1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Yeh FL, Wang Y, Tom I, Gonzalez LC, Sheng M. TREM2 binds to apolipoproteins, including APOE and CLU/APOJ, and thereby facilitates uptake of amyloid-beta by microglia. Neuron. 2016; 91:328-340. [DOI] [PubMed] [Google Scholar]
- 6. Xing J, Titus AR, Humphrey MB. The TREM2-DAP12 signaling pathway in Nasu-Hakola disease: A molecular genetics perspective. Res Rep Biochem. 2015; 5:89-100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Jay TR, von Saucken VE, Landreth GE. TREM2 in neurodegenerative diseases. Mol Neurodegener. 2017; 12:56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Serrano-Pozo A, Frosch MP, Masliah E, Hyman BT. Neuropathological alterations in Alzheimer disease. Cold Spring Harb Perspect Med. 2011; 1:a006189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Guerreiro R, Wojtas A, Bras J, et al. TREM2 variants in Alzheimer's disease. N Engl J Med. 2013; 368:117-127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Yuan P, Condello C, Keene CD, Wang Y, Bird TD, Paul SM, Luo W, Colonna M, Baddeley D, Grutzendler J. TREM2 haplodeficiency in mice and humans impairs the microglia barrier function leading to decreased amyloid compaction and severe axonal dystrophy. Neuron. 2016; 90:724-739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wang Y, Ulland TK, Ulrich JD, et al. TREM2-mediated early microglial response limits diffusion and toxicity of amyloid plaques. J Exp Med. 2016; 213:667-675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bird TD, Koerker RM, Leaird BJ, Vlcek BW, Thorning DR. Lipomembranous polycystic osteodysplasia (brain, bone, and fat disease): A genetic cause of presenile dementia. Neurology. 1983; 33:81-86. [DOI] [PubMed] [Google Scholar]
- 13. Mirra SS, Heyman A, McKeel D, Sumi SM, Crain BJ, Brownlee LM, Vogel FS, Hughes JP, van Belle G, Berg L. The Consortium to Establish a Registry for Alzheimer's Disease (CERAD). Part II. Standardization of the neuropathologic assessment of Alzheimer's disease. Neurology. 1991; 41:479-486. [DOI] [PubMed] [Google Scholar]
- 14. Braak H, Alafuzoff I, Arzberger T, Kretzschmar H, Del Tredici K. Staging of Alzheimer disease-associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol. 2006; 112:389-404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kleinberger G, Yamanishi Y, Suárez-Calvet M, et al. TREM2 mutations implicated in neurodegeneration impair cell surface transport and phagocytosis. Sci Transl Med. 2014; 6:243ra86. [DOI] [PubMed] [Google Scholar]
- 16. Iwata A, Chen XH, McIntosh TK, Browne KD, Smith DH. Long-term accumulation of amyloid-β in axons following brain trauma without persistent upregulation of amyloid precursor protein genes. J Neuropathol Exp Neurol. 2002; 61:1056-1068. [DOI] [PubMed] [Google Scholar]
- 17. Jay TR, Hirsch AM, Broihier ML, Miller CM, Neilson LE, Ransohoff RM, Lamb BT, Landreth GE. Disease progression-dependent effects of TREM2 deficiency in a mouse model of Alzheimer's disease. J Neurosci. 2017; 37:637-647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Jack CR, Jr, Knopman DS, Jagust WJ, Petersen RC, Weiner MW, Aisen PS, Shaw LM, Vemuri P, Wiste HJ, Weigand SD, Lesnick TG, Pankratz VS, Donohue MC, Trojanowski JQ. Tracking pathophysiological processes in Alzheimer's disease: An updated hypothetical model of dynamic biomarkers. Lancet Neurol. 2013; 12:207-216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ghezzi L, Carandini T, Arighi A, et al. Evidence of CNS β-amyloid deposition in Nasu-Hakola disease due to the TREM2 Q33X mutation. Neurology. 2017; 89:2503-2505. [DOI] [PubMed] [Google Scholar]
- 20. Haure-Mirande JV, Audrain M, Fanutza T, Kim SH, Klein WL, Glabe C, Readhead B, Dudley JT, Blitzer RD, Wang M, Zhang B, Schadt EE, Gandy S, Ehrlich ME. Deficiency of TYROBP, an adapter protein for TREM2 and CR3 receptors, is neuroprotective in a mouse model of early Alzheimer's pathology. Acta Neuropathol. 2017; 134:769-788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Jiang T, Tan L, Zhu XC, Zhou JS, Cao L, Tan MS, Wang HF, Chen Q, Zhang YD, Yu JT. Silencing of TREM2 exacerbates tau pathology, neurodegenerative changes, and spatial learning deficits in P301S tau transgenic mice. Neurobiol Aging. 2015; 36:3176-3186. [DOI] [PubMed] [Google Scholar]