

Abstract
Animal Toll-like receptors (TLRs) have evolved through a pattern of duplication and divergence. Whereas mammalian TLRs directly recognize microbial ligands, Drosophila Tolls bind endogenous ligands downstream of both developmental and immune signaling cascades. Here, we find that most Toll genes in Drosophila evolve slowly with little gene turnover (gains/losses), consistent with their important roles in development and indirect roles in microbial recognition. In contrast, we find that the Toll-3/4 genes have experienced an unusually rapid rate of gene gains and losses, resulting in lineage-specific Toll-3/4s and vastly different gene repertoires among Drosophila species, from zero copies (e.g., D. mojavensis) to nineteen copies (e.g., D. willistoni). In D. willistoni, we find strong evidence for positive selection in Toll-3/4 genes, localized specifically to an extracellular region predicted to overlap with the binding site of Spätzle, the only known ligand of insect Tolls. However, because Spätzle genes are not experiencing similar selective pressures, we hypothesize that Toll-3/4s may be rapidly evolving because they bind to a different ligand, akin to TLRs outside of insects. We further find that most Drosophila Toll-3/4 genes are either weakly expressed or expressed exclusively in males, specifically in the germline. Unlike other Toll genes in D. melanogaster, Toll-3, and Toll-4 have apparently escaped from essential developmental roles, as knockdowns have no substantial effects on viability or male fertility. Based on these findings, we propose that the Toll-3/4 genes represent an exceptionally rapidly evolving lineage of Drosophila Toll genes, which play an unusual, as-yet-undiscovered role in the male germline.
Keywords: immunity; gene duplication; phylogenomics; positive selection; gene gain and loss, Toll, Toll-like receptor, TLR
Introduction
Toll and Toll-like genes encode type I transmembrane receptors that are critical for both innate immunity and development in animal genomes (Valanne et al. 2011; Lindsay and Wasserman 2014; De Nardo 2015). Across the animal kingdom, Toll receptor repertoires have diversified by a series of gene duplications followed by neofunctionalization, subfunctionalization, or pseudogenization (Roach et al. 2005; Temperley et al. 2008; Huang et al. 2011; Buckley and Rast 2012). This diversification has allowed the Toll superfamily of proteins to recognize a variety of extracellular and endosomal stimuli, triggering a transcriptional response by NF-κB proteins. Within mammals, the functions of Toll-like receptors (TLRs) have been extensively studied in the context of innate immunity. Each receptor is thought to bind a distinct set of conserved microbial molecules. For example, TLR4 recognizes bacterial lipopolysaccharide (Poltorak et al. 1998; Hoshino et al. 1999), whereas TLR11 recognizes profilin from protozoa (Yarovinsky et al. 2005). Similarly, TLR3 and TLR7 recognize incoming viruses within endosomes via recognition of double-stranded DNA or single-stranded DNA, respectively (Alexopoulou et al. 2001; Heil et al. 2004). Some TLRs present in most mammals have been lost in the human lineage, including the TLR11/12 family, altering the repertoire of microbial recognition pathways present in humans (Roach et al. 2005).
While TLRs play many important roles in mammals, this superfamily of receptors was first discovered in Drosophila, with the prototypical Toll (or Toll-1) receptor (Anderson et al. 1985). Within insects, the Toll-1 pathway responds to Gram-positive bacterial and fungal pathogens, while also playing critical roles in embryonic development (Lindsay and Wasserman 2014). The mechanism of microbial recognition differs significantly between mammalian and insect Toll pathways (Valanne et al. 2011). Whereas mammalian TLRs directly bind to microbial molecules to initiate signaling, insect Toll-1 recognizes these cues indirectly through an extracellular proteolytic cascade that results in the processing of the Toll-1 endogenous ligand, Spätzle, to its active form (Morisato and Anderson 1994). Processed, extracellular Spätzle ligands are then bound by Toll-1 to propagate the signal inside of the cell.
The diversity of functions encoded by the Toll-like genes in mammals has spurred evolutionary and functional analyses of these proteins in diverse animal genomes, including insects. Many insect Toll paralogs are ancient, with orthologs present in distantly related arthropods (Christophides et al. 2002; Zou et al. 2007; Gerardo et al. 2010; Palmer and Jiggins 2015). There have also been lineage-specific duplications of Toll family genes as well as lineage-specific losses (Christophides et al. 2002; Zou et al. 2007; Gerardo et al. 2010; Palmer and Jiggins 2015). Some of these lineage-specific Tolls are important for pathogen recognition and host fitness. For instance, Toll-11 paralogs found only in mosquitos can protect the host from Plasmodium falciparum infection (Redmond et al. 2015). Nevertheless, most lineage-restricted Tolls remain poorly studied. Because insect Tolls have diversified in parallel evolutionary trajectories to mammalian TLRs, experimentally tractable insects like Drosophila are promising model systems to understand how Toll families diversify their signaling repertoires over time.
Among insects, the Toll repertoire has been best analyzed in Drosophila melanogaster, which has nine Toll paralogs (named Toll-1 through Toll-9) (Tauszig et al. 2000). Each of these paralogs encodes proteins with an N-terminal region comprised of leucine-rich repeats (LRRs) and cysteine-rich LRR variants (termed LRRNT and LRRCT), a central transmembrane domain, and a C-terminal, cytoplasmic TIR domain involved in signal transduction. Most of the Drosophila Toll paralogs have been less well characterized than the originally identified Toll-1 gene. Nevertheless, experimental strategies involving characterization of mutant, knockdown and over-expression phenotypes have revealed that most Drosophila Toll paralogs (Toll-1, -2, -5, -6, -7, -8, and -9) participate in some aspect of development (Anderson et al. 1985; Tauszig et al. 2000; Yagi et al. 2010). Four Toll genes (Toll-1, -5, -7, and -9) have also been implicated in immune signaling (Lemaitre et al. 1996; Tauszig et al. 2000; Ooi et al. 2002; Bettencourt et al. 2004; Nakamoto et al. 2012). In contrast, no functions or phenotypes have been ascribed to the Drosophila Toll-3 (also known as MstProx) or Toll-4 genes.
The Toll gene family has also been studied to identify the selective pressures that have acted upon Drosophila immune genes. These approaches can pinpoint particular paralogs and even particular amino acids that are rapidly evolving. Some of these broad surveys have identified weak signatures of positive selection in Toll-1 (Schlenke and Begun 2003; Sackton et al. 2007; Han et al. 2013) and Toll-5 (Schlenke and Begun 2003), but these signatures were not always robust enough to pass statistical tests (Sackton et al. 2007; Obbard et al. 2009; Han et al. 2013). Analyses of Toll-3 and Toll-4 have demonstrated similar weak signatures of positive selection by a pairwise McDonald–Kreitman test (Obbard et al. 2009) or maximum likelihood methods (Han et al. 2013). However, several of these previous studies of Drosophila immune gene evolution excluded Toll-3 and Toll-4 genes or analyzed only a small fraction of the alignment due to difficulties in identifying and/or aligning orthologs (Sackton et al. 2007; Han et al. 2013).
Here, we investigate the evolution of Toll-3 and Toll-4 genes via phylogenomic analyses of Drosophila genomes. Our analyses confirm and extend previous findings that most Toll paralogs are generally present in a single copy in Drosophila genomes (Heger and Ponting 2007) and largely evolve under purifying selection (Sackton et al. 2007; Obbard et al. 2009; Han et al. 2013). In contrast, we find that the Toll-3/4 lineage evolves rapidly, undergoing recurrent episodes of gene loss and gene gain, as well as strong positive selection in some Drosophila species. As a result of this dynamic evolution, the number of Toll-3/4 genes in Drosophila genomes varies from zero to 19 (including pseudogenes). Although they remain functionally uncharacterized, we find that many of these lineage-specific Toll-3/4 paralogs are expressed in males, specifically in testes. Additionally, unlike other Toll genes, Toll-3/4s have apparently escaped from essential developmental roles, as knockdowns have no substantial effects on D. melanogaster viability or male fertility. We further divide Toll genes of insects into 3 evolutionary classes, with genes like Toll-3/4s representing the most rapidly evolving class. Based on our findings, we hypothesize that Toll-3/4 genes are a highly unusual class of Toll genes, participating in a still-undiscovered function in the male germline.
Results
The Prolific Toll-3/4 Gene Family of Drosophila
To compile a detailed compendium of all Toll paralogs, we first identified all Tolls from a well-assembled, well-annotated set of 12 Drosophila species genomes (Drosophila 12 Genomes Consortium et al. 2007), using blastn and tblastn searches with D. melanogaster Toll-1 to -9 as queries. We extracted the amino acid sequences from the TIR domain of all Toll proteins, generated multiple alignments, and then carried out phylogenetic analyses (fig. 1A;supplementary fig. S1, Supplementary Material online). In the resulting phylogeny, we found that all of the Drosophila Toll homologs could be unambiguously assigned to one of 8 clades. For seven of these clades (Toll-1, -2, -5, -6, -7, -8, and -9), Toll genes clustered tightly at the ends of long branches. Consistent with previous findings, this tree suggested that the TIR domains diverged from each other long before the origin of the Drosophila genus, and that they have been strongly conserved among the 12 Drosophila species since that time. The majority of Toll genes also rarely duplicated, remaining at single copy in most species. Exceptions were Toll-2 which has 2 copies in D. sechellia, Toll-8 which has 2 copies in D. grimshawi, and Toll-9, which has 2 copies in the D. yakuba genome (fig. 1B;supplementary fig. S1, Supplementary Material online).
Fig. 1.

Diversity of Toll family proteins in 12 Drosophila species. (A) Phylogeny constructed from the TIR domain from 135 Toll family genes using PhyML. Colored branches are those with unambiguous D. melanogaster orthologs, as defined by tree topology and synteny. Selected internal nodes are labeled with posterior probability, and all nodes with support <0.5 have been collapsed. Root based on Gerardo et al. (2010). (B) Number of Toll subfamily genes+pseudogenes. Number of Toll-3/4 genes is highlighted in red. Loci identified by blastn and tblastn and assigned to Toll family based on alignments, phylogeny, and synteny. Dmel, D. melanogaster; Dsim, D. simulans; Dsec, D. sechellia; Dere, D. erecta; Dyak, D. yakuba; Dana, D. ananassae; Dpse, D. pseudoobscura; Dper, D. persimilis; Dwil, D. willistoni, Dvir, D. virilis; Dmoj, D. mojavensis; Dgri, D. grimshawi.
In contrast, we found that Toll-3 and Toll-4 genes did not appear in distinct clades, but were instead both found within a single clade that we term the “Toll-3/4 clade”, which was most closely related to the Toll-1 and Toll-5 clades (fig. 1A). In addition to the annotated Toll-3 and Toll-4 orthologs, we identified many Toll-3/4 genes from other Drosophila species that do not have direct orthologs in the D. melanogaster genome, but are instead related to the Toll-3/Toll-4 common ancestor. We refer to these genes as “Toll-3/4 genes” to disambiguate them from the bona fide Toll-3 and Toll-4 orthologs we find in some species more closely related to D. melanogaster. In some lineages, the Toll-3/4 family is greatly expanded, with as many as 19 different Toll-3/4 genes or pseudogenes in the genome (fig. 1B). This implies that some Drosophila species encode more Toll-3/4 genes than the total repertoire of all other Toll genes combined. This pattern of prolific gains is also accompanied by unusual losses. For example, we were unable to identify any Toll-3/4 genes from D. mojavensis or D. grimshawi, despite finding orthologs from the seven remaining Toll families in these genomes. However, we were able to identify two Toll-3/4 genes from D. virilis, implying that Toll-3/4s were present in the last common ancestor of the 12 species. To confirm the loss of Toll-3/4 genes in D. mojavensis and D. grimshawi, we used the virilis Toll-3/4 genes as queries in tblastn searches of these two genomes, but the only hits were to other Toll genes (e.g. Toll-1, -2, and -5 to -9). Similarly, we queried the genomes of several more distantly related fly species—Drosophila busckii, Bactrocera oleae, Hermiteia illucens, Phortica variegata, Sarcophaga bullata, Musca domestica, and Glossina morsitans morsitans—to determine when Toll-3/4s arose, but did not identify any additional homologs. We therefore infer that the Toll-3/4 family must have arisen early in the evolution of the Drosophila genus and has subsequently been independently lost at least twice during Drosophila evolution—from both the D. mojavensis and the D. grimshawi lineages.
Extensive Turnover and Independent Diversification of Toll-3/4s
We confirmed and extended our phylogenetic findings by examining the syntenic location of each of the identified Toll genes within the 12 Drosophila genomes. We found that most Tolls are present in a shared syntenic location in each of the 12 Drosophila genomes with only occasional duplication to a distant genomic locus (fig. 2; supplementary fig. S2, Supplementary Material online). In contrast, we found that the Toll-3/4 family genes are found in numerous, unshared syntenic loci (fig. 2). For instance, we found that the Toll-4 gene is in the same syntenic location in species as divergent as D. melanogaster and D. ananassae but not in D. pseudoobscura or more distantly related species. This implies that Toll-4 originated in its syntenic location in the common ancestor of D. melanogaster and D. ananassae, after the divergence from D. pseudoobscura. Curiously, D. virilis also has a Toll-3/4 gene adjacent to the melanogaster Toll-4 syntenic locus, raising the possibility that the D. virilis Toll-3/4 gene has been retained in its ancestral location since prior to the D. melanogaster/D. virilis divergence.
Fig. 2.

Toll-3/4 genes are rapidly gained and lost from multiple syntenic loci. The Toll-5 (Tehao) locus is representative of most Toll paralogs that are conserved in their syntenic locus across Drosophila spp. In contrast, Toll-3/4 genes are gained and lost from multiple genomic regions. Only those loci containing more than one Toll-3/4 gene across the 12 species are depicted. Neighboring genes used to mark a shared syntenic locus are colored and labeled. Dotted arrows indicate regions where a gene is encoded within an intron and/or on the opposite strand of another gene. "Distant locus" refers to genomic regions that are on different scaffolds or are more than 1.5 Mb away in order to show loci that have moved during chromosome rearrangements.
Toll-3 (also called MstProx) appears to be younger and present in even fewer species than Toll-4. Most D. melanogaster have an intact Toll-3, but there is also a segregating premature stop codon allele that is present in ∼2% of lines in the Drosophila Genetic Reference Panel collection (Mackay et al. 2012), as well as in the D. melanogaster reference genome. We found intact Toll-3 genes in the same, syntenic locus in D. melanogaster, D. simulans, and D. sechellia but also identified clear signs of pseudogenization in D. erecta and D. yakuba, where the homologous sequences were disrupted by many SNPs and deletions, resulting in numerous stop codons in all reading frames. Consequently, these loci were easily identified by their nucleotide similarity (blastn) but not by their translated sequences (tblastn). We found no traces of the Toll-3 gene in the syntenic locus of more diverged species. Thus, Toll-3/4 turnover is observable over even short time scales, reflecting an ancient and ongoing dynamic of gene gain and loss.
To understand Toll-3/4 evolutionary dynamics with better resolution, we constructed a phylogenetic tree based on an amino acid alignment of Toll-3/4 proteins from 12 Drosophila species and additional melanogaster group species (fig. 3). We obtained these sequences by blast queries to published genomes. We also performed PCR to obtain additional Toll-3/4 sequences from species whose genomes have not been sequenced (see Materials and Methods). The maximum-likelihood based phylogeny revealed that the Toll-3/4 genes have diversified independently—forming separate, distinct clades—within the melanogaster group, the obscura group, the willistoni group and the virilis group. Outside of the melanogaster group, we found no direct orthologs of Toll-3 or Toll-4, consistent with our analyses of the syntenic loci (fig. 2). Furthermore, although most Drosophila species possess Toll-3/4 family genes, only closely related species share individual Toll-3/4 orthologs, and many Drosophila lineages have private, species-specific repertoires of these genes. For example, within the melanogaster group, we found that D. yakuba encodes four Toll-4-like genes (fig. 3). In contrast, we found D. kikkawai has no Toll-4 ortholog but instead four Toll-3-like genes, including an ortholog of D. melanogaster Toll-3.
Fig. 3.

Phylogeny based on amino acid alignment of 61 Toll-3/4 genes. Pink or red branches indicate genes in the Toll-4 or Toll-3 syntenic loci, respectively. Internal nodes labeled with posterior probability. Genes designated as Toll4.x were identified from intergenic regions in the current annotations, and therefore do not have a gene id number. **GF22704 is a withdrawn gene annotation, but was manually re-annotated from the gDNA sequence. ***Toll-4.obs1 merges two adjacent gene models: GA24352 and GA32586.
In more distantly-related lineages such as the obscura group (D. pseudoobscura and D. persimilis) and willistoni group, we similarly found many Toll-3/4 loci per species, each encoding either a single gene or a cluster of tandem duplicates of genes or pseudogenes (fig. 2). These loci also appeared to be lineage specific. We found two syntenic loci of Toll-3/4 genes encoding up to 6 genes that were unique to the obscura group. For ease of reference, we refer to these as Toll-3/4.obs1 to 6 (see fig. 3 for corresponding Flybase gene designations). We also found six distinct Toll-3/4 genes in two locations unique to the D. willistoni genome (Toll-3/4.wil1 to 6). Oddly, one genomic locus appears to have independently acquired nonorthologous Toll-3/4 genes in both D. willistoni (Toll-3/4.wil7 to 9) and D. pseudoobscura (Toll-3/4.obs7 to 8), owing to a chromosome rearrangement in the obscura group that linked the neighboring gene CG16825 to another locus, CG11236 (figs. 2 and 3). We found additional, isolated Toll-3/4 genes or pseudogenes in D. ananassae, D. virilis, and D. willistoni that are not schematized in figure 2. These isolated genes are distant from other Toll-3/4s, and are in genomic loci that do not encode Toll-3/4 genes in any of the other well-annotated Drosophila genomes (fig. 3). We conclude from this pattern of diversification that most Toll-3/4 genes are quite young and have undergone a rapid birth-and-death process of independent duplication and recurrent pseudogenization. We emphasize that this birth-and-death process within Drosophila spp. seems to be highly accelerated for the Toll-3/4 paralogs, but not for any of the other Toll genes.
What Is the Mechanism for Recurrent Birth of Toll-3/4 Genes?
We next investigated whether the repeated duplications of Toll-3/4 genes might be due to their residence within genomic regions that are hotspots for duplication or copy number variants (CNVs). If so, then Toll-3/4 diversity may be driven by mutational bias, independent of Toll-3/4 function(s). Genome-wide surveys of CNVs have recently been performed within diverse D. melanogaster populations (Zichner et al. 2013; Cardoso-Moreira et al. 2016) as well as in D. simulans and D. yakuba (Rogers et al. 2014). Within these datasets, Toll-6, Toll-7, and Toll-9 within D. melanogaster had CNVs that partially duplicated portions of the gene. In contrast, none of the Toll-1, -2, -3, -4, -5, or -8 genes had CNVs within coding regions, although several had variation within introns. No segregating duplications were detected in D. simulans or D. yakuba that overlapped any Toll loci. Therefore, it does not appear that Toll-3/4 diversity can be explained by their residence within previously mapped duplication hotspots.
To understand the mechanism of the rapid Toll-3/4 duplications, we looked in detail at the D. yakuba paralogs of Toll-4, which we refer to as Toll-4.yak1 to yak4. The yak1 and yak2 genes lie within to the Toll-4 syntenic locus, while the other Toll-4 copies reside elsewhere on chromosome 2L. Notably, the D. yakuba sister species D. erecta has only a single pseudogene copy of Toll-4 that resides in the syntenic locus, indicating that the D. yakuba duplications have likely occurred during the approximately 8 My since the D. yakuba–D. erecta divergence (Tamura et al. 2004). We hypothesized that this recent origin of Toll-4 duplications in D. yakuba might leave genomic evidence of the mechanism of duplication, especially because these genes are not found in complex multigene clusters as they are in D. pseudoobscura and D. willistoni.
The D. yakuba gene Toll-4.yak1 in the ancestral locus contains 4 coding exons, as is true for the syntenic Toll-4 genes in other melanogaster group species. However, for Toll-4.yak2, Toll-4.yak3, and Toll-4.yak4 we only found sequences corresponding to exons 3 and 4 of the parental, syntenic gene, while exons 1 and 2 were apparently missing (fig. 4). An investigation of the nearby genomic region revealed that a DNAREP1_DM transposable element, also called DINE-1 (Yang et al. 2006), resides within intron 2 of Toll-4.yak1. Additional DNAREP1_DM elements flank both sides of the Toll-4.yak2 and Toll-4.yak3 genes and are also present 5’ of the Toll-4.yak4 gene. We infer that these DNAREP1_DM elements may have provided regions of sequence homology that allowed for exons 3 and 4 of Toll-4 to be copied elsewhere on the chromosome through aberrant recombination. Since exons 3 and 4 encode the majority of the Toll-4 sequence (including LRRs, a transmembrane domain, and the TIR domain), these recombination events led to the origin of novel Toll-4 paralogs with diversified N-termini once new start codons were acquired postduplication. We note that this mechanism of repeat-mediated duplication via nonallelic recombination has the potential not only to rapidly copy Toll-3/4 genes around the genome, but also to rapidly delete these genes via recombination of adjacent repeats. We expect that the Toll-3/4 repertoire we observe in modern Drosophila genomes is therefore merely a snapshot of the many additional copies of these genes that have likely existed in the history of these species, or that might still exist in unsampled strains.
Fig. 4.
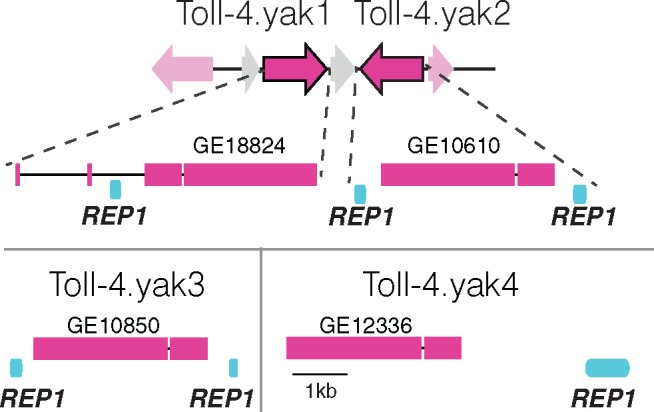
Recent Toll-4 duplications in D. yakuba likely mediated by repetitive elements. Toll-4.yak1 resides in the syntenic Toll-4 locus and has four exons (pink boxes). DNAREP1_DM repetitive elements (REP1, light blue) lie within intron 2 of Toll-4.yak1 and flank Toll-4 duplicates elsewhere on chromosome 2L.
Toll-3/4 Genes Show Male-Specific Expression
We next investigated whether the partial Toll-4 duplications we observed in D. yakuba were expressed or whether they were unexpressed pseudogenes. We analyzed the expression of the Toll-4 duplicates in four RNA-seq datasets from whole, adult D. yakuba (Chen et al. 2014). The Toll-4.yak4 locus lies within the intron of another gene, which meant that we were not confident in assigning mapped reads to Toll-4.yak4 as opposed to its overlapping gene. However, we did observe low level Toll-4.yak1, yak2, and yak3 transcription in the adult male samples, albeit at different levels in the two biological replicates (0.40 < FPKM < 0.90 in replicate 1 and 0.05 < FPKM < 0.2 in replicate 2; supplementary material S1, Supplementary Material online). Notably, little to no expression was observed in either of the whole female samples (FPKM < 0.02). We interpret these results to mean that even partial Toll-4 duplications have the potential to produce novel genes that are expressed, especially in males.
To investigate if male-biased expression is a general feature of Toll-3/4 genes, we examined the expression of all Toll family genes in publically available RNA-seq datasets for D. melanogaster in addition to the following species that have experienced Toll-3/4 duplications: D. kikkawai, D. pseudoobscura, D. willistoni, and D. virilis (supplementary material S1, Supplementary Material online). Toll-1 is a maternally deposited gene that controls dorsal-ventral embryonic patterning, and is therefore highly expressed in embryos, ovaries, and adult females of all species examined. It is also expressed broadly in multiple adult tissues, as are Toll-2, -5, -6, -7, -8, and -9 at varying levels.
In contrast, many Toll-3/4 duplicates were not detectably expressed. However, in all cases where any expression of Toll-3/4 genes was detected, it was predominantly found in males, especially in testes (supplementary material S1, Supplementary Material online). In D. melanogaster, low level Toll-3 and Toll-4 RNA expression has been observed in larvae, pupae, imaginal discs, whole adult males, and testes (Graveley et al. 2011). Late in embryogenesis, Toll-4 expression was also observed by in situ hybridization in a minority of putative lymph gland precursor cells (Kambris et al. 2002). In D. pseudoobscura, we found that 6 of the 9 Toll-3/4 genes were expressed (FPKM > 0.05), all of which were male-specific and predominantly expressed in testes. While most of these had relatively low expression around FPKM = 1, Toll-3/4.obs3, was expressed considerably higher with an average FPKM of 9.7.
In D. willistoni, we confidently detected expression from only 4 of the 19 Toll-3/4 genes (Toll-3/4.wil2, 3, 4, and 17; supplementary material S1, Supplementary Material online). These genes were expressed in male abdomens but not female abdomens, presumably due to expression in the male reproductive system. In all cases, Toll-3/4 genes were expressed at low levels around FPKM = 1, perhaps reflecting expression in a small minority of cells. Toll-3/4.wil2, 3, and 4 genes appeared to be full-length, with predicted LRRs, a transmembrane domain, and a C-terminal TIR domain. Curiously, Toll-3/4.wil17 also showed evidence of expression despite having a much smaller open reading frame than other Tolls, and was predicted to encode only a secretion signal and a few LRRs.
For D. kikkawai and D. virilis, none of the Toll-3/4s were expressed above FPKM = 0.1, although we note that these RNA-seq experiments analyzed mRNA from whole males and whole females and therefore lacked sensitivity to detect low-level, tissue-specific transcripts. Indeed, when we individually analyzed the expression of two of the D. kikkawai Toll-3/4s by RT-PCR, we found that one was male- and testes-specific, while the other was not expressed (fig. 5).
Fig. 5.

Drosophila kikkawai Toll-4 duplicates include both testis-specific genes and those without detectable expression. PCR from D. kikkawai genomic DNA (g) or cDNA from whole adults (W), heads (H), testes (T), ovaries (O), carcass without gonads (C), or larvae (L). Gene-specific primers were designed across small introns, as demonstrated by the slight size shift between genomic DNA and cDNA. Expression of ribosomal gene Rp49 is shown as a control.
We also queried previous datasets of genes expressed in testes and ovaries of Drosophila spp. In D. ananassae, Toll-4.ana1 and Toll-4.ana2 showed low expression but were significantly higher expressed in testes than in ovaries, while Toll-3 expression was not detected in any tissue. In D. pseudoobscura, Toll-3/4.obs2, Toll-3/4.obs3, and Toll-3/4.obs6 were all expressed and were at higher levels in testes than in ovaries, while the remaining Toll-3/4s were not detected (VanKuren and Vibranovski 2014).
The lack of detectable expression of many Toll-3/4 genes is consistent with their highly dynamic evolution, including complete loss in some species. Alternatively, these genes might only be expressed under specific conditions (e.g., under pathogen infection), which have yet to be examined, especially outside of D. melanogaster. However, when they are clearly expressed, Toll-3/4s were transcribed exclusively in males, predominantly in testes.
Drosophila melanogaster Toll-3/4s Have Minimal Impact on Viability or Male Fertility
We next used the genetic tools available in D. melanogaster to investigate male viability and fertility in flies with perturbed Toll-3/4 expression. Using crosses with Gal4 driver lines, we knocked down or overexpressed Toll family transcripts, either across the whole fly (using Act5C-Gal4) or within the male germline (using vasa- or bam-Gal4). Because transgene lines vary in their knockdown efficiency, for both Toll-3 and Toll-4 we tested 2–3 independent RNAi lines with each Gal4 driver. The very low endogenous levels of D. melanogaster Toll-3/4 transcripts precluded experimental validation of changes in expression upon RNAi-induced knockdown, but we were nevertheless able to assess the phenotypes of these flies.
Control experiments with whole-fly perturbation of Toll-1 and -5 yielded expected viability defects. Overexpression of Toll-1 was lethal in early developmental stages whereas Toll-5 knockdown yielded viable adult flies, but these all died within 7 days post-eclosure. In contrast, all Toll-3/4 knockdown and overexpression flies were viable without obvious developmental or viability defects, consistent with previous reports (Yagi et al. 2010). The number of knockdown flies obtained from these crosses did not differ from Mendelian expectations, and there was no sex bias among the progeny in any cross (for all crosses P > 0.1 by χ2 test, n > 130 offspring counted per cross) suggesting that these genes do not play an essential role in viability, sex-specific or otherwise. This apparent release of Toll-3/4 genes from essential developmental roles is also consistent with our observation that some Drosophila spp. have lost all of their Toll-3/4 genes.
The lack of an essential role in development does not necessarily imply that these genes are unimportant for host fitness. In particular, the impacts of Toll-3/4s on male fertility have not previously been investigated. To test if the knockdown or overexpression flies had fertility defects, we crossed males of each genotype to female flies from the yw strain and counted the resulting offspring. As a control, we crossed yw males to yw females. In these experiments, all knockdown males sired viable adult offspring and did so at approximately the same levels as control males (supplementary fig. S3, Supplementary Material online). There was a minor but statistically significant decrease in offspring number when Toll-3 was knocked down with the vasa germline Gal4 driver (P < 0.05 for each RNAi line by 2-tailed t-test), but this decrease was not observed when males were knocked down with the other two Gal4 drivers. The Toll-3 and Toll-4 overexpression males had no observable viability or fertility defects (data not shown). In summary, we observed mild, if any, impact of Toll-3 or Toll-4 knockdown or overexpression on male viability or fertility in D. melanogaster.
Do Toll-3/4 Genes Evolve under Positive Selection?
Given the lack of an obvious role for Toll-3/4 genes in viability and fertility, we next asked if these genes are evolving under neutral processes, or alternatively if they exhibited evolutionary signatures of adaptation or constraint. Previous analyses have identified weak signatures of positive selection in Toll-3 and Toll-4 using maximum likelihood methods (Han et al. 2013) and in D. melanogaster/D. simulans Toll-4 based on a McDonald–Kreitman test (Obbard et al. 2009). We therefore investigated whether Drosophila Toll genes had generally evolved under positive selection, first by comparing pairwise amino acid identity of the D. melanogaster Tolls with their orthologs from D. simulans, D. yakuba, and D. ananassae (table 1). We found that Toll-2, -6, -7, and -8 were most highly conserved at >90% amino acid (aa) identity in all comparisons, whereas Toll-1, -5, and -9 were less conserved (>70% aa identity), with most of this divergence arising in their extracellular domains. Strikingly, Toll-4 was much more diverged—only 47% aa identity between D. melanogaster and D. ananassae. Toll-4 also has a higher gene-wide dN/dS compared with the other Toll genes, particularly in the extracellular domain (pairwise dN/dS in table 1, see supplementary material S2, Supplementary Material online for dN/dS along each branch). We found a similar divergence and elevation of dN/dS for Toll-3 between D. simulans and D. melanogaster. We did not find gene-wide signatures of dN/dS >1 for any of the Toll genes, but such gene-wide analyses have limited power to detect positive selection when it occurs at a minority of sites.
Table 1.
Toll-3 and Toll-4 Are More Diverged from Their Orthologs Than Other Tolls.
| Gene Name | %aa Identity | dN/dS Full Gene | dN/dS Extracellular | dN/dS Intracellular |
|---|---|---|---|---|
| Toll | 97.2/ 87.5/ 72.2 | 0.07/ 0.19/ 0.08 | 0.08/ 0.15/ 0.09 | 0.05/ 0.28/ 0.03 |
| Toll-2 | 99.7/ 98.8/ 92.9 | 0.01/ 0.02/ 0.02 | 0.01/ 0.02/ 0.02 | 0.00/ 0.00/ 0.01 |
| Toll-3 | 90.3/ NA/ NA | 0.31/ NA/ NA | 0.31/ NA/ NA | 0.19/ NA/ NA |
| Toll-4 | 91.5/ 72.2/ 47.2 | 0.44/ 0.48/ 0.19 | 0.47/ 0.52/ 0.12 | 0.14/ 0.15/ 0.08 |
| Toll-5 | 96.9/ 92.7/ 76.7 | 0.07/ 0.08/ 0.06 | 0.10/ 0.11/ 0.08 | 0.02/ 0.00/ 0.00 |
| Toll-6 | 99.3/ 99.0/ 93.6 | 0.02/ 0.02/ 0.01 | 0.02/ 0.01/ 0.01 | 0.04/ 0.02/ 0.02 |
| Toll-7 | 99.4/ 98.6/ 90.4 | 0.03/ 0.01/ 0.01 | 0.03/ 0.01/ 0.03 | 0.00/ 0.02/ 0.03 |
| Toll-8 | 99.3/ 98.4/ 92.3 | 0.03/ 0.02/ 0.02 | 0.03/ 0.03/ 0.02 | 0.03/ 0.02/ 0.03 |
| Toll-9 | 95.8/ 91.8/ 74.1 | 0.13/ 0.12/ 0.09 | 0.15/ 0.13/ 0.11 | 0.06/ 0.07/ 0.03 |
Note.—Pairwise percent amino acid identity and dN/dS between the D. melanogaster gene and its ortholog in the syntenic locus in D. simulans/D. yakuba/D. ananassae. NA indicates that neither D. yakuba nor D. ananassae has a syntenic copy of Toll-3.
To overcome this limitation, we then proceeded to a more in-depth analysis of positive selection using the maximum likelihood-based program PAML (Yang 1997). A previous PAML analysis had identified a signature of positive selection on the Toll-1 gene in Drosophila spp., but not on any other Toll paralogs (Sackton et al. 2007). A second paper found weak evidence of positive selection for Toll-1, -3, and -4, but none of these signals passed a false discovery rate test (Han et al. 2013). As both of these results were part of large-scale screens for positively selected genes, we decided to revisit the analyses in more depth. When we analyzed the Toll-1 orthologs from 12 Drosophila genomes, we found that the gene annotation for D. sechellia was truncated, and the D. persimilis Toll-1 was split into two adjacent gene models. We manually extended the D. sechellia annotation and excluded the D. persimilis gene from analysis. Subsequently, we constructed a multiple alignment and a phylogenetic tree of Toll-1. To examine signatures of positive selection, we used the codeml program in the PAML suite, which employs maximum likelihood methods to estimate the rate of synonymous (dS) and nonsynonymous (dN) substitutions at each codon to determine if there is significant evidence of positive selection (i.e., dN/dS > 1). More specifically, the NSsites test compares the likelihood of models in which positive selection is not permitted (M7: dN/dS < 1; M8a: dN/dS ≤ 1) to one that permits positive selection at a subset of amino acid sites (M8: dN/dS > 1) to evaluate whether the latter is a better fit to the sequence data. In contrast to previous work, we did not find evidence for Toll-1 positive selection (PAML M7 vs. M8 P = 0.27, M8a vs. M8 P = 0.48). We speculate that gene annotation errors may have clouded previous analyses of Toll-1 evolution. We performed the same analyses for Toll-2, -5, -6, -7, -8, and -9, and also found no evidence of positive selection across the 12 Drosophila genomes.
We then turned our attention to the Toll-3/4 genes, which were more challenging to analyze due to their rapid diversification. To obtain enough orthologs of Toll-3 and Toll-4 for these analyses, we queried additional closely related species of the melanogaster group whose genomes had been recently sequenced but not yet annotated: D. bipectinata, D. kikkawai, D. ficusphila, D. elegans, D. takahashii, D. suzukii, D. biarmipes, and D. eugracilis. Since Toll-3 had been lost in many of the species we analyzed, we used degenerate primers to additionally amplify and sequence the Toll-3 genomic locus from D. pseudotakahashii. We manually annotated the Toll-3/4 coding sequences using an iterative method of blast searches and alignments, generating relatively high-confidence gene models for the majority of Toll-3/4 genes from the melanogaster group (see Materials and Methods). We find that the Toll-3/4s vary significantly in their size, specifically in the expansion and contraction of the number of LRR domains. Analyses based on multiple-alignments are not able to assess whether this change in LRR domains is driven by drift or positive selection, so we focused our analyses on substitutions within the well-aligned regions of the Toll-3/4s.
Using phylogenetics and shared synteny analysis, we confirmed that all of the putative Toll-3/4 orthologs we obtained corresponded to the Toll-3 and Toll-4 clades (fig. 3). We constructed full-length alignments of the Toll-3 and Toll-4 orthologs, ensuring that we analyzed only homologous sequences by pruning the alignment to eliminate the N-terminus of Toll-4 genes (exons 1 and 2). Analyses of positive selection can be misled by recombination or gene conversion among paralogs, as this can result in different evolutionary histories for different regions of a gene. To avoid these complications, we used the program GARD on all alignments to test for evidence of recombination. No significant breakpoints were detected for Toll-3 or -4. We then analyzed alignments of Toll-3 and Toll-4 with PAML as described above (table 2). We found that Toll-3 did not show evidence of positive selection and we observed only a weak signature of positive selection in Toll-4 in the melanogaster group species (P-value =0.05 for M7–M8 comparison and P =0.29 for M8a–M8 comparison). A modest proportion (3.7%) of sites in the Toll-4 alignment were predicted to evolve under positive selection, but their average dN/dS (or “omega”) values were only slightly higher than 1 (1.34). Indeed, no individual sites were identified as having evolved under recurrent positive selection with a high posterior probability (>0.95) according to the Bayes Empirical Bayes (BEB) test. These findings are consistent with previous studies on fewer species that have found only a modest signature of positive selection in Toll-4.
Table 2.
PAML Analyses of Toll Family Alignments.
| Genes | # Sequences | M7 versus M8 P-Value | M8a versus M8 P-Value | # Positively Selected Sites (BEB >95%) | Highest Omega in M8 | % Sites dN/dS>1 |
|---|---|---|---|---|---|---|
| Toll-3 | 13 | 0.49 | 0.72 | – | – | – |
| Toll-4: 136–3267 | 13 | 0.05 | 0.29 | 0 | 1.34 | 3.7 |
| Obscura Toll-3/4s: 1–1584 | 12 | 0.02 | 0.03 | 1 | 4.72 | 0.6 |
| Obscura Toll-3/4s: 1585–2763 | 12 | 0.05 | 0.27 | 0 | 3.05 | 1.0 |
| Obscura Toll-3/4s: 2764–3180 | 12 | 0.20 | 0.16 | – | – | – |
| Willistoni Toll-3/4s: 1–1308 | 13 | 1.2×10−38 | 4.4×10−31 | 28 | 3.74 | 16.1 |
| Willistoni Toll-3/4s: 1309–1593 | 13 | 0.43 | 0.99 | – | – | – |
| Willistoni Toll-3/4s: 1594–1833 | 13 | 0.30 | 0.6 | – | – | – |
| Willistoni Toll-3/4s: 1834–3339 | 13 | 4.6×10−4 | 0.26 | 0 | 1.33 | 7.6 |
Note.—Obscura and willistoni alignments were each divided into subsections before analysis, and the coordinates for each section prior to gblocks masking are listed. BEB refers to the Bayes Empirical Bayes test.
When we analyzed the Toll-3/4 paralogs present in the obscura group (D. pseudoobscura and D. persimilis) as above, GARD analyses identified 2 significant recombination breakpoints. We therefore split the alignment into three segments and analyzed each separately. As with Toll-4 in the melanogaster group, we found modest evidence suggestive of positive selection in two of the three segments (together comprising the extracellular and transmembrane domains; table 2), with only the most N-terminal segment significant in both the M7–M8 and M8a–M8 comparisons. Thus, for the melanogaster and obscura group Toll-3/4s we find only weak, if any, statistical evidence in favor of positive selection.
The Toll-3/4 genes from D. willistoni showed a strikingly different signature. From an alignment of 13 D. willistoni Toll-3/4s, we again detected multiple recombination breakpoints by GARD; we used PAML to analyze each segment separately. We found very strong evidence of positive selection in the Toll-3/4 genes of the D. willistoni genome (P-values <10−30 for both M7–M8 and M8a–M8 comparisons, table 2). In the most N-terminal segment (bp1-1308), 16% of sites were predicted to have evolved under positive selection with an average dN/dS of 3.74. Twenty-four individual sites were identified by BEB analyses with a posterior probability greater than 0.95. After manual inspection of these sites, we conservatively excluded residues that were near gaps or gblocks-masked regions, obtaining a set of 20 high-confidence sites of positive selection in the D. willistoni Toll-3/4s. We also used the PARRIS program implemented under the HyPhy suite (see Materials and Methods) to analyze the most N-terminal segment of the D. willistoni Toll-3/4 alignment. This is a more conservative test of positive selection, because it accounts for recombination and synonymous site variation. Even under this more stringent test, we found robust evidence of positive selection having acted on D. willistoni (P-value = 10−5), specifically in the N-terminal region of Toll-3/4s. We therefore conclude that Toll-3/4 genes in the D. willistoni genome show unambiguous, strong signatures of positive selection, unlike the Toll-3/4 genes in other lineages.
Positive Selection in Toll-3/4 Proteins in D. willistoni Overlaps Predicted Spätzle-Binding Pocket
We next investigated where the positively selected sites in the D. willistoni Toll-3/4s were likely positioned in the protein using Phyre2 (Kelley et al. 2015). This program predicts the structure of an input amino acid sequence through comparisons to protein crystal structure databases. Phyre2 modeled the extracellular region of D. willistoni Toll-3/4 GK28112 (Toll-3/4.wil15) on a structure of D. melanogaster Toll-1 bound to its ligand Spätzle (c4lxrA; Parthier et al. 2014). This structural model included amino acids 127–847 of GK28112 and consisted of a cysteine-rich LRRCT domain as well as an arc of LRRs. When we mapped the positively selected residues onto the model, we found that these residues clustered in a discrete patch on the underside of the LRR arc (fig. 6), despite being scattered throughout the primary sequence of the protein. Seven additional positively selected residues fell in the unmodeled region 0-126, which Phyre2 could not suitably align to c4lxrA. When we compared the positively selected residues with those bound by Spätzle in the c4lxrA structure, we found that the positively selected positions were flanking and partially overlapping the Spätzle-binding interface (fig. 6B and C).
Fig. 6.

Positively selected sites are predicted to form a discrete patch that partly overlaps the Spätzle binding site. (A) Phyre2 predictions of D. willistoni GK28112 residues 127–847, colored according to conservation with other wilistoni Toll-3/4s (white to blue). Space filling residues (dark blue) within the leucine-rich repeats (LRRs) are those identified as positively selected by PAML. Additional positively selected residues were found in region 1 to 126, which was not included in the Phyre2 model. Inset cartoon represents the crystal structure of D. melanogaster Toll (gray) bound to its ligand Spätzle (cyan), showing regions of Toll-Spätzle contact (orange boxes). Light gray portions of the Toll crystal structure were not used in the Phyre2 model of GK28112. (B) View of the underside of the LRR arch of the GK28112 model, with predicted Spätzle-binding residues in orange. (C) Same view as (B), with positively selected residues as space filling spheres. The 16 positively selected residues in the modeled region overlapped the N-terminal portion of the Spätzle binding pocket and seven were homologous to residues that bind Spätzle in the Toll crystal structure.
In Drosophila, the only known ligands of Tolls are the endogenous Spätzle proteins, which exist as six paralogs in D. melanogaster (Morisato and Anderson 1994; Parker et al. 2001; Valanne et al. 2011). If the Spätzle ligands were driving the rapid evolution of Toll-3/4s, we hypothesized that we would see a correlated expansion and diversification of Spätzle genes, specifically in lineages with abundant Toll-3/4s. To test this hypothesis, we again queried the 12 Drosophila genomes to look at Spätzle diversity. Contrary to our hypothesis, Spätzle 1–6 were found in single copy in nearly all genomes queried (supplementary fig. S5, Supplementary Material online). Previous surveys detected weak signatures of positive selection on Spätzle by McDonald–Kreitman test (Obbard et al. 2009) and PAML [(Han et al. 2013) but not (Sackton et al. 2007)] similar to those observed for Toll-1 and -4, but nothing akin to the strong signatures we observed for the D. willistoni Toll-3/4s. We therefore hypothesize that the Toll-3/4 proteins in D. willistoni may have rapidly evolved in response to different rapidly evolving ligand, or alternatively, in response to an unknown antagonist.
Discussion
Diverse repertoires of animal TLRs have evolved through a pattern of duplication and divergence to recognize a variety of microbial ligands. Although some TLRs bind to invariant microbial molecules and are broadly conserved, these receptors can also show signs of intermittent positive selection in multiple lineages, including the cell-surface TLRs of mammals (Barreiro et al. 2009; Areal et al. 2011; Quach et al. 2013), most TLRs in birds (Alcaide and Edwards 2011; Grueber et al. 2014), and the greatly expanded TLR repertoire of echinoderms (Buckley and Rast 2012). It has been suggested that such diversification enables these TLRs to adapt to novel or rapidly changing microbial ligands.
Unlike in other animals, insect TLRs have been shown to recognize microbial ligands indirectly, downstream of extracellular signaling cascades that proteolytically cleave Spätzle, Toll’s endogenous ligand. To a large extent, the constrained evolution of Toll family genes in Drosophila is consistent with their previously defined, indirect role in microbial recognition or development, which would require conservation rather than evolutionary novelty.
Our analyses identify the poorly studied Toll-3/4 genes as exceptional among the Drosophila Tolls in several respects. First, unlike other Toll genes that have been shown to be important for development, we find that Toll-3 and Toll-4 are largely dispensable for viability and fertility in D. melanogaster. This finding is in contrast to their closest D. melanogaster paralogs, Toll-1 and Toll-5; perturbation of either leads to viability defects.
Second, we found that Toll-3/4 genes in many lineages have experienced an unusually rapid cycle of gene gain and loss. This process is occurring simultaneously in multiple Drosophila species, resulting in many private, lineage-specific Toll-3/4s. These types of diverse and rapidly evolving gene families are particularly challenging to analyze in large-scale screens for positive selection due to difficulties with gene annotation and assignments of orthology, necessitating the detailed, manual curation we have undertaken in this report. We also observed several lineages where the Toll-3/4 genes were relatively static, as well as several that have lost Toll-3/4s all together. Intriguingly, human TLRs that are localized to the cell surface such as TLR1, 5, and 10 also have high frequencies of circulating, likely deleterious alleles (up to 10%), while additionally showing evidence of positive selection and even adaptive sweeps through the human population (Barreiro et al. 2009). It has been suggested that these TLRs may alternate between essential, protective roles and redundant, nonessential functions depending on the microbial pressures experienced by host populations. It seems likely that Toll-3/4 evolution is also driven by strong selective pressures that are experienced only sporadically, or only in certain lineages.
A third unusual feature of Toll-3/4s is their restricted expression to males, largely to the male germline. This pattern of male germline-specific expression is common to very recently duplicated Drosophila genes (Assis and Bachtrog 2013), and our findings are consistent with the “out of testis” model for the evolution of new genes (Kaessmann 2010). This male-biased expression was observed for Toll-3/4s of different ages from many species, including D. melanogaster, D. kikkawai, D. yakuba, D. pseudoobscura, and D. willistoni, suggesting that it is a shared feature of Toll-3/4s. In several species, the level of expression remained low. In D. melanogaster, low, tissue-specific Toll-3/4 expression has generally hindered detailed study of these receptors (Tauszig et al. 2000; Kambris et al. 2002). Even so, one study detected Toll-4 proteins in the mature sperm from dissected spermatheca of mated females via mass-spectroscopy, showing that these proteins are synthesized and transmitted to females upon mating (Wasbrough et al. 2010). Given that we did not observe detectable expression from many Toll-3/4s, we infer that these are either pseudogenes, that they are genes expressed only in restricted tissues or conditions, or that the RNA-seq assays did not sequence deeply enough to detect these low-level transcripts. Still, several of the D. pseudoobscura Toll-3/4s such as Toll4.obs3 are robustly expressed and are therefore reasonable candidates for functional studies.
Finally, while other Toll paralogs are strongly conserved and show no evidence for positive selection, we find strong evidence for Toll-3/4 positive selection within D. willistoni, which encodes the largest number of Toll-3/4s. These evolutionary signatures are localized specifically to a region of the LRRs predicted to overlap with the Spätzle binding site. Although we did not detect robust evidence for positive selection for Toll-3/4 genes from either the melanogaster or obscura groups, we note that these Toll-3/4 genes are still the most unusual among the Drosophila Tolls in terms of their degree of divergence and rate of gene turnover. Indeed, the evolution of the Drosophila Toll-3/4 gene family that we describe here more closely resembles the dynamic evolution and diversification of TLRs in distant animal lineages than that of other Drosophila Toll genes.
The forces driving the rapid evolution of Toll-3/4s remain a mystery. One of the major bottlenecks in our understanding is the lack of a functional role attributable to these genes. In the model species D. melanogaster, Toll-3 or Toll-4 knockdown or overexpression has not been reported to cause obvious phenotypes in viability, morphology, or antimicrobial gene expression (Ooi et al. 2002; Yagi et al. 2010; Nakamoto et al. 2012; Samaraweera et al. 2013). Our results are consistent with previous reports and we additionally find little, if any, impact of Toll-3/4s on viability or male fertility. Although the study of most genes, including other Tolls, is enabled by experiments in D. melanogaster, the idiosyncrasy of Toll-3/4 evolution suggests that D. melanogaster may not be the best system to study their function. Since the birth of Toll-3 during the early evolution of the melanogaster group, the branches leading to D. melanogaster have not experienced any Toll-3/4 gains or losses, and these genes show, at most, modest signatures of positive selection. Because the Toll-3/4 gene family is evolving most rapidly in species like D. willistoni and D. pseudoobscura, expanding functional experiments to these species may reveal more robust phenotypes.
Still, the strong evidence of positive selection on D. willistoni Toll-3/4s specifically within the ligand-binding pocket provides a clue as to the pressures driving Toll-3/4 evolution. Rapid turnover at this interface implies that these Toll-3/4s are under selection to rapidly alter their binding affinity and/or specificity. What molecular interactions could be driving these changes? The ligands of most Toll paralogs have not been experimentally determined, but because Toll-1 binds to Spätzle, it is generally assumed that other Toll paralogs bind to Spätzle family proteins as well. Yet, all of the Spätzle paralogs are rarely duplicated and highly conserved, which means that none are likely causing the rapid evolution of Toll-3/4s. Instead, we hypothesize that D. willistoni Toll-3/4 evolution is driven by one of two possible scenarios—either these receptors are evolving to evade binding by an unknown antagonist molecule, or they are evolving to bind and recognize a novel, rapidly evolving ligand.
At a larger scale, what functions are these receptors likely to perform? Because these genes have not been experimentally well-characterized, all hypotheses about the functions of Toll-3/4 receptors are necessarily speculative. Even so, there are several contexts in which Toll-3/4s could function in a lineage-specific manner in the male germline that would be consistent with their rapid evolution. First, they may play a subtle role in male reproduction. Testis-expressed genes are among the most rapidly evolving in animal genomes, due to selective pressures such as sperm competition, sexual selection, and competitive interactions between sperm and the female reproductive system (Chapman 2001; Swanson and Vacquier 2002; Wolfner 2011). Because these reproductive pressures have a strong impact on organismal fitness, even small changes in fertility could be subject to selection. The observation that Toll-4 from D. melanogaster is transmitted to females upon mating (Wasbrough et al. 2010) is consistent with a potential role in reproduction. Alternatively, if Toll-3/4s operate in an immune context, they may respond to widespread, vertically-transmitted bacteria such as Wolbachia that reduce the fertility of infected males (O'Neill and Karr 1990; Fujita et al. 2011). In this scenario, we predict that Toll-3/4s would localize to Wolbachia-containing vacuoles within the host cytoplasm, which have been observed by electron microscopy (Callaini et al. 1994; Fischer et al. 2014). Although previous studies have found that Wolbachia do not broadly activate host immunity through antimicrobial peptide expression (Bourtzis et al. 2000), they do alter host immune responses by unknown signaling pathways (Teixeira et al. 2008), where Toll-3/4s could perhaps play a role.
Beyond the highly unusual evolutionary trajectory of Toll-3/4s, our analysis allows us to revisit previous genomic surveys of Toll gene repertoires to propose that the Toll family genes of insects fall into three distinct evolutionary classes. The first of these consists of the highly conserved Toll-2, -6, -7, and -8 genes (table 1). Toll-6, -7, and -8 are ancient genes, with one-to-one orthologs present across insects and even in some crustaceans (Christophides et al. 2002; Zou et al. 2007; Gerardo et al. 2010; Palmer and Jiggins 2015). Although Toll-2 is a relatively recent duplicate of Toll-7, it appears to evolve under similar constraints within Drosophila spp. The strong conservation of this first class suggests these genes predominantly play essential roles in insect physiology and development (Yagi et al. 2010; but see Nakamoto et al. 2012). The second class of Toll genes consists of Toll-1, -5, and -9, which evolve much faster than the first class, particularly in their extracellular domains (table 1). All three of these genes have been implicated in microbial sensing (Lemaitre et al. 1996; Tauszig et al. 2000; Bettencourt et al. 2004; but see Narbonne-Reveau et al. 2011), and so it is possible that this receptor variation has been driven by evolutionary arms races with pathogens. Still, the evolution of Toll-1, -5, and -9 remains somewhat constrained, likely due to the additional developmental roles of these receptors (Yagi et al. 2010). We propose that the Toll-3/4 genes of Drosophila are examples of a third, extremely rapidly evolving class of insect Tolls that are characterized by their lineage-specificity and evolutionary flexibility rather than by conservation and straightforward orthology. Toll-3/4s have apparently been released from their essential, developmental constraints (as evidenced by their frequent losses and lack of a gross effect on viability or fertility upon knockdown in D. melanogaster) to become much more genetically labile than their ancestral counterparts in the Toll-1/5 lineage. The Toll-1/5 lineage is ancient (Gerardo et al. 2010) and has repeatedly spawned additional, species-specific duplicates in insects as diverse as mosquitos, wasps, beetles, and aphids (Christophides et al. 2002; Zou et al. 2007; Gerardo et al. 2010). We hypothesize that some of these Toll-1/5 lineage-specific duplicates across insects have also been released from their essential, developmental roles to follow the rapid evolutionary path traversed by the Toll-3/4 receptors in Drosophila. In this manner, the third class of insect Tolls shares evolutionary characteristics with TLRs in other animal lineages, where the receptors can evolve to optimize their immune roles with few developmental constraints. Further characterization of Toll-3/4s promises to reveal the selective pressures driving their rapid evolution, and potentially novel, noncanonical functions for TLRs in insects.
Materials and Methods
Toll Sequences and Gene Model Annotations
Sequences for the Toll paralogs were obtained from publically available genome assemblies. These included the 12 Drosophila genomes that have been well-annotated: D. melanogaster, D. simulans, D. sechellia, D. erecta, D. yakuba, D. ananassae, D. persimilis, D. pseudoobscura, D. willistoni, D. virilis, D. mojavensis, and D. grimshawi (Drosophila 12 Genomes Consortium et al. 2007). We also obtained Toll-3 and Toll-4 sequences from the following assembled but unannotated genomes of melanogaster group species: D. biarmipes, D. eugracilis, D. takahashii, D. elegans, D. ficusphila, D. kikkawai, D. bipectinata and D. suzukii (Chiu et al. 2013; Chen et al. 2014). We examined the genome of D. rhopaloa for Toll-3/4 genes as well, but found only Toll-3/4 pseudogenes, which we excluded from further analyses. We searched the genomes from the following outgroup fly species as well, but did not identify any Toll-3/4 homologs: D. busckii, B. oleae, H. illucens, P. variegata, S. bullata, M. domestica, and G. morsitans morsitans (International Glossina Genome Initiative 2014; Scott et al. 2014; Vicoso and Bachtrog 2015).
To identify additional melanogaster group orthologs of Toll-3, we used degenerate primers to amplify and sequence the syntenic locus from D. pseudotakahashii (#14022.0301.01) via PCR. We used NEB Phusion polymerase and the following primers: GCGCTTCATTTCAYTCCTTTG and GCTACAGGGTCCRCAGAA. This reaction yielded a single band of approximately 8kb. This product was gel extracted and analyzed by Sanger sequencing. Sequences obtained in our study have been deposited into Genbank (KY451958).
We identified Toll loci from genome assemblies via blastn and tblastn, using the D. melanogaster Tolls as queries. For Tolls-1, -2, -5, -6, -7, -8, and -9, the annotated transcripts across the 12 Drosophila genomes usually generated high-quality alignments with few gaps. However, in two species, D. sechellia and D. persimilis, the Toll gene model was incorrect. The D. sechellia Toll GM10345 was truncated due to a single base pair deletion in the assembly that caused a frame shift and early stop codon. We manually corrected this deletion (based on the D. simulans homologous sequence) to recover an intact, full length Toll sequence. The D. persimilis Toll had been split into two adjacent gene models, GL27223 and GL27224. We used the TIR domain from GL27224 to construct the tree of Toll genes (fig. 1).
Nearly, all of the Toll-3/4 loci required extensive manual corrections of the gene models, particularly at the N-termini. The incorrect gene models were likely due to the rapid evolution of these genes and the limited transcriptome data available for these loci. We attempted to construct more accurate Toll-3/4 gene models through a manual, homology-guided annotation of the blastn- and tblastn-identified genomic regions. For each locus, we began by constructing a preliminary gene model that combined previous annotations (if any were available) with additional genomic regions we identified through blastn/tblastn from D. melanogaster Toll-3/4s. These blast-identified regions were extended into the surrounding genomic DNA to include the largest contiguous open reading frame that was bounded by the most plausible start, stop, and/or splice sites. We then constructed multiple alignments of preliminary Toll-3/4 gene models at the nucleotide and amino acid levels using MAFFT v7.017 (Katoh et al. 2002). Within these alignments, we identified regions that were conserved among most Toll-3/4s but missing from a single sequence, searched the adjacent genomic locus for the missing homologous sequence, and altered the exon boundaries accordingly. We also surveyed the genomic loci for large open reading frames that could represent additional or extended exons, and retained these regions in the gene model if the resulting transcript aligned well with other Toll-3/4s. This process of constructing alignments and adjusting gene models was performed iteratively until no further changes could improve the alignability of the sequences. We note that this conservative annotation approach could potentially lead to an underestimate of the rate of Toll-3/4 evolution by minimizing changes in gene models that are not shared among Toll-3/4s.
We identified multiple degenerated fragments of Toll-3/4s that were severely truncated or that had acquired multiple stop codons along their length. However, we also found some Toll-3/4s that carried a single nonsense mutation while the rest of the gene was highly conserved, for example D. melanogaster Toll-3 (also known as MstProx) and D. yakuba Toll-4.yak1 (also known as GE18824). The D. melanogaster Toll-3 mutation is known to be segregating in wild populations, and is found at approximately 2% of the population in the Drosophila Genetic Reference Panel (Mackay et al. 2012). To test if the same was true of Toll-4.yak1, we sequenced this region from D. yakuba lines Tai15 and Tai18 as above using the following primers: TACTCGCTCGCATACCCATT and ACGCAGCGCAACAGAAAAAT. Both D. yakuba lines were homozygous for a single base pair insertion relative to the reference genome that resulted in a full length, intact gene. Therefore we manually corrected the sequences for these two genes to eliminate the nonsense mutations for downstream analyses.
Alignments and Phylogenies
We constructed alignments of Toll family amino acid sequences using the multiple alignment program MAFFT v7.017 (Katoh et al. 2002). To obtain alignments of the corresponding nucleotide sequences for use in positive selection analyses, we used pal2nal v14 (Suyama et al. 2006).
To construct a phylogeny across multiple Toll families (fig. 1), we used the amino acid sequences of the intracellular TIR domains, as this is the most conserved and alignable region. The boundaries of TIR domain sequences were identified using hmmscan from HMMER v3.1 and aligned as above.
We constructed the Toll-3/4 tree (fig. 3) from a larger section of the multiple protein alignment. We defined “full length” Toll-3/4 proteins as those predicted to contain an intact TIR domain and multiple LRRs by SMART (Schultz et al. 1998). Any loci that lacked open reading frames containing these domains were designated as “pseudogenes”. We aligned 61 full length sequences as above, then used Gblocks v0.91b (Talavera and Castresana 2007) to remove poorly aligned or extremely diverged regions of the alignment. We used these Gblocks parameters: Minimum number of sequences for a conserved position = 31/61, minimum number of sequences for a flanking position = 51/61; maximum number of contiguous nonconserved positions = 8, minimum length of a block = 5, gap positions allowed with half of sequences. The resulting trimmed alignment included 417 amino acid positions, representing 28% of the original alignment including the TIR domains.
Phylogenies were built using PhyML (Guindon and Gascuel 2003) using the HKY85 substitution model, estimated transition/transversion ratio, estimated gamma distribution, and approximate likelihood ratio tests. Branches with support less than 0.50 (or 50% support) were collapsed.
Finding Repetitive Elements with UCSC
We used the UCSC genome browser to examine repetitive elements near the D. yakuba Toll-3/4s. This database includes repeats identified by RepeatMasker (A.F.A. Smit, R. Hubley & P. Green RepeatMasker at http://repeatmasker.org; last accessed June 2, 2017) based on the Repbase database of repetitive elements (Jurka 2000). We then delineated the boundaries of the identified DNAREP1_DM elements in the D. yakuba genomic sequence relative to the exons/introns of the various D. yakuba Toll-3/4 paralogs.
RNA-Seq Analysis
We analyzed RNA-seq reads from the Drosophila modENCODE project (Chen et al, 2014) to look at expression in whole males and whole females of D. yakuba (SRR166820, SRR166821, SRR768435, SRR768436), D. kikkawai (SRR346732, SRR346730), and D. virilis (SRR166836, SRR166837, SRR768439, SRR768440). For D. pseudoobscura, we used reads from modENCODE series GSE31302 which included the following samples: Whole males, whole females, ovaries, testes, male carcass [without testes], and female carcass [without ovaries] (SRR166830, SRR166831, SRR166828, SRR166829, SRR330563, SRR330564, SRR330561, SRR330562, SRR330559, SRR330560, SRR330557, SRR330558). For D. willistoni, we used head, thorax and abdomen samples from Meisel et al. (2012) (SRP008012). The RNA-seq data derived from the following fly strains, which were also used for the reference genomes: D. yakuba Drosophila Species Stock Center (DSSC) #14021-0261.01, D. kikkawai DSSC #14028-0561.14, D. virilis DSSC #15010-1051.87, and D. pseudoobscura DSSC #14011-0121.94.
We used the following pipeline to trim and map the reads and to obtain FPKM expression values. We trimmed the reads for quality using Trimmomatic (Bolger et al. 2014). For paired end reads, we then discarded all those that were no longer paired after trimming. We mapped the reads using Tophat2 (Kim et al. 2013) against these genomes: dpse_r3.04, dyak_r1.05, droWil1, Dkik_2.0, and dvir_r1.06. We restricted the maximum number of alignments allowed to 2 to reduce the likelihood of reads from one Toll-3/4 paralog mapping to other paralogs and giving faulty signals of expression. This conservative approach limited our ability to detect expression from Toll-3/4 genes in species with very close paralogs, such as in D. yakuba and D. willistoni (nearest paralog pairwise nucleotide identity of 94.7% and 96.6%, respectively), but was very useful in D. kikkawai (85.9% identity), D. virilis (71.1% identity), and D. pseudoobscura (67.4% identity). We generated FPKM values using Cufflinks (Trapnell et al. 2010) with quartile normalization and effective length correction, using the reference annotations as a guide to the gene boundaries. Because most Toll-3/4 genes are incorrectly annotated or missing from reference annotations, we manually added or altered the annotations before running Cufflinks. We used gene-wise counts for the FPKM values. Occasionally, Cufflinks did not report FPKM values for the annotated gene model, but instead assigned those reads to a new gene model that was slightly longer or shorter than the annotation; in these instances, we used the FPKM values for the overlapping, nearly identical Cufflinks gene model.
Drosophila melanogaster expression values were obtained from modENCODE via Flybase (Graveley et al. 2011).
RT-PCR Experiments
We investigated the expression of the D. kikkawai Toll-3 duplicates by PCR on cDNA from D. kikkawai tissues of males and females (strain DSSC #14028-0561.00), including whole adults, heads, ovaries, testes, and the remaining carcasses after dissection of gonadal tissues. RNA was extracted, treated with TURBO DNase (Ambion), and used for cDNA synthesis (SuperScript III, Invitrogen). To test for cross-contamination of the samples with genomic DNA, “No RT” control samples for each tissue were also generated in which reverse transcriptase was excluded from the reaction. We used the following primers to examine the expression of Toll4.kik2 (AGATCAAAGACTGCGACCGT and GGTTCATATGGCCCCTCGAT) and Toll4.kik7 (ACATCTACGCTCCGGAAACC and AGGCAGTCAGGTTCTCAAAGG). We could not reliably measure the expression levels of the other D. kikkawai paralogs by RT-PCR due to their high sequence similarity to each other, which resulted in primer cross-reactivity. Toll4.kik2 and Toll4.kik7 primer sets were tested in parallel on the cDNA and on the “No RT” samples, and no bands were observed from the “No RT” reactions. To ensure the quality of the D. kikkawai cDNA, we also used ribosomal Rp49 primers on all cDNA samples, and observed similar amplification from all reactions.
Testing the Viability and Fertility of Toll-3/4 Knockdown and Overexpression Flies
The following Gal4 driver lines of D. melanogaster were used to generate Toll-3 and Toll-4 knockdown and overexpression flies: yw; Act5C-Gal4/TM6 BL3954, vas-Gal4/FM7c (m507) (Carreira-Rosario et al. 2016), w; bam-Gal4 VP16 III (Chen and McKearin 2003). The following RNAi lines were used to knockdown Toll-3: GD31513 and BL28526; or Toll-4: KK102642, GD47966, BL28543. For Toll-5 knockdown, we used GD839. The UAS-Toll-1, UAS-Toll-3, and UAS-Toll-4 lines used for overexpression were a gift from Dr Y. Tony Ip (Yagi et al. 2010).
To generate knockdown or overexpression flies, we crossed virgin females from the Gal4 driver lines to males from each of the RNAi or UAS overexpression lines on day 0. After 5–7 days, we removed the adults from the vials. On day 14–18 after setting up the crosses, we counted all adult offspring and collected the knockdown or overexpression males. To test the fertility of these males, we crossed them to 3–5 day old yw virgin females, and counted the resulting offspring. As a control, we crossed the same virgin flies to yw males. We set up the fertility crosses with two different methods, but did not observe large fertility defects with either method. For the Act5C-Gal4 crosses, we crossed single males and females in each of 10 replicate vials. We allowed the flies to mate and lay eggs for 5.5 days, then removed the adults. We discarded vials where one or both flies had died during the 5.5-day period. Eighteen days after setting up the fertility crosses, we counted all eclosed adult offspring. For the vasa- and bam-Gal4 crosses, we crossed 3 males with 3 yw virgins per vial and allowed them to mate and lay eggs for 3.5 days before removing them. We counted the eclosed adult offspring 14–17 days after setting up the cross.
Tests of Positive Selection
We tested for positive selection in multiple alignments of Toll-3/4s, as well as all other Toll family genes. Due to the extensive sequence diversity within Toll-3/4s, alignments of these proteins from all Drosophila spp. contained many gaps and poorly aligned regions. We therefore tested for positive selection on smaller Toll-3/4 subclades, attempting to balance the improved statistical power from including more sequences with the improved alignment quality from excluding highly diverged sequences. Obscura group Toll-3/4.obs2 genes were excluded because they were significantly shorter than the other Toll-3/4s, as was D. pseudoobscura Toll-4.obs9 GA32237, a partial gene on a small scaffold. The Toll-3/4s included in each subclade are listed in supplementary figure S3, Supplementary Material online and the alignments used are presented in supplementary materials S2–S5, Supplementary Material online. The subclades considered are listed in table 2.
For each amino acid alignment, we use pal2nal to obtain the corresponding nucleotide alignment and removed the poorly aligned regions with Gblocks as described above. Toll-3 and -4 retained 95%–97% of the positions in the alignment following Gblocks curation. The D. willistoni and obscura group Toll-3/4s were less well-aligned and so retained 90% and 63% of alignment positions following Gblocks, respectively. We uploaded each alignment to the Datamonkey server (Pond and Frost 2005) and ran the automated substitution model selection program. To test for recombination, we ran each alignment through GARD (Kosakovsky Pond 2006) using a general discrete model for site-to-site rate variation and 3 rate classes. If significant breakpoints were detected, we divided the alignments into multiple sections at these breakpoints and re-ran GARD on each section until no further significant breakpoints were detected. For Toll-3 and -4, there were no significant breakpoints, while the D. willistoni alignment was divided into 4 sections and the Obscura alignment was divided into 3 sections. All following analyses were performed on each alignment section separately.
We generated PhyML trees as above, which we used to run PAML and PARRIS. For each initial PAML run, we used a starting omega of 0.4 and a codon frequency model of F3x4. For the D. willistoni alignment that showed some evidence of positive selection, we also reran PAML with varied codon models (1/61 or F3X4) and starting omegas (up to 1.5) and found that the results were robust to changes in the model parameters. We examined the positions of the codons identified as positively selected in our alignments under the BEB criteria, a single residue in the obscura alignment and 24 positions in the D. willistoni N-terminal alignment. We did not consider any residues immediately adjacent to Gblocks curated regions, as these may represent low-quality regions of the alignment, leaving 20 high confidence positions.
For PARRIS, we performed all analyses within the HyPhy suite, using the codon substitution model 010012 as recommended by the HyPhy model selection algorithm.
Structural Predictions
To identify where the 20 D. willistoni positions were likely found in the Toll-3/4 protein, we used Phyre2 (Kelley et al. 2015). This algorithm uses remote homology searches and secondary structure predictions of a query amino acid sequence against a database of known crystal structures. For satisfactory regions of alignment, the program threads the query sequence onto a backbone of the known structure and models in loops and side chains to yield a structural prediction. We used the D. willistoni protein GK28112 as a query. The Phyre2 hits included TIR domains and multiple mammalian TLRs in addition to a co-crystal structure of the extracellular domain of D. melanogaster Toll bound to the Spätzle ligand (4lxr, Parthier et al., 2014). Given the phylogenetic proximity, we focused on the GK28112 prediction derived from the D. melanogaster Toll structure, which modeled residues 127–847 of GK28112. This region included 16 of the 20 positively selected residues; the remaining sites were in the N-terminal 0–126 region not modeled by Phyre2. We used the Phyre2 alignment of GK28112 and D. melanogaster Toll to map both the positively selected residues and the previously identified Spätzle-interacting residues onto the structure using Chimera (Pettersen et al. 2004). We also used the Phyre2 alignment as the seed for a consensus alignment of 4lxr, GK28112, and 19 other D. willistoni Toll-3/4s, generated using Geneious, which was then used to color the structural model by level of conservation.
Supplementary Material
Supplementary data are available at Molecular Biology and Evolution online.
Supplementary Material
Acknowledgements
The authors wish to thank L. Kursel for the use of D. kikkawai cDNA and A. Molaro, L. Kursel, and J. Young for comments on the manuscript. We are supported by a Damon Runyon Postdoctoral Fellowship (T.C.L.) and grant NIH R01 GM074108 (H.S.M.). H.S.M. is an Investigator of the Howard Hughes Medical Institute.
References
- Alcaide M, Edwards SV.. 2011. Molecular evolution of the Toll-like receptor multigene family in birds. Mol Biol Evol. 28:1703–1715. [DOI] [PubMed] [Google Scholar]
- Alexopoulou L, Holt AC, Medzhitov R, Flavell RA.. 2001. Recognition of double-stranded RNA and activation of NF-kappaB by Toll-like receptor 3. Nature 413:732–738. [DOI] [PubMed] [Google Scholar]
- Anderson KV, Jürgens G, Nüsslein-Volhard C.. 1985. Establishment of dorsal-ventral polarity in the Drosophila embryo: genetic studies on the role of the Toll gene product. Cell 42:779–789. [DOI] [PubMed] [Google Scholar]
- Areal H, Abrantes J, Esteves PJ.. 2011. Signatures of positive selection in Toll-like receptor (TLR) genes in mammals. BMC Evol Biol. 11:368.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Assis R, Bachtrog D.. 2013. Neofunctionalization of young duplicate genes in Drosophila. Proc Natl Acad Sci USA. 110:17409–17414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barreiro LB, Ben-Ali M, Quach H, Laval G, Patin E, Pickrell JK, Bouchier C, Tichit M, Neyrolles O, Gicquel B, et al. 2009. Evolutionary dynamics of human Toll-like receptors and their different contributions to host defense. PLoS Genet. 5:e1000562.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bettencourt R, Tanji T, Yagi Y, Ip YT.. 2004. Toll and Toll-9 in Drosophila innate immune response. J Endotoxin Res. 10:261–268. [DOI] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, Usadel B.. 2014. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30:2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourtzis K, Pettigrew MM, O'Neill SL.. 2000. Wolbachia neither induces nor suppresses transcripts encoding antimicrobial peptides. Insect Mol Biol. 9:635–639. [DOI] [PubMed] [Google Scholar]
- Buckley KM, Rast JP.. 2012. Dynamic evolution of toll-like receptor multigene families in echinoderms. Front Immunol. 3:136.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callaini G, Riparbelli MG, Dallai R.. 1994. The distribution of cytoplasmic bacteria in the early Drosophila embryo is mediated by astral microtubules. J Cell Sci. 107(Pt 3): 673–682. [DOI] [PubMed] [Google Scholar]
- Cardoso-Moreira M, Arguello JR, Gottipati S, Harshman LG, Grenier JK, Clark AG.. 2016. Evidence for the fixation of gene duplications by positive selection in Drosophila. Genome Res. 266:787–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carreira-Rosario A, Bhargava V, Hillebrand J, Kollipara RK, Ramaswami M, Buszczak M.. 2016. Repression of Pumilio protein expression by Rbfox1 promotes germ cell differentiation. Dev Cell 36:562–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman T. 2001. Seminal fluid-mediated fitness traits in Drosophila. Heredity 87:511–521. [DOI] [PubMed] [Google Scholar]
- Chen D, McKearin DM.. 2003. A discrete transcriptional silencer in the bam gene determines asymmetric division of the Drosophila germline stem cell. Development 130:1159–1170. [DOI] [PubMed] [Google Scholar]
- Chen ZX, Sturgill D, Qu J, Jiang H, Park S, Boley N, Suzuki AM, Fletcher AR, Plachetzki DC, FitzGerald PC, et al. 2014. Comparative validation of the D. melanogaster modENCODE transcriptome annotation. Genome Res. 24:1209–1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu JC, Jiang X, Zhao L, Hamm CA, Cridland JM, Saelao P, Hamby KA, Lee EK, Kwok RS, Zhang G, et al. 2013. Genome of Drosophila suzukii, the spotted wing drosophila. G3 3:2257–2271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christophides GK, Zdobnov E, Barillas-Mury C, Birney E, Blandin S, Blass C, Brey PT, Collins FH, Danielli A, Dimopoulos G, et al. 2002. Immunity-related genes and gene families in Anopheles gambiae. Science 298:159–165. [DOI] [PubMed] [Google Scholar]
- De Nardo D. 2015. Toll-like receptors: activation, signalling and transcriptional modulation. Cytokine 74:181–189. [DOI] [PubMed] [Google Scholar]
- Drosophila 12 Genomes Consortium, Clark AG, Eisen MB, Smith DR, Bergman CM, Oliver B, MARKOW TA, Kaufman TC, Kellis M, Gelbart W, et al. 2007. Evolution of genes and genomes on the Drosophila phylogeny. Nature 450:203–218. [DOI] [PubMed] [Google Scholar]
- Fischer K, Beatty WL, Weil GJ, Fischer PU.. 2014. High pressure freezing/freeze substitution fixation improves the ultrastructural assessment of Wolbachia endosymbiont – filarial nematode host interaction. PLoS ONE 9:e86383.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita Y, Mihara T, Okazaki T, Shitanaka M, Kushino R, Ikeda C, Negishi H, Liu Z, Richards JS, Shimada M.. 2011. Toll-like receptors (TLR) 2 and 4 on human sperm recognize bacterial endotoxins and mediate apoptosis. Hum Reprod. 26:2799–2806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerardo NM, Altincicek B, Anselme C, Atamian H, Barribeau SM, de Vos M, Duncan EJ, Evans JD, Gabaldon T, Ghanim M, et al. 2010. Immunity and other defenses in pea aphids, Acyrthosiphon pisum. Genome Biol. 11:R21.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graveley BR, Brooks AN, Carlson JW, Duff MO, Landolin JM, Yang L, Artieri CG, van Baren MJ, Boley N, Booth BW, et al. 2011. The developmental transcriptome of Drosophila melanogaster. Nature 471:473–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grueber CE, Wallis GP, Jamieson IG.. 2014. Episodic positive selection in the evolution of avian Toll-like receptor innate immunity genes. PLoS ONE 9:e89632.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guindon SXP, Gascuel O.. 2003. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol. 52:696–704. [DOI] [PubMed] [Google Scholar]
- Han M, Qin S, Song X, Li Y, Jin P, Chen L, Ma F.. 2013. Evolutionary rate patterns of genes involved in the Drosophila Toll and Imd signaling pathway. BMC Evol Biol. 13(1–1): [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heger A, Ponting CP.. 2007. Evolutionary rate analyses of orthologs and paralogs from 12 Drosophila genomes. Genome Res. 17:1837–1849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heil F, Hemmi H, Hochrein H, Ampenberger F, Kirschning C, Akira S, Lipford G, Wagner H, Bauer S.. 2004. Species-specific recognition of single-stranded RNA via toll-like receptor 7 and 8. Science 303:1526–1529. [DOI] [PubMed] [Google Scholar]
- Hoshino K, Takeuchi O, Kawai T, Sanjo H, Ogawa T, Takeda Y, Takeda K, Akira S.. 1999. Cutting edge: Toll-like receptor 4 (TLR4)-deficient mice are hyporesponsive to lipopolysaccharide: evidence for TLR4 as the Lps gene product. J Immunol. 162:3749–3752. [PubMed] [Google Scholar]
- Huang Y, Temperley ND, Ren L, Smith J, Li N, Burt DW.. 2011. Molecular evolution of the vertebrate TLR1 gene family - a complex history of gene duplication, gene conversion, positive selection and co-evolution. BMC Evol Biol. 11:149.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- International Glossina Genome Initiative. 2014. Genome sequence of the tsetse fly (Glossina morsitans): vector of African trypanosomiasis. Science 344:380–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurka J. 2000. Repbase update: a database and an electronic journal of repetitive elements. Trends Genet. 16:418–420. [DOI] [PubMed] [Google Scholar]
- Kaessmann H. 2010. Origins, evolution, and phenotypic impact of new genes. Genome Res. 20:1313–1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kambris Z, Hoffmann JA, Imler J-L, Capovilla M.. 2002. Tissue and stage-specific expression of the Tolls in Drosophila embryos. Gene Exp Patterns 2:311–317. [DOI] [PubMed] [Google Scholar]
- Katoh K, Misawa K, Kuma K-I, Miyata T.. 2002. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 30:3059–3066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley LA, Mezulis S, Yates CM, Wass MN, Sternberg MJE.. 2015. The Phyre2 web portal for protein modeling, prediction and analysis. Nat Protocols 10:845–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL.. 2013. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and genefusions. Genome Biol. 14:R36.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosakovsky Pond SL. 2006. Automated phylogenetic detection of recombination using a genetic algorithm. Mol Biol Evol. 23:1891–1901. [DOI] [PubMed] [Google Scholar]
- Lemaitre B, Nicolas E, Michaut L, Reichhart JM, Hoffmann JA.. 1996. The dorsoventral regulatory gene cassette spätzle/Toll/cactus controls the potent antifungal response in Drosophila adults. Cell 86:973–983. [DOI] [PubMed] [Google Scholar]
- Lindsay SA, Wasserman SA.. 2014. Conventional and non-conventional Drosophila Toll signaling. Dev Comp Immunol. 42:16–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackay TFC, Richards S, Stone EA, Barbadilla A, Ayroles JF, Zhu D, Casillas S, Han Y, Magwire MM, Cridland JM, et al. 2012. The Drosophila melanogaster genetic reference panel. Nature 482:173–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meisel RP, Malone JH, Clark AG.. 2012. Disentangling the relationship between sex-biased gene expression and X-linkage. Genome Res. 22:1255–1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morisato D, Anderson KV.. 1994. The spätzle gene encodes a component of the extracellular signaling pathway establishing the dorsal-ventral pattern of the Drosophila embryo. Cell 76:677–688. [DOI] [PubMed] [Google Scholar]
- Nakamoto M, Moy RH, Xu J, Bambina S, Yasunaga A, Shelly SS, Gold B, Cherry S.. 2012. Virus recognition by Toll-7 activates antiviral autophagy in Drosophila. Immunity 36:658–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narbonne-Reveau K, Charroux B, Royet J.. 2011. Lack of an antibacterial response defect in Drosophila Toll-9 mutant. PLoS ONE 6:e17470.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Neill SL, Karr TL.. 1990. Bidirectional incompatibility between conspecific populations of Drosophila simulans. Nature 348:178–180. [DOI] [PubMed] [Google Scholar]
- Obbard DJ, Welch JJ, Kim K-W, Jiggins FM.. 2009. Quantifying adaptive evolution in the Drosophila immune system. PLoS Genet. 5:e1000698.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ooi JY, Yagi Y, Hu X, Ip YT.. 2002. The Drosophila Toll-9 activates a constitutive antimicrobial defense. EMBO Rep. 3:82–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer WJ, Jiggins FM.. 2015. Comparative genomics reveals the origins and diversity of arthropod immune systems. Mol Biol Evol. 32:2111–2129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker JS, Mizuguchi K, Gay NJ.. 2001. A family of proteins related to Spätzle, the toll receptor ligand, are encoded in the Drosophila genome. Proteins 45:71–80. [DOI] [PubMed] [Google Scholar]
- Parthier C, Stelter M, Ursel C, Fandrich U, Lilie H, Breithaupt C, Stubbs MT.. 2014. Structure of the Toll-Spatzle complex, a molecular hub in Drosophila development and innate immunity. Proc Natl Acad Sci USA. 111:6281–6286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE.. 2004. UCSF Chimera–a visualization system for exploratory research and analysis. J Comput Chem. 25:1605–1612. [DOI] [PubMed] [Google Scholar]
- Poltorak A, He X, Smirnova I, Liu MY, Van Huffel C, Du X, Birdwell D, Alejos E, Silva M, Galanos C, et al. 1998. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science 282:2085–2088. [DOI] [PubMed] [Google Scholar]
- Pond SLK, Frost SDW.. 2005. Datamonkey: rapid detection of selective pressure on individual sites of codon alignments. Bioinformatics 21:2531–2533. [DOI] [PubMed] [Google Scholar]
- Quach H, Wilson D, Laval G, Patin E, Manry J, Guibert J, Barreiro LB, Nerrienet E, Verschoor E, Gessain A, et al. 2013. Different selective pressures shape the evolution of Toll-like receptors in human and African great ape populations. Hum Mol Genet. 22:4829–4840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redmond SN, Eiglmeier K, Mitri C, Markianos K, Guelbeogo WM, Gneme A, Isaacs AT, Coulibaly B, Brito-Fravallo E, Maslen G, et al. 2015. Association mapping by pooled sequencing identifies TOLL 11 as a protective factor against Plasmodium falciparum in Anopheles gambiae. BMC Genomics 16:779.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roach JC, Glusman G, Rowen L, Kaur A, Purcell MK, Smith KD, Hood LE, Aderem A.. 2005. The evolution of vertebrate Toll-like receptors. Proc Natl Acad Sci USA. 102:9577–9582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers RL, Cridland JM, Shao L, Hu TT, Andolfatto P, Thornton KR.. 2014. Landscape of Standing Variation for Tandem Duplications in Drosophila yakuba and Drosophila simulans. Mol Biol Evol. 31:1750–1766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sackton TB, Lazzaro BP, Schlenke TA, Evans JD, Hultmark D, Clark AG.. 2007. Dynamic evolution of the innate immune system in Drosophila. Nat Genet. 39:1461–1468. [DOI] [PubMed] [Google Scholar]
- Samaraweera SE, O'Keefe LV, Price GR, Venter DJ, Richards RI.. 2013. Distinct roles for Toll and autophagy pathways in double-stranded RNA toxicity in a Drosophila model of expanded repeat neurodegenerative diseases. Hum Mol Genet. 22:2811–2819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlenke TA, Begun DJ.. 2003. Natural selection drives Drosophila immune system evolution. Genetics 164:1471–1480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz J, Milpetz F, Bork P, Ponting CP.. 1998. SMART, a simple modular architecture research tool: identification of signaling domains. Proc Natl Acad Sci USA. 95:5857–5864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott JG, Warren WC, Beukeboom LW, Bopp D, Clark AG, Giers SD, Hediger M, Jones AK, Kasai S, Leichter CA, et al. 2014. Genome of the house fly, Musca domestica L., a global vector of diseases with adaptations to a septic environment. Genome Biol. 15:92.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suyama M, Torrents D, Bork P.. 2006. PAL2NAL: robust conversion of protein sequence alignments into the corresponding codon alignments. Nucleic Acids Res. 34:W609–W612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanson WJ, Vacquier VD.. 2002. The rapid evolution of reproductive proteins. Nat Rev Genet. 3:137–144. [DOI] [PubMed] [Google Scholar]
- Talavera G, Castresana J.. 2007. Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Systematic Biol. 56:564–577. [DOI] [PubMed] [Google Scholar]
- Tamura K, Subramanian S, Kumar S.. 2004. Temporal patterns of fruit fly (Drosophila) evolution revealed by mutation clocks. Mol Biol Evol 21:36–44. [DOI] [PubMed] [Google Scholar]
- Tauszig S, Jouanguy E, Hoffmann JA, Imler J-L.. 2000. Toll-related receptors and the control of antimicrobial peptide expression in Drosophila. Proc Natl Acad Sci USA. 97:10520–10525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teixeira L, Ferreira Á, Ashburner M.. 2008. The bacterial symbiont Wolbachia induces resistance to RNA viral infections in Drosophila melanogaster. PLoS Biol. 6:e2.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Temperley ND, Berlin S, Paton IR, Griffin DK, Burt DW.. 2008. Evolution of the chicken Toll-like receptor gene family: a story of gene gain and gene loss. BMC Genomics 9:62.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, Pachter L.. 2010. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol. 28:516–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valanne S, Wang JH, Ramet M.. 2011. The Drosophila Toll signaling pathway. J Immunol. 186:649–656. [DOI] [PubMed] [Google Scholar]
- VanKuren NW, Vibranovski MD.. 2014. A novel dataset for identifying sex-biased genes in Drosophila. J Genomics 2:64–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vicoso B, Bachtrog D.. 2015. Numerous transitions of sex chromosomes in Diptera. PLoS Biol. 13:e1002078.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wasbrough ER, Dorus S, Hester S, Howard-Murkin J, Lilley K, Wilkin E, Polpitiya A, Petritis K, Karr TL.. 2010. The Drosophila melanogaster sperm proteome-II (DmSP-II). J Proteomics 73:2171–2185. [DOI] [PubMed] [Google Scholar]
- Wolfner MF. 2011. Precious essences: female secretions promote sperm storage in Drosophila. PLoS Biol. 9:e1001191.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yagi Y, Nishida Y, Ip YT.. 2010. Functional analysis of Toll-related genes in Drosophila. Dev Growth Differ. 52:771–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H-P, Hung T-L, You T-L, Yang T-H.. 2006. Genomewide comparative analysis of the highly abundant transposable element DINE-1 suggests a recent transpositional burst in Drosophila yakuba. Genetics 173:189–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z. 1997. PAML: a program package for phylogenetic analysis by maximum likelihood. Comput Appl Biosci. 13:555–556. [DOI] [PubMed] [Google Scholar]
- Yarovinsky F, Zhang D, Andersen JF, Bannenberg GL, Serhan CN, Hayden MS, Hieny S, Sutterwala FS, Flavell RA, Ghosh S, et al. 2005. TLR11 activation of dendritic cells by a protozoan profilin-like protein. Science 308:1626–1629. [DOI] [PubMed] [Google Scholar]
- Zichner T, Garfield DA, Rausch T, Stutz AM, Cannavo E, Braun M, Furlong EEM, Korbel JO.. 2013. Impact of genomic structural variation in Drosophila melanogaster based on population-scale sequencing. Genome Res. 23:568–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou Z, Evans JD, Lu Z, Zhao P, Williams M, Sumathipala N, Hetru C, Hultmark D, Jiang H.. 2007. Comparative genomic analysis of the Tribolium immune system. Genome Biol. 8:R177.. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.