

Abstract
Background
During myocardial ischemia/reperfusion (MI/R) injury, there is extensive release of immunogenic metabolites that activate cells of the innate immune system. These include ATP and AMP, which upregulate chemotaxis, migration, and effector function of early infiltrating inflammatory cells. These cells subsequently drive further tissue devitalization. Mesenchymal stromal cells (MSCs) are a potential treatment modality for MI/R because of their powerful anti‐inflammatory capabilities; however, the manner in which they regulate the acute inflammatory milieu requires further elucidation. CD73, an ecto‐5′‐nucleotidase, may be critical in regulating inflammation by converting pro‐inflammatory AMP to anti‐inflammatory adenosine. We hypothesized that MSC‐mediated conversion of AMP into adenosine reduces inflammation in early MI/R, favoring a micro‐environment that attenuates excessive innate immune cell activation and facilitates earlier cardiac recovery.
Methods and Results
Adult rats were subjected to 30 minutes of MI/R injury. MSCs were encapsulated within a hydrogel vehicle and implanted onto the myocardium. A subset of MSCs were pretreated with the CD73 inhibitor, α,β‐methylene adenosine diphosphate, before implantation. Using liquid chromatography/mass spectrometry, we found that MSCs increase myocardial adenosine availability following injury via CD73 activity. MSCs also reduce innate immune cell infiltration as measured by flow cytometry, and hydrogen peroxide formation as measured by Amplex Red assay. These effects were dependent on MSC‐mediated CD73 activity. Finally, through echocardiography we found that CD73 activity on MSCs was critical to optimal protection of cardiac function following MI/R injury.
Conclusions
MSC‐mediated conversion of AMP to adenosine by CD73 exerts a powerful anti‐inflammatory effect critical for cardiac recovery following MI/R injury.
Keywords: cardioprotection, cell therapy, inflammation, ischemia, ischemia reperfusion injury, mesenchymal stem cell, myocardial inflammation
Subject Categories: Cell Therapy, Inflammation, Ischemia, Mechanisms, Stem Cells
Clinical Perspective
What Is New?
Our study identifies a novel mechanism by which mesenchymal stromal cells regulate excessive inflammation by increasing local adenosine bioavailability through CD73 enzymatic function when implanted following myocardial ischemia/reperfusion injury.
What Are the Clinical Implications?
This work adds to the growing mechanistic understanding of how cell‐based therapy is beneficial in cardiovascular disease states, and may assist in establishing optimal use of such therapies in further preclinical research.
Introduction
When cardiac cells die following myocardial ischemia/reperfusion (MI/R) injury, they release intracellular metabolites into the interstitial space that rapidly recruit and activate cells of the innate immune system.1, 2, 3, 4, 5 This cycle of cell recruitment is often self‐amplified and sustained in MI/R and leads to further cell necrosis, scar formation, and cardiac dysfunction.6, 7, 8, 9, 10, 11 Adenine nucleotides such as ATP and AMP are potent danger‐associated molecular patterns that trigger chemotactic migration, vessel wall adhesion, and pro‐inflammatory cytokine production by early infiltrating leukocytes such as neutrophils and macrophages.12, 13, 14, 15 However, these adenine nucleotides can be dephosphorylated to adenosine. While better recognized for its extensive role in the regulation of vascular tone,16 adenosine also acts through adenosine receptors on immune cells to exert counter‐regulatory signals that inhibit chemotaxis, leukocyte adhesion, and reactive oxygen species (ROS) formation. Additionally, adenosine signaling can promote angiogenic activity, anti‐inflammatory cytokine secretion, and leukocytic efferocytosis of apoptotic cells in a manner that is protective of the heart during MI/R.17, 18, 19, 20, 21
In this study, we examined whether mesenchymal stromal cells (MSCs) could curtail recruitment of innate immune cells through localized production of adenosine in a preclinical model of MI/R injury. Unlike the brief changes in tissue levels created by exogenously administered adenosine22 or adenosine‐receptor agonists,23 MSCs are capable of continuously and rapidly metabolizing ATP and AMP to adenosine by the actions of surface membrane ecto‐5′‐nucleotidases CD39 and CD73, respectively.24, 25, 26 Increased bioavailability of adenosine may subsequently favor an anti‐inflammatory microenvironment that mitigates the destructive potential of the innate immune response.24, 25, 26, 27 We found that implanted MSCs increase adenosine bioavailability through the action of CD73 and that this mechanism is critical to reducing early innate immune cell infiltration and ROS formation while protecting cardiac function following MI/R injury.
Methods
The data, analytic methods, and study materials will be/have been made available to other researchers for purposes of reproducing the results or replicating the procedure.28
Experimental Animals
Sprague‐Dawley rats (Strain 400, 250–350 g, male, 10–12 weeks; Charles River, Raleigh, NC) were used. This study conforms to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health and was approved by the Institutional Animal Care and Use Committee at Emory University School of Medicine. All procedures for animal care were conducted in accordance with institutional guidelines.
MI/R Injury Model
Rats were subjected to MI/R injury by transient ligation of the left anterior descending artery or to sham operation with ligation as previously described.29, 30, 31 Rats were anesthetized with 2.0% isoflurane gas (Diamondback Drugs, Scottsdale, AZ), intubated, and mechanically ventilated with a rodent respirator. The animal was placed in right lateral decubitus position, given 0.03 mg/kg buprenorphine (Diamondback Drugs) subcutaneously, and the chest cavity was opened by left thoracotomy to expose the heart under sterile surgical technique. After removal of the pericardium by gentle dissection, the left anterior descending artery was visualized directly and 5‐0 silk suture used to ligate the vessel ≈2 mm below its emergence from the left atrial appendage. Temporary occlusion was achieved by passing suture ends through sterile polyethylene 10 tubing so that it abutted the left anterior descending artery en face and clamping with hemostat. Occlusion of the vessel was confirmed by myocardial blanching and hypokinesis of the left ventricular (LV) free wall. Following 30 minutes of ischemic injury, the ligation was released and reperfusion confirmed by return of color to the LV myocardium. At the time of reperfusion, experimental therapy was provided. Rats that died during ischemic insult were excluded from the experiment. All sham‐injured animals had the same procedure performed without ligation.
Isolation and Culture of Rat MSCs
MSCs were isolated and enriched from rat bone marrow in a manner previously described.32 Rats were anesthetized by carbon monoxide asphyxiation. Hind limbs were removed and cleaned of connective tissue. Minimum Essential Medium Eagle, Alpha (Sigma‐Aldrich, St. Louis, MO) was used to flush marrow and cells were passed through a 70‐μm strainer. Cells were spun at 200g, 4°C for 5 minutes followed by aspiration of supernatant. Pellet was resuspended in 25 mL of Minimum Essential Medium Eagle, Alpha supplemented with 10% fetal bovine serum/1% penicillin‐streptomycin and plated on a 10‐cm culture dish at 37°C and 5% carbon dioxide incubator. Cells were cultured for 1 week and MSC identity was confirmed by isolation of CD29+CD90+CD73+CD45− surface markers.
Preparation of Biomaterial‐Supported MSCs
One million allogeneic rat MSCs were encapsulated in ultrapure alginate as previously described.33 Capsules were then localized to the anterior wall of the myocardium with a poly(ethylene)glycol hydrogel. Cell viability 24 hours after implantation was assessed by extracting MSCs and performing live‐dead assay. For the purposes of this article, we are using the term encapsulated MSCs (eMSCs) to describe combined use of alginate encapsulation and poly(ethylene)glycol‐mediated implantation for simplicity. This strategy of combined biomaterial encapsulation was performed as a validated method to improve retention and viability of MSCs at the site of implant in order to obviate known obstacles to cell delivery.33, 34 A subset of MSCs was pretreated with the specific CD73 inhibitor,35 α,β‐methylene adenosine diphosphate (APCP), as previously described,24 before encapsulation and implantation.
Quantification of Adenosine From MSC Culture Media for Liquid Chromatography/Mass Spectrometry
For in vitro experiments, rat MSCs were cultured in 24‐well plates at a density of 2.0×105/cm2 in 24‐well plates for 1 hour at 37°C with or without 50 μmol/L AMP (Sigma) in phosphate‐free incubation buffer with or without 100 μmol/L APCP as previously described.24 Culture media was collected and diluted 1:100 before liquid chromatography/mass spectrometry (LC/MS).
Preparation of Myocardial Tissue for LC/MS
Rat hearts were harvested at 1 and 3 days post MI/R injury. Hearts were rapidly perfused with adenosine deaminase inhibitor, EHNA hydrochloride (5 μmol/L, Sigma), and ligated from its attachment to the great vessels. Atria were excised just below the level of the appendages and the right ventricular free wall was removed. LV tissue was then snap frozen in liquid nitrogen, and kept at −80°C until further processing. Preparation of myocardial tissue for LC/MS was performed as previously described with the following modifications.36, 37 Frozen tissue was homogenized in prechilled acetonitrile/methanol/water (1:2:2, vol/vol/vol). Crude lysate was then heated for 10 minutes at 60°C, cooled on ice for 10 minutes, and then centrifuged at 2350g for 90 minutes at 4°C. Supernatant was isolated and further purified by centrifugation at 21 000g for 3 minutes through a 35‐μm filter. Supernatant was then diluted 1:1000 in double‐distilled H2O to prevent mass overload and then taken to LC/MS for metabolite analysis.
Quantification of Adenosine via LC/MS
The experiments were performed on Thermo‐FTMS Ultra with a Thermo Dionex Ultimate 3000 dual pump and a Shimadzu SIL‐20AC HT autosampler with a CBM20 controller. The instrument was controlled with Xcalibur and Chomeleon Xpress software supplied by Thermo. The mass spectra/mass spectra were acquired in the ion trap portion of the FTMS‐Ultra, using a center of mass collision energy of 35 V and an isolation window of 1 to 268 atomic mass units (AMU). The source parameters were capillary temperature 275°C, sheath gas 40 arbitrary units, electrospray ionization (ESI) voltage 5 kV, capillary voltage 35 V, and tube lens 110 V. A 10‐μL injection was used for each sample with 2 analyses for each sample. The chromatography used an Ascentis Express C18 15 cm × 2.1 mm, 2.7 μm (Cat#53825‐U) column with solvent A as 0.1% formic acid in water and B as acetonitrile. All solvents were LC/MS grade. The gradient used is shown in Table S1. The data were examined by integration of the peak caused by the 268 to 136 amu transition in a reconstructed ion chromatogram using Quant browser in Xcalibur software. An external standard curve from 1000 to 10 pmol was created for each analysis group for quantitation. All values were then standardized to adenosine content from sham‐injured animals.
Immune Cell Quantification by Flow Cytometry
Leukocytes were isolated from whole LV myocardium 1 day following MI/R as previously described with some modifications.30, 38, 39, 40, 41 Rats were anesthetized and perfused with 20 mL of ice cold phosphate‐buffered saline (PBS) to exclude circulating blood cells. The atrial cavities and free right ventricular wall were rapidly removed and the remaining LV rapidly dissected and minced with fine scissors. Tissue was enzymatically digested in 10 mL of solution containing collagenase type II (from Clostridium histolyticum; Sigma), and DNase I (0.002%, Grade I from bovine pancreas; Roche, Laval, Quebec) dissolved in Hanks balanced salt solution for 40 minutes at 37°C under gentle agitation at 100 rotations per minute. Following digestion, cellular material was kept at 4°C for the remainder of procession. The tissue was passed through a 70‐μm cell strainer and centrifuged at 600g for 5 minutes. The cellular pellet was resuspended in 30% Percoll (Sigma‐Aldrich, Oakville, ON, Canada) solution and overlaid onto a 70% Percoll solution and spun at 1300g for 20 minutes. The leukocyte‐enriched fraction was removed from interphase and washed with red blood cell lysis buffer. Enriched cells were aliquoted into suspensions, and labeled with the following fluorescent markers: dead stain‐AmCyan (eBioscience, #65‐0866‐14), anti‐CD45‐PE (Biolegend, #202207), anti‐myeloperoxidase‐FITC (Abcam, ab90812), anti‐CD68‐APC (Miltenyi Biotec, REA 237), and anti‐CD163‐RPE (Bio‐rad, MCA342PE). Cells were fixed with 4% paraformaldehyde and permeabilized using BD perm/wash buffer (#554723). Flow cytometry analysis was performed on LSR II instrument (BD Biosciences) and analyzed using the Cytobank Community online web tool.
Quantification of Myocardial Hydrogen Peroxide Formation
Hydrogen Peroxide (H2O2) formation was measured using Amplex Red Assay (Invitrogen, Carlsbad, CA) as previously described.42, 43 Briefly, rat hearts were perfused with ice‐cold PBS and rapidly excised 24 hours following injury. MI/R injured myocardial tissue was incubated with Amplex Red (100 μmol/L) and horseradish peroxidase (1 U/mL) for 30 minutes at 37°C in Krebs‐HEPES buffer protected from light. Fluorescence was measured (530‐nm excitation/590‐nm emission) and subtracted from background. H2O2 formation was normalized to wet weight of tissue.
Quantification of Myocardial Dysfunction
Evaluation of myocardial dysfunction was performed by transthoracic echocardiography at baseline and days 1, 2, 7, 14, and 28 following MI/R as previously described.44, 45 Rats were first anesthetized using 2% isoflurane and placed on a staging platform ventral side up with chest wall fur removed. The concentration of isoflurane was adjusted between 1% and 3% to maintain a heart rate between 330 and 360 beats/min during imaging sessions. Cardiac image sequences were acquired using a Vevo3100 Imaging platform at a frame rate of 310 frames per second using a 13‐ to 24‐mHz ultra‐high‐frequency linear array transducer (MX250, VisualSonics Inc., Toronto, ON, Canada). Images for speckle‐tracking analysis were acquired in the parasternal long‐axis views and included LV anterior and inferior wall from base to apex with visualization of the mitral valve, aorta, and papillary muscles.45 Two‐dimensional echocardiographic images obtained without significant shadowing artifact in parasternal long‐axis were analyzed using 2‐dimensional speckle‐tracking software (Vevostrain Analysis; VisualSonics Inc). Global longitudinal strain (GLS) and global longitudinal strain rate were determined by average peak strain and strain rate, respectively, across all segments of the myocardium. Data were measured over 3 heartbeats and averaged.46
Assessment of Viability From Explanted eMSCs
eMSCs with or without APCP pretreatment were explanted from animals at days 1 and 3 following MI/R injury. MSC containing alginate capsules were separated from polyethylene glycol adhesive by rinsing explants with 10 mL of saline, mincing, and digesting in 2 mL of 0.1% collagenase type II (Worthington, Cat# LS004176) in PBS for 10 minutes at 37°C while shaking at 60 rpm. Capsules were transferred into 2 mL of saline, stained with live/dead kit (Invitrogen, Cat# L3224) by following manufacturer's specifications. Maximal intensity projection images were obtained on a Zeiss LSM 510 Meta confocal microscope at ×10 magnification.
Human MSCs
Human MSCs satisfying the accepted standard definition were obtained from the Emory Personalized Immunotherapy Core (EPIC). All cellular products from the EPIC core are compliant with the US Food and Drug Administration phase I Good Manufacturing Process standards.
Human Neutrophil Isolation
Human neutrophils were obtained from donors as previously described.47 All studies involving drawing of human blood were approved by the Emory University Institutional Review Board and were in accordance with institutional guidelines. All donors gave consent before blood draw. Ten to 15 mL of blood was drawn in an EDTA tube, overlaid on 5 mL lympholyte cell separation media (Cedarlane Labs #CL5070; Burlington), and centrifuged at 500g for 35 minutes at 20°C. Neutrophils were collected from the interphase and diluted into 10 mL of Hanks balanced salt solution without calcium/magnesium. Suspension was spun at 350g for 10 minutes at 20°C and supernatant was removed. Pellet was washed with red cell lysis buffer, centrifuged at 250g for 5 minutes using minimal brake, and supernatant was removed. Two percent human serum albumin in 500 μL Hanks balanced salt solution without calcium/magnesium was used to resuspend the pellet.
Detection of ROS Formation by Neutrophils Cultured In Vitro
Freshly isolated human neutrophils were seeded onto 96‐well plates at a density of 1×105 cells/well in 30 μL of RPMI‐1640 media. For MSC co‐culture experiments, wells were preseeded with human MSCs 1×104 cells/well. Inflammatory stimulation was invoked by addition of 10 ng/mL of tumor necrosis factor α (Peprotech, Rocky Hill, NJ) and 2 μmol/L of N‐formyl‐methionyl‐leucyl‐phenylalanine (Sigma) for 2 hours. For experiments requiring detection of H2O2 formation, Amplex Red (Invitrogen, Carlsbad, CA) mix was added and fluorescence read with filter setting at 530‐nm excitation/590‐nm emission. For experiments assessing superoxide formation, hydrocyanine‐3 (LI‐COR, Lincoln, NE) was added and fluorescence read with filter setting at 540‐nm excitation/570‐nm emission by Synergy H1 Plate Reader (Biotek, Winooski, VT).
Statistical Analysis
Values are presented as mean±SEM. Data among multiple groups were compared using 1‐way ANOVA followed by Tukey's post hoc analysis. For myocardial dysfunction, all data were analyzed without multiple testing correction, as differences between treatment groups, but not within treatment groups over time, were sought at specific time points. Data between 2 groups were compared using unpaired 2‐tailed t test. A P<0.05 was considered significant. All statistical analysis was performed on GraphPad Prism 5.0 (GraphPad Prism Software Inc, San Diego, CA,).
Results
MSCs Support Myocardial Adenosine Bioavailability Acutely Following MI/R via CD73 Activity
Adenosine bioavailability is an important determinant of tissue‐level inflammation. Here, we used LC/MS to measure adenosine production. One day following MI/R injury, there was significant loss of adenosine bioavailability in animals treated with PBS or hydrogel vehicle control compared with those treated with eMSCs. We used a highly specific nonhydrolyzable CD73 antagonist, APCP,35 to deactivate CD73 activity in a subset of eMSCs before delivery. After treatment with APCP, eMSCs were unable to increase adenosine bioavailability compared with PBS or hydrogel vehicle controls (relative adenosine compared with sham per mg LV tissue, MI/R+PBS 0.68±0.11; MI/R+eMSCs 1.32±0.17; MI/R+APCP‐eMSCs 0.53±0.06, Figure 1A). At day 3 post MI/R injury, all treatment groups had significant reduction in adenosine bioavailability compared with sham, but there were no observable differences among any of the treatment groups (relative adenosine compared with sham per mg LV tissue, MI/R+PBS 0.34±0.15; MI/R+eMSCs 0.33±0.04; MI/R+APCP‐eMSCs 0.40±0.06, Figure 1B).
Figure 1.
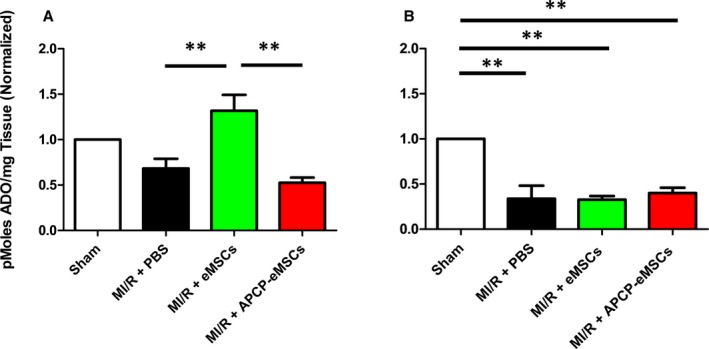
Implantation of eMSCs temporarily increases myocardial bioavailability of ADO following MI/R injury by CD73 activity. A, LC/MS analysis of LV tissue 1 day following MI/R injury demonstrates significantly higher levels of ADO 1 day following injury compared with PBS control. This elevated level of ADO, however, is not observed in animals treated with APCP‐eMSCs. ADO levels 1 day after MI/R were not significantly different in any treatment group from sham injury. B, ADO content of LV tissue 3 days after injury demonstrated significant loss of ADO bioavailability in all treatment groups compared with sham injury. No difference was detected among the PBS, eMSC, or APCP‐eMSC treatment groups. Values normalized to sham injury. Data are expressed as mean±SEM, n=5 to 6, **P<0.005. One‐way ANOVA, Tukey post hoc test. ADO indicates adenosine; APCP‐eMSCs, α,β‐methylene adenosine diphosphate pretreated eMSCs; eMSCs, encapsulated mesenchymal stromal cells; LC/MS, liquid chromatography/mass spectrometry; LV, left ventricle; MI/R, myocardial ischemia/reperfusion; PBS, phosphate‐buffered saline.
MSCs Require Both CD73 Activity and Exogenous AMP to Produce Extracellular Adenosine In Vitro
To determine which factors were critical to MSC‐mediated adenosine production, we performed LC/MS on MSC culture media. LC/MS on growth media without cells or the culture media derived from MSCs demonstrated that no detectable levels of adenosine were produced. However, upon supplementation of media with AMP, MSCs were able to generate ≈10 000 nmol of adenosine per 1×106 cells. This capacity to convert AMP to adenosine was significantly impaired when MSCs were pretreated with APCP (adenosine per 1×106 MSCs, media only nondetectable; MSCs nondetectable; MSCs+AMP 10 700±1670 nmol; MSCs+AMP+APCP 1610±250 nmol, Figure 2).
Figure 2.
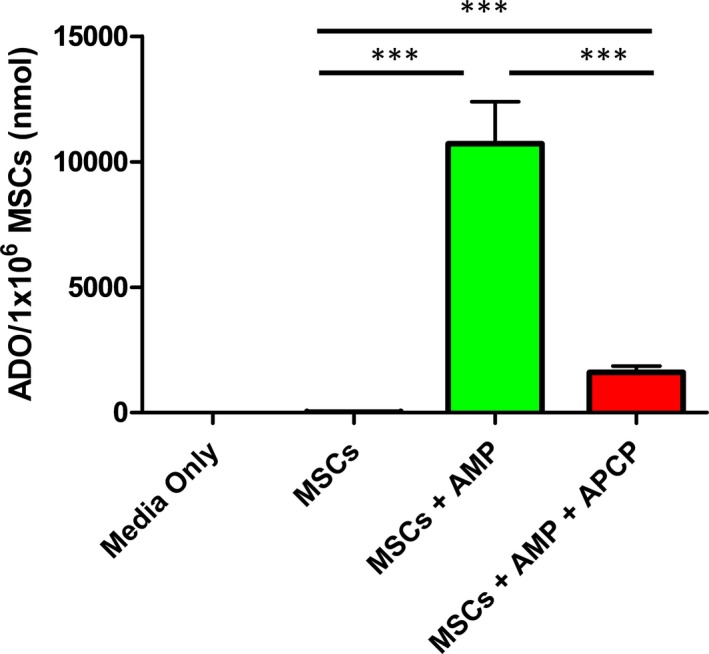
MSCs require exogenous AMP and depend on CD73 activity to produce extracellular ADO in vitro. LC/MS quantification of ADO levels from MSC culture media demonstrate the necessity of supplemental AMP to generate ADO. This production of ADO is largely inhibited by co‐treatment with APCP. Data are expressed as mean±SEM, n=6, ***P<0.0005. One‐way ANOVA, Tukey post hoc test. ADO indicates adenosine; AMP, adenosine monophosphate; APCP, α,β‐methylene adenosine diphosphate; LC/MS, liquid chromatography/mass spectrometry; MSCs, mesenchymal stromal cells.
MSCs Attenuate Early Innate Immune Cell Infiltration Following MI/R Injury via CD73 Activity
Innate immune cells such as neutrophils and macrophages are critical cell mediators of early inflammation following MI/R. They are responsible for amplifying pro‐inflammatory cytokines, generating ROS, and exacerbating cardiac dysfunction.10, 30, 48 We used flow cytometry to determine the number of immune cells within the LV tissue 1 and 3 days after MI/R. Gating strategy is demonstrated in Figure S1. Encapsulated MSCs inhibited infiltration of myocardium by CD45+ white blood cells (number of white blood cells ×103 per LV, sham 559±106; MI/R+PBS 4239±364; MI/R+eMSCs 1771±153; MI/R+APCP‐eMSCs 3979±360, Figure 3A), MPOhigh neutrophils (number of neutrophils ×103 per LV, sham 214±27; MI/R+PBS 2300±334; MI/R+eMSCs 1022±126; MI/R+APCP‐eMSCs 2301±354, Figure 3B) and CD68high macrophages (number of macrophages ×103 per LV, sham 208±53; MI/R+PBS 1417±88; MI/R+eMSCs 507±26; MI/R+APCP‐eMSCs 1427±231, Figure 3C) compared with PBS‐treated animals at 24 hours. Furthermore, at 24 hours eMSCs pretreated with APCP lost their ability to inhibit total leukocyte, neutrophil, and macrophage infiltration compared with eMSCs that were not pretreated with APCP. Hydrogel vehicle had no effect on immune cell infiltration at 24 hours compared with PBS‐treated animals in all populations examined including CD45+ leukocytes (number of white blood cells ×103 per LV, MI/R+PBS 4240±360; MI/R+hydrogel 3390±370, Figure S2A), MPOhigh neutrophils (number of neutrophils ×103 per LV, MI/R+PBS 2300±334; MI/R+hydrogel 2146±217, Figure S2B), and CD68high macrophages (number of macrophages ×103 per LV, MI/R+PBS 1417±88; MI/R+hydrogel 1414±180, Figure S2C). Enumeration of immune cells 3 days after injury demonstrated that implantation of eMSCs reduced total CD45+ leukocytes (number of white blood cells ×103 per LV, sham 302±62; MI/R+PBS 3170±575; MI/R+eMSCs 1110±213; MI/R+APCP‐eMSCs 1530±501, Figure 3D) and CD68high macrophages (number of macrophages ×103 per LV, sham 102±15; MI/R+PBS 196±277; MI/R+eMSCs 680±105; MI/R+APCP‐eMSCs 870±347, Figure 3F). Animals treated with APCP‐eMSCs did not have significant effect on leukocyte‐ and macrophage‐specific infiltration compared with eMSCs at 3 days post MI/R. MPOhigh neutrophil numbers diminished to levels seen in sham‐injured rats in all treatment groups by day 3 post‐MI/R (number of neutrophils ×103 per LV, sham 102±44; MI/R+PBS 192±55; MI/R+eMSCs 225±121; MI/R+APCP‐eMSCs 172±35, Figure 3E) and were not significantly different.
Figure 3.
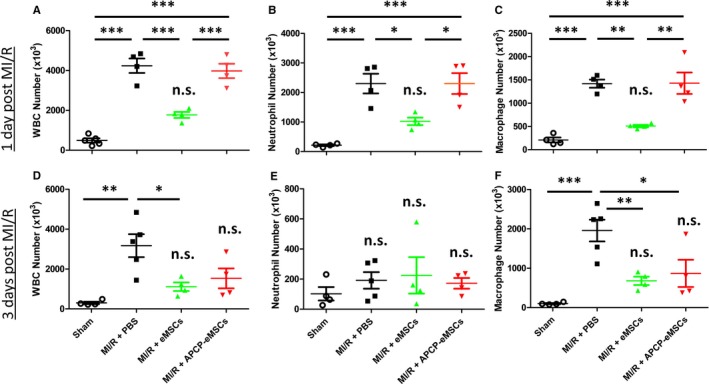
Implantation of eMSCs reduces infiltration of innate immune cells at 1 and 3 days after myocardial MI/R injury. Quantification of innate immune cell subtypes by flow cytometry in animals treated with PBS, eMSCs, or APCP‐eMSC. Animals treated with eMSCs had significantly reduced infiltration of (A) CD45+ WBCs, (B) MPO high neutrophils, and (C) CD68high macrophages compared with PBS 1 day following injury. Animals treated with APCP‐eMSCs had no significant reduction of any cell type quantified compared with PBS control 1 day after injury. Implantation of eMSCs also lead to sustained reduction in overall (D) CD45+ WBC infiltration compared with PBS‐treated controls 3 days after MI/R injury. None of the treatment groups had significantly different (E) MPO high neutrophil infiltration compared with sham injury at 3 days. F, CD68high macrophage infiltration was significantly reduced by eMSC‐treated animals compared with PBS control. APCP‐eMSC‐treated animals also did not demonstrate significant differences compared with sham at 3 days following injury. Data are expressed as mean±SEM, n=4 to 5, *P<0.05, **P<0.005, ***P<0.0005, n.s. indicates not significant compared with sham. One‐way ANOVA, Tukey post hoc test. APCP‐eMSCs indicates α,β‐methylene adenosine diphosphate pretreated eMSCs; eMSCs, encapsulated mesenchymal stromal cells; MI/R, myocardial ischemia/reperfusion injury; MPO, myeloperoxidase; PBS, phosphate‐buffered saline; WBCs, white blood cells.
MSCs Require CD73 Activity to Prevent Increases in Myocardial H2O2 Formation Following MI/R
ROS are danger‐associated molecular patterns that both trigger and arise from cellular stress, serving as an activation signal for the innate immune response.10, 49 Their formation triggers endogenous pattern recognition receptors such as Toll‐like receptors that activate innate immunity in mammalian tissue.11, 50 In vitro, MSCs reduce neutrophil ROS production including generation of H2O2 and superoxide when in coculture with isolated neutrophils from human blood. These findings have been previously reported in the literature and independently verified by our lab (Figure S3).51 These findings are purportedly because of signaling between adenosine and the adenosine A2a‐receptor, which regulates NADPH‐oxidase assembly in immune cells.52 To determine whether MSCs were able to disrupt the formation of ROS in vivo, we measured H2O2 formation 1 day following MI/R. Significant increases in myocardial H2O2 formation were observed in PBS‐ or hydrogel‐treated animals when compared with sham‐injured controls. In contrast, treatment of animals with eMSCs prevented significant increases in ROS formation compared with sham. Furthermore, APCP‐eMSCs were unable to prevent significant increases in ROS formation (relative H2O2 formation compared with sham per mg of LV tissue, MI/R+PBS 4.46±0.63; MI/R+hydrogel 4.32±1.47; MI/R+eMSCs 2.59±0.35; MI/R+APCP‐eMSCs 3.44±0.51, Figure 4).
Figure 4.
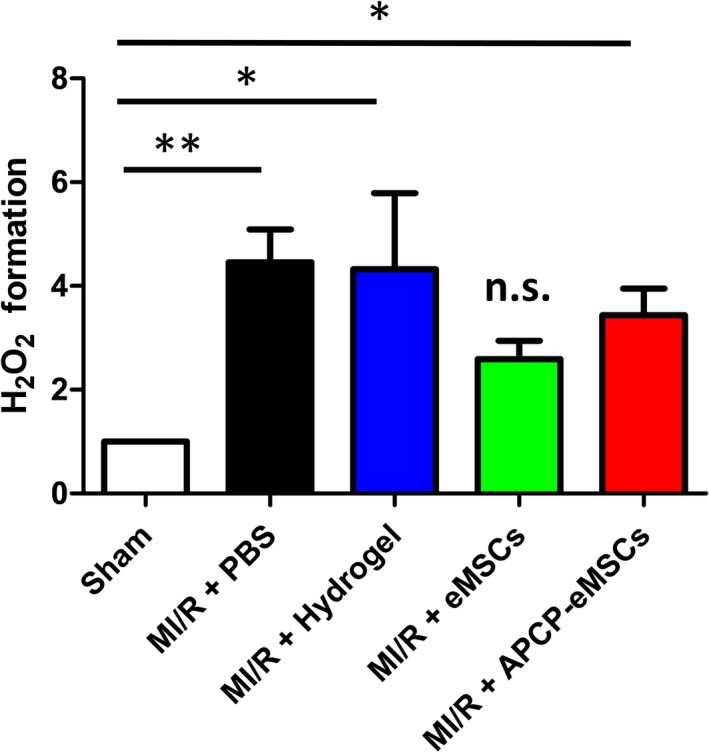
eMSCs prevent an increase in H2O2 formation via CD73 activity 1 day after MI/R injury. H2O2 formation per mg of LV myocardial tissue was measured by Amplex Red assay in animals treated with PBS, hydrogel vehicle, eMSCs, or APCP‐eMSC. Values normalized to sham treatment. n=6 to 7. Data are expressed as mean±SEM. *P<0.05, **P<0.005, n.s. indicates not significant compared with sham. One‐way ANOVA, Tukey post hoc test. APCP‐eMSCs indicates α,β‐methylene adenosine diphosphate pretreated eMSCs; eMSC, encapsulated mesenchymal stromal cells; H2O2, hydrogen peroxide; LV, left ventricle; MI/R, myocardial ischemia/reperfusion; PBS, phosphate‐buffered saline.
MSCs Require CD73 to Protect Cardiac Function Following MI/R
We examined whether CD73 activity was required for protection of hemodynamic function using 2‐dimensional and speckle‐tracking echocardiography before injury and at days 1, 2, 7, 14, and 28 following MI/R. We chose to perform strain analysis because of its higher sensitivity in detecting injury or recovery in myocardial function compared with fractional shortening. Animals given eMSCs with active CD73 function had significant improvements in GLS at days 7, 14, and 28 (GLS 7 days after injury, %, MI/R+PBS 16.5±1.2; MI/R+hydrogel 17.5±0.7; MI/R+eMSCs 25.1±0.9; MI/R+APCP‐eMSCs 14.4±1.5, Figure 5A) (GLS 14 days after injury, %, MI/R+PBS 16.0±1.9; MI/R+hydrogel 15.8±0.9; MI/R+eMSCs 25.0±1.4; MI/R+APCP‐eMSCs 16.7±1.2, Figure 5A), (GLS 28 days after injury, %, MI/R+PBS 15.9±1.3; MI/R+hydrogel 18.1±1.6; MI/R+eMSCs 27.1±1.5; MI/R+APCP‐eMSCs 19.2±1.4, Figure 5A). Global longitudinal strain rate was significantly improved in eMSC‐treated animals compared with PBS and hydrogel vehicle‐treated controls at days 14 and 28 (global longitudinal strain rate 14 days after injury, 1/s, MI/R+PBS 3.2±0.4; MI/R+hydrogel 4.1±0.5; MI/R+eMSCs 6.0±0.2; MI/R+APCP‐eMSCs 3.3±0.6, Figure 5B) (global longitudinal strain rate 28 days after injury, 1/s, MI/R+PBS 3.6±0.4; MI/R+hydrogel 3.8±0.4; MI/R+eMSCs 5.5±0.2; MI/R+APCP‐eMSCs 4.0±0.4, Figure 5B). Ejection fraction at day 28 in eMSC‐treated animals was significantly improved compared with PBS and hydrogel vehicle controls (ejection fraction 28 days after injury, %, MI/R+PBS 57.0±1.8; MI/R+hydrogel 57.2±1.7; MI/R+eMSCs 68.2±2.3; MI/R+APCP‐eMSCs 58.6±3.3, Figure 5C). There were no significant differences among the PBS, hydrogel vehicle control, and APCP‐eMSC groups at any time points measured.
Figure 5.
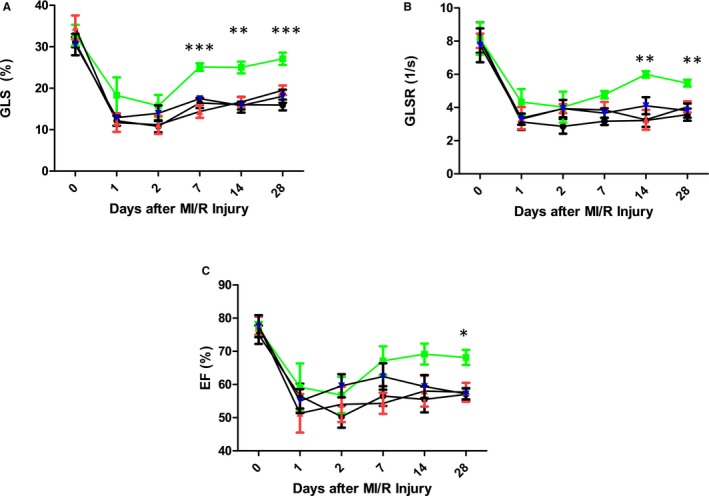
eMSCs require CD73 activity to promote recovery in cardiac function following MI/R injury. Animals treated with PBS, hydrogel vehicle control, eMSCs, or APCP‐eMSCs were assessed by speckle‐tracking echocardiography at baseline and at days 1, 2, 7, 14, and 28. A, GLS was improved from days 7 to 28, (B) GLSR was improved from days 7 to 28, and (C) EF was improved at day 28 for animals treated with eMSCs compared with all other treatment groups. APCP‐eMSC‐treated animals had no significant improvement in cardiac function compared with PBS or hydrogel vehicle control. Data are expressed as mean±SEM. n=5 to 6, *P<0.05, **P<0.005, ***P<0.0005 for MI/R+eMSCs compared with all other treatment groups. One‐way ANOVA, Tukey post hoc test. APCP‐eMSCs indicates α,β‐methylene adenosine diphosphate pretreated eMSCs; EF, ejection fraction; eMSCs, encapsulated MSCs; GLS, global longitudinal strain; GLSR, global longitudinal strain rate; MI/R, myocardial ischemia/reperfusion; PBS, phosphate‐buffered saline.
Pretreatment of MSCs With APCP Does Not Adversely Affect Short‐Term Postimplant Viability
Although off‐target effects have not previously been reported, it is unclear whether inhibition of CD73 function using the pharmacologic inhibitor APCP had an effect on MSC viability. We performed live/dead cell imaging on explanted eMSC capsules at days 1 and 3 post MI/R. Maximal intensity projections show similar viability from all recovered eMSCs capsules with or without APCP pretreatment at days 1 and 3 following MI/R injury (viability %, eMSCs 1 day 66.4±1.4; APCP‐eMSCs 1 day 63.0±2.5; eMSCs 3 day 65.6±1.6; APCP‐eMSCs 3 day 63.1±1.4, Figure 6).
Figure 6.
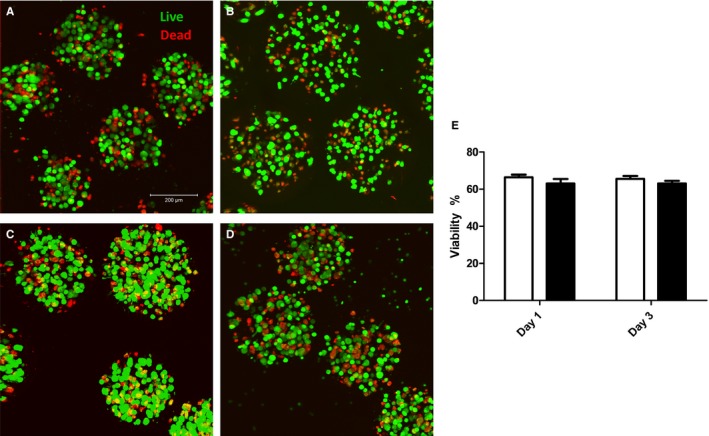
Pretreatment of eMSCs with CD73 inhibitor, APCP, does not decrease cell viability following implantation onto MI/R injured myocardium. Live/dead fluorescent imaging of eMSCs explanted from MI/R injured hearts after (A) 1 day without APCP pretreatment, (B) 1 day with APCP pretreatment, (C) 3 days without APCP pretreatment, and (D) 3 days with APCP pretreatment. E, Viability was not statistically different between eMSCs with our without APCP pretreatment at days 1 or 3. Data are expressed as mean±SEM, n=6. Unpaired t test. APCP indicates α,β‐methylene adenosine diphosphate; eMSCs, encapsulated mesenchymal stromal cells; MI/R, myocardial ischemia/reperfusion.
Discussion
The innate immune response is vital for stabilizing early cardiac injury, but the signaling cascade is often amplified by the release of danger‐associated molecular patterns from damaged cells, leading to worsening of MI/R injury.11 We demonstrate for the first time that MSCs reduce acute inflammation associated with innate immunity in vivo by increasing adenosine bioavailability via surface ecto‐5′‐nucleotidase CD73. While MSCs have also been shown to use cytokines, exosomes, and other means to regulate inflammation, our data suggest that purinergic signaling is also important and powerful. First, we show that MSCs increase bioavailability of adenosine shortly following MI/R through a CD73‐dependent mechanism. Second, we show that MSC‐mediated adenosine production inhibits innate immune cell infiltration within injured myocardium. Third, we show that MSC‐mediated adenosine prevents significant increases in ROS formation following injury. While this effect is likely attributable to reduction of ROS formation from several cell sources (ie, myocytes and fibroblasts), neutrophils are a known significant source of H2O2 and superoxide in vivo.53 Finally, we show that MSCs require CD73‐mediated adenosine production to improve cardiac function following MI/R injury. We also validated that the use of the pharmacologic inhibitor APCP significantly impairs the capacity to metabolize AMP to adenosine, without causing appreciable adverse effect on MSC viability postimplantation. Together, these findings suggest that MSCs convert pro‐inflammatory AMP to anti‐inflammatory adenosine, subsequently triggering an “off” signal for the deleterious effects of activated innate immune cells.
CD73 is an important ecto‐5′‐nucleotidase in cardiovascular disease in both animals and humans. Transgenic mice with CD73 knockout have increased susceptibility to MI/R injury because of capillary dysfunction and innate immune cell extravasation.54 Upon treatment with adenosine‐receptor agonist or soluble CD73, these transgenic mice exhibit similar response to injury compared with wild‐type controls.54 Interestingly, a phenotype in humans with CD73 deficiency has also been identified. These individuals have premature peripheral arterial calcifications and are affected by debilitating claudication.55 The findings illustrate a crucial role of CD73‐mediated adenosine production in regulating vascular inflammation. MSC therapy may be beneficial in certain cases by delivering a functionally robust, CD73‐enriched population of cells that maintain their viability and activity in vulnerable tissue. Purinergic metabolism may work in conjunction with other described mechanisms such as paracrine factors, mitochondrial/organelle transfer, and extracellular vesicle formation to cultivate an anti‐inflammatory, pro‐regenerative microenvironment56; however, the relative importance and interaction between these mechanisms in cardiac disease remain unresolved.
Although extensively recognized for its ability to induce reactive hyperemia, effects on vasomotor tone do not sufficiently explain the cardioprotective function of adenosine in MI/R. Many adenine nucleotides including ATP and AMP are potent vasodilators, yet they serve no benefit in regulating MI/R injury.16 Furthermore, the vasodilation seen in coronary blood flow with adenosine has been previously reported to be dependent on upregulation of NADPH‐oxidase–mediated ROS formation in coronary endothelial cells.57, 58 In light of our findings that MSCs are capable of increasing adenosine levels while simultaneously preventing significant increases in ROS formation, it seems unlikely that vasodilation is the primary therapeutic mechanism at play. Rather, it seems plausible that adenosine may exert cardioprotection by stimulation of adenosine A2a receptors on neutrophils and macrophages.59, 60, 61 While a number of adenosine receptor isoforms exist, stimulation of the adenosine A2a receptor pathway may predominate when biological concentrations of adenosine increase.62 Activation of the adenosine A2a‐receptor signaling pathway is implicated in phosphorylation of cyclic‐AMP responsive element‐binding protein, which in turn inhibits nuclear factor‐κβ through competitive binding at DNA transcriptional promoter sites.63 Competitive inhibition of nuclear factor‐κβ by cyclic‐AMP responsive element‐binding protein is known as 1 important downstream mechanism for disrupting activation of innate immunity.64 Alternatively, the A2b receptor has also been implicated for its role in ischemic preconditioning in a mouse model of MI/R, suggesting that CD73‐mediated adenosine production by MSCs could act on a number of downstream targets.65
In humans, therapies aimed at adenosine signaling in MI/R injury have not demonstrated clinical efficacy, with recent meta‐analysis of randomized trials demonstrating no benefit of adjunctive adenosine therapy following revascularization.22 Investigations studying the use of adenosine‐receptor agonist therapy for MI/R have also been tested extensively in experimental models59, 60, 61 but have failed in clinical trials.23 Failure of these trials may be attributable to short‐term bolus administration of adenosine or adenosine‐receptor agonists during the peri‐infarct period, which may be of too short duration to counterbalance the sustained and self‐amplifying inflammatory response. In fact, previous authors have demonstrated that the therapeutic benefit of adenosine injection in a model of MI/R is dependent on repeated injections, which may prohibit its clinical feasibility.66 Exogenously administered adenosine has an extremely limited biological half‐life, and so use of biomaterial‐supported MSCs may be a novel approach to promote constitutive conversion of AMP to adenosine during the critical hours that follow MI/R injury.33, 67
Interestingly, our findings demonstrate that even though MSCs sharply increase adenosine production 24 hours after injury, the effect is lost at 72 hours. This surprising result may be reflective of the necessity for MSCs to metabolize extracellular AMP in order to produce adenosine, as we have demonstrated in Figure 2. Myocyte necrosis is known to peak within the first 24 hours of injury,68 leading to high levels of ATP and AMP being released from myocardial cells, which in turn can be converted to adenosine by MSCs. By day 3, however, cell necrosis decreases substantially and may deprive MSCs of the extracellular AMP substrate necessary to continue generating adenosine. Despite this time‐limited effect, adenosine appears to be a potent signaling molecule within the first 24 hours of injury, which serves to blunt the rapid and robust infiltration of circulating leukocytes, a process that has been well documented to propagate myocardial necrosis.2, 53 While the duration of effect for APCP on MSCs after in vivo implantation is not completely clear, Figure 3 would suggest it is temporary since innate immune cell infiltration is significantly reduced at day 1 but not at day 3, by eMSC‐treated animals compared with those treated with APCP‐eMSCs post‐MI/R injury. These findings further support the hypothesis that even a transient reduction of activated early immune cells post‐MI/R may lead to significant long‐term improvements in cardiac function injury.6, 7 This study further highlights the important role of MSCs in regulating the complex interplay between cellular necrosis and innate immunity within the first 24 hours following injury.
Our study has certain limitations. The first is the relatively small sample size. While the authors determined that the sample sizes used in this study were sufficient to detect a significant difference between the eMSC group versus vehicle controls, it may have been underpowered to detect differences between the APCP‐eMSC or hydrogel group and PBS control, and thus may not represent true lack of effect. Second, we have not yet determined whether adenosine signaling pathways are activated on target innate immune cells, which would be important in validating the mechanism of action described here. Finally, our findings would be strengthened by genetic knockdown of CD73 expression. Although the authors have explored use of a CD73‐deficient MSC line, current evidence suggests that CD73 may be critical for cell proliferation.69, 70 Knockdown of this enzyme may limit MSC growth kinetics in vitro, which presents unique challenges for expansion before implantation, and thus requires further study.
Here, we have detailed a new powerful mechanism of the immunomodulatory potential of MSCs in a preclinical model of MI/R injury. Identifying key enzymes, such as CD73, as we have here, and establishing their temporal importance in regulation of the acute and chronic inflammatory cascade post‐MI/R will be invaluable for informing optimal design cell‐based therapeutic strategies to support myocardial salvage, cardioprotection, and conversion of the cellular microenvironment to one that favors early regeneration over inflammation in order to impart improved outcomes. Recent advances in cellular genetic editing techniques such as CRISPR‐Cas9 could be harnessed to upregulate or augment intrinsic MSCs enzyme function before implantation in order to achieve maximal benefit. Alternatively, the growing field of exosomal/hydrogel engineering has shown growing promise as a novel, efficacious disease treatment strategy.71, 72 With increasing mechanistic insight, these treatment strategies may converge and be used to guide further studies aimed at engineering the optimal use of cells or cell components for future iterations of cell‐based therapy in MI/R injury.
Sources of Funding
This study was supported by American Heart Association Scientist Development Grant (14SDG18530001) to Levit, and by the National Heart, Lung, and Blood Institute of the NIH under award number T32HL007745 to Shin.
Disclosures
None.
Supporting information
Table S1. Liquid Chromatography/Mass Spectrometry Utilized Ascentis Column With 0.1% Formic Acid in Water as Solvent (A) and Acetonitrile as Solvent (B)
Figure S1. Gating strategy to identify cell subpopulations that comprise the innate immune system.
Figure S2. Hydrogel delivery vehicle does not cause reduced innate immune cell infiltration compared with PBS control.
Figure S3. MSCs inhibit neutrophil‐mediated reactive oxygen species formation in vitro. Human neutrophils were cultured with RPMI with and without MSC co‐culture and stimulated with inflammatory TNFα/fMLP for inflammatory stimulation.
Acknowledgments
The authors of this study would like to thank the Emory Pediatric Flow Cytometry Core, the Emory Personalized Immunotherapy Core, and the Emory Mass Spectrometry Center in Chemistry for their technical support during this project.
(J Am Heart Assoc. 2018;7:e006949 DOI: 10.1161/JAHA.117.006949.)29331956
References
- 1. Hausenloy DJ, Yellon DM. Myocardial ischemia‐reperfusion injury: a neglected therapeutic target. J Clin Invest. 2013;123:92–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Yellon DM, Hausenloy DJ. Myocardial reperfusion injury. N Engl J Med. 2007;357:1121–1135. [DOI] [PubMed] [Google Scholar]
- 3. de Haan JJ, Smeets MB, Pasterkamp G, Arslan F. Danger signals in the initiation of the inflammatory response after myocardial infarction. Mediators Inflamm. 2013;2013:206039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. De Servi S, Mazzone A, Ricevuti G, Fioravanti A, Bramucci E, Angoli L, Stefano G, Specchia G. Granulocyte activation after coronary angioplasty in humans. Circulation. 1990;82:140–146. [DOI] [PubMed] [Google Scholar]
- 5. Serrano CV Jr, Ramires JA, Venturinelli M, Arie S, D'Amico E, Zweier JL, Pileggi F, da Luz PL. Coronary angioplasty results in leukocyte and platelet activation with adhesion molecule expression. Evidence of inflammatory responses in coronary angioplasty. J Am Coll Cardiol. 1997;29:1276–1283. [DOI] [PubMed] [Google Scholar]
- 6. Litt MR, Jeremy RW, Weisman HF, Winkelstein JA, Becker LC. Neutrophil depletion limited to reperfusion reduces myocardial infarct size after 90 minutes of ischemia. Evidence for neutrophil‐mediated reperfusion injury. Circulation. 1989;80:1816–1827. [DOI] [PubMed] [Google Scholar]
- 7. Romson JL, Hook BG, Kunkel SL, Abrams GD, Schork MA, Lucchesi BR. Reduction of the extent of ischemic myocardial injury by neutrophil depletion in the dog. Circulation. 1983;67:1016–1023. [DOI] [PubMed] [Google Scholar]
- 8. Mehta J, Dinerman J, Mehta P, Saldeen TG, Lawson D, Donnelly WH, Wallin R. Neutrophil function in ischemic heart disease. Circulation. 1989;79:549–556. [DOI] [PubMed] [Google Scholar]
- 9. Bidouard JP, Duval N, Kapui Z, Herbert JM, O'Connor SE, Janiak P. SSR69071, an elastase inhibitor, reduces myocardial infarct size following ischemia‐reperfusion injury. Eur J Pharmacol. 2003;461:49–52. [DOI] [PubMed] [Google Scholar]
- 10. Duilio C, Ambrosio G, Kuppusamy P, DiPaula A, Becker LC, Zweier JL. Neutrophils are primary source of O2 radicals during reperfusion after prolonged myocardial ischemia. Am J Physiol Heart Circ Physiol. 2001;280:H2649–H2657. [DOI] [PubMed] [Google Scholar]
- 11. Mann DL. The emerging role of innate immunity in the heart and vascular system: for whom the cell tolls. Circ Res. 2011;108:1133–1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ayna G, Krysko DV, Kaczmarek A, Petrovski G, Vandenabeele P, Fesus L. ATP release from dying autophagic cells and their phagocytosis are crucial for inflammasome activation in macrophages. PLoS One. 2012;7:e40069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chen Y, Yao Y, Sumi Y, Li A, To UK, Elkhal A, Inoue Y, Woehrle T, Zhang Q, Hauser C, Junger WG. Purinergic signaling: a fundamental mechanism in neutrophil activation. Sci Signal. 2010;3:ra45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Elliott MR, Chekeni FB, Trampont PC, Lazarowski ER, Kadl A, Walk SF, Park D, Woodson RI, Ostankovich M, Sharma P, Lysiak JJ, Harden TK, Leitinger N, Ravichandran KS. Nucleotides released by apoptotic cells act as a find‐me signal to promote phagocytic clearance. Nature. 2009;461:282–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. McDonald B, Pittman K, Menezes GB, Hirota SA, Slaba I, Waterhouse CC, Beck PL, Muruve DA, Kubes P. Intravascular danger signals guide neutrophils to sites of sterile inflammation. Science. 2010;330:362–366. [DOI] [PubMed] [Google Scholar]
- 16. Burnstock G. Purinergic signaling in the cardiovascular system. Circ Res. 2017;120:207–228. [DOI] [PubMed] [Google Scholar]
- 17. Headrick JP, Lasley RD. Adenosine receptors and reperfusion injury of the heart. Handb Exp Pharmacol. 2009;193:189–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kumar V, Sharma A. Adenosine: an endogenous modulator of innate immune system with therapeutic potential. Eur J Pharmacol. 2009;616:7–15. [DOI] [PubMed] [Google Scholar]
- 19. Sullivan GW, Lee DD, Ross WG, DiVietro JA, Lappas CM, Lawrence MB, Linden J. Activation of A2A adenosine receptors inhibits expression of alpha 4/beta 1 integrin (very late antigen‐4) on stimulated human neutrophils. J Leukoc Biol. 2004;75:127–134. [DOI] [PubMed] [Google Scholar]
- 20. Sullivan GW, Rieger JM, Scheld WM, Macdonald TL, Linden J. Cyclic AMP‐dependent inhibition of human neutrophil oxidative activity by substituted 2‐propynylcyclohexyl adenosine A(2A) receptor agonists. Br J Pharmacol. 2001;132:1017–1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. van der Hoeven D, Wan TC, Auchampach JA. Activation of the A(3) adenosine receptor suppresses superoxide production and chemotaxis of mouse bone marrow neutrophils. Mol Pharmacol. 2008;74:685–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Su Q, Nyi TS, Li L. Adenosine and verapamil for no‐reflow during primary percutaneous coronary intervention in people with acute myocardial infarction. Cochrane Database Syst Rev. 2015:Cd009503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kopecky SL, Aviles RJ, Bell MR, Lobl JK, Tipping D, Frommell G, Ramsey K, Holland AE, Midei M, Jain A, Kellett M, Gibbons RJ. A randomized, double‐blinded, placebo‐controlled, dose‐ranging study measuring the effect of an adenosine agonist on infarct size reduction in patients undergoing primary percutaneous transluminal coronary angioplasty: the ADMIRE (Am P579 Delivery for Myocardial Infarction REduction) study. Am Heart J. 2003;146:146–152. [DOI] [PubMed] [Google Scholar]
- 24. Chen X, Shao H, Zhi Y, Xiao Q, Su C, Dong L, Liu X, Li X, Zhang X. CD73 pathway contributes to the immunosuppressive ability of mesenchymal stem cells in intraocular autoimmune responses. Stem Cells Dev. 2016;25:337–346. [DOI] [PubMed] [Google Scholar]
- 25. Sattler C, Steinsdoerfer M, Offers M, Fischer E, Schierl R, Heseler K, Daubener W, Seissler J. Inhibition of T‐cell proliferation by murine multipotent mesenchymal stromal cells is mediated by CD39 expression and adenosine generation. Cell Transplant. 2011;20:1221–1230. [DOI] [PubMed] [Google Scholar]
- 26. Regateiro FS, Cobbold SP, Waldmann H. CD73 and adenosine generation in the creation of regulatory microenvironments. Clin Exp Immunol. 2013;171:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Barletta KE, Ley K, Mehrad B. Regulation of neutrophil function by adenosine. Arterioscler Thromb Vasc Biol. 2012;32:856–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Replication data for: ADO production by MSCs in MI/R. Harvard Dataverse; 2017. DOI: 10.7910/DVN/AFYKXF.
- 29. Tokudome S, Sano M, Shinmura K, Matsuhashi T, Morizane S, Moriyama H, Tamaki K, Hayashida K, Nakanishi H, Yoshikawa N, Shimizu N, Endo J, Katayama T, Murata M, Yuasa S, Kaneda R, Tomita K, Eguchi N, Urade Y, Asano K, Utsunomiya Y, Suzuki T, Taguchi R, Tanaka H, Fukuda K. Glucocorticoid protects rodent hearts from ischemia/reperfusion injury by activating lipocalin‐type prostaglandin D synthase‐derived PGD2 biosynthesis. J Clin Invest. 2009;119:1477–1488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Yan X, Anzai A, Katsumata Y, Matsuhashi T, Ito K, Endo J, Yamamoto T, Takeshima A, Shinmura K, Shen W, Fukuda K, Sano M. Temporal dynamics of cardiac immune cell accumulation following acute myocardial infarction. J Mol Cell Cardiol. 2013;62:24–35. [DOI] [PubMed] [Google Scholar]
- 31. Wu Y, Yin X, Wijaya C, Huang MH, McConnell BK. Acute myocardial infarction in rats. J Vis Exp. 2011;48:2464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zhang L, Chan C. Isolation and enrichment of rat mesenchymal stem cells (MSCs) and separation of single‐colony derived MSCs. J Vis Exp. 2010;37:1852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Levit RD, Landazuri N, Phelps EA, Brown ME, Garcia AJ, Davis ME, Joseph G, Long R, Safley SA, Suever JD, Lyle AN, Weber CJ, Taylor WR. Cellular encapsulation enhances cardiac repair. J Am Heart Assoc. 2013;2:e000367 DOI: 10.1161/JAHA.113.000367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Taylor DA, Chandler AM, Gobin AS, Sampaio LC. Maximizing cardiac repair: should we focus on the cells or on the matrix? Circ Res. 2017;120:30–32. [DOI] [PubMed] [Google Scholar]
- 35. Zhang B. CD73: a novel target for cancer immunotherapy. Cancer Res. 2010;70:6407–6411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Jia X, Fontaine BM, Strobel F, Weinert E. A facile and sensitive method for quantification of cyclic nucleotide monophosphates in mammalian organs: basal levels of eight cNMPs and identification of 2′,3′‐cIMP. Biomolecules. 2014;4:1070–1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Pani AK, Jiao Y, Sample KJ, Smeyne RJ. Neurochemical measurement of adenosine in discrete brain regions of five strains of inbred mice. PLoS One. 2014;9:e92422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ma Y, Chiao YA, Clark R, Flynn ER, Yabluchanskiy A, Ghasemi O, Zouein F, Lindsey ML, Jin YF. Deriving a cardiac ageing signature to reveal MMP‐9‐dependent inflammatory signalling in senescence. Cardiovasc Res. 2015;106:421–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ma Y, Yabluchanskiy A, Iyer RP, Cannon PL, Flynn ER, Jung M, Henry J, Cates CA, Deleon‐Pennell KY, Lindsey ML. Temporal neutrophil polarization following myocardial infarction. Cardiovasc Res. 2016;110:51–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Shiraishi M, Shintani Y, Shintani Y, Ishida H, Saba R, Yamaguchi A, Adachi H, Yashiro K, Suzuki K. Alternatively activated macrophages determine repair of the infarcted adult murine heart. J Clin Invest. 2016;126:2151–2166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Cotter MJ, Muruve DA. Isolation of neutrophils from mouse liver: a novel method to study effector leukocytes during inflammation. J Immunol Methods. 2006;312:68–78. [DOI] [PubMed] [Google Scholar]
- 42. Lyle AN, Deshpande NN, Taniyama Y, Seidel‐Rogol B, Pounkova L, Du P, Papaharalambus C, Lassegue B, Griendling KK. Poldip2, a novel regulator of Nox4 and cytoskeletal integrity in vascular smooth muscle cells. Circ Res. 2009;105:249–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Weber DS, Rocic P, Mellis AM, Laude K, Lyle AN, Harrison DG, Griendling KK. Angiotensin II‐induced hypertrophy is potentiated in mice overexpressing p22phox in vascular smooth muscle. Am J Physiol Heart Circ Physiol. 2005;288:H37–H42. [DOI] [PubMed] [Google Scholar]
- 44. Koshizuka R, Ishizu T, Kameda Y, Kawamura R, Seo Y, Aonuma K. Longitudinal strain impairment as a marker of the progression of heart failure with preserved ejection fraction in a rat model. J Am Soc Echocardiogr. 2013;26:316–323. [DOI] [PubMed] [Google Scholar]
- 45. Andrews TG, Lindsey ML, Lange RA, Aune GJ. Cardiac assessment in pediatric mice: strain analysis as a diagnostic measurement. Echocardiography. 2014;31:375–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ishizu T, Seo Y, Kameda Y, Kawamura R, Kimura T, Shimojo N, Xu D, Murakoshi N, Aonuma K. Left ventricular strain and transmural distribution of structural remodeling in hypertensive heart disease. Hypertension. 2014;63:500–506. [DOI] [PubMed] [Google Scholar]
- 47. Oh H, Siano B, Diamond S. Neutrophil isolation protocol. J Vis Exp. 2008;17:745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. van den Akker F, Deddens JC, Doevendans PA, Sluijter JP. Cardiac stem cell therapy to modulate inflammation upon myocardial infarction. Biochem Biophys Acta. 2013;1830:2449–2458. [DOI] [PubMed] [Google Scholar]
- 49. Weiss SJ. Tissue destruction by neutrophils. N Engl J Med. 1989;320:365–376. [DOI] [PubMed] [Google Scholar]
- 50. Frantz S, Kelly RA, Bourcier T. Role of TLR‐2 in the activation of nuclear factor kappaB by oxidative stress in cardiac myocytes. J Biol Chem. 2001;276:5197–5203. [DOI] [PubMed] [Google Scholar]
- 51. Raffaghello L, Bianchi G, Bertolotto M, Montecucco F, Busca A, Dallegri F, Ottonello L, Pistoia V. Human mesenchymal stem cells inhibit neutrophil apoptosis: a model for neutrophil preservation in the bone marrow niche. Stem Cells. 2008;26:151–162. [DOI] [PubMed] [Google Scholar]
- 52. Sharma AK, LaPar DJ, Stone ML, Zhao Y, Mehta CK, Kron IL, Laubach VE. NOX2 activation of natural killer T cells is blocked by the adenosine A(2A) receptor to inhibit lung ischemia‐reperfusion injury. Am J Respir Crit Care Med. 2016;193:988–999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Vinten‐Johansen J. Involvement of neutrophils in the pathogenesis of lethal myocardial reperfusion injury. Cardiovasc Res. 2004;61:481–497. [DOI] [PubMed] [Google Scholar]
- 54. Antonioli L, Pacher P, Vizi ES, Haskó G. CD39 and CD73 in immunity and inflammation. Trends Mol Med. 2013;19:355–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. St. Hilaire C, Ziegler SG, Markello TC, Brusco A, Groden C, Gill F, Carlson‐Donohoe H, Lederman RJ, Chen MY, Yang D, Siegenthaler MP, Arduino C, Mancini C, Freudenthal B, Stanescu HC, Zdebik AA, Chaganti RK, Nussbaum RL, Kleta R, Gahl WA, Boehm M. NT5E mutations and arterial calcifications. N Engl J Med. 2011;364:432–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Spees JL, Lee RH, Gregory CA. Mechanisms of mesenchymal stem/stromal cell function. Stem Cell Res Ther. 2016;7:125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Zhou Z, Rajamani U, Labazi H, Tilley SL, Ledent C, Teng B, Mustafa SJ. Involvement of NADPH oxidase in A2A adenosine receptor‐mediated increase in coronary flow in isolated mouse hearts. Purinergic Signal. 2015;11:263–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. El‐Awady MS, Rajamani U, Teng B, Tilley SL, Mustafa SJ. Evidence for the involvement of NADPH oxidase in adenosine receptor‐mediated control of coronary flow using A(1) and A(3) knockout mice. Physiol Rep. 2013;1:e00070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Dai W, Hale SL, Nayak R, Kloner RA. ATL 313, a selective A(2A) adenosine receptor agonist, reduces myocardial infarct size in a rat ischemia/reperfusion model. Open Cardiovasc Med J. 2009;3:166–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Kis A, Baxter GF, Yellon DM. Limitation of myocardial reperfusion injury by AMP579, an adenosine A1/A2A receptor agonist: role of A2A receptor and Erk1/2. Cardiovasc Drugs Ther. 2003;17:415–425. [DOI] [PubMed] [Google Scholar]
- 61. Meng H, McVey M, Perrone M, Clark KL. Intravenous AMP 579, a novel adenosine A(1)/A(2a) receptor agonist, induces a delayed protection against myocardial infarction in minipig. Eur J Pharmacol. 2000;387:101–105. [DOI] [PubMed] [Google Scholar]
- 62. Linden J. Regulation of leukocyte function by adenosine receptors. Adv Pharmacol. 2011;61:95–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Haskó G, Pacher P. A(2A) receptors in inflammation and injury: lessons learned from transgenic animals. J Leukoc Biol. 2008;83:447–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Wen AY, Sakamoto KM, Miller LS. The role of the transcription factor CREB in immune function. J Immunol. 2010;185:6413–6419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Eckle T, Krahn T, Grenz A, Kohler D, Mittelbronn M, Ledent C, Jacobson MA, Osswald H, Thompson LF, Unertl K, Eltzschig HK. Cardioprotection by ecto‐5′‐nucleotidase (CD73) and A2B adenosine receptors. Circulation. 2007;115:1581–1590. [DOI] [PubMed] [Google Scholar]
- 66. Yetgin T, Uitterdijk A, te Lintel Hekkert M, Merkus D, Krabbendam‐Peters I, van Beusekom HM, Falotico R, Serruys PW, Manintveld OC, van Geuns RJ, Zijlstra F, Duncker DJ. Limitation of infarct size and no‐reflow by intracoronary adenosine depends critically on dose and duration. JACC Cardiovasc Interv. 2015;8:1990–1999. [DOI] [PubMed] [Google Scholar]
- 67. Landazuri N, Levit RD, Joseph G, Ortega‐Legaspi JM, Flores CA, Weiss D, Sambanis A, Weber CJ, Safley SA, Taylor WR. Alginate microencapsulation of human mesenchymal stem cells as a strategy to enhance paracrine‐mediated vascular recovery after hindlimb ischaemia. J Tissue Eng Regen Med. 2012;10:222–232. [DOI] [PubMed] [Google Scholar]
- 68. Anversa P, Cheng W, Liu Y, Leri A, Redaelli G, Kajstura J. Apoptosis and myocardial infarction. Basic Res Cardiol. 1998;93(suppl 3):8–12. [DOI] [PubMed] [Google Scholar]
- 69. Zhang B. CD73 promotes tumor growth and metastasis. Oncoimmunology. 2012;1:67–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Wang L, Fan J, Thompson LF, Zhang Y, Shin T, Curiel TJ, Zhang B. CD73 has distinct roles in nonhematopoietic and hematopoietic cells to promote tumor growth in mice. J Clin Invest. 2011;121:2371–2382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Kamerkar S, LeBleu VS, Sugimoto H, Yang S, Ruivo CF, Melo SA, Lee JJ, Kalluri R. Exosomes facilitate therapeutic targeting of oncogenic KRAS in pancreatic cancer. Nature. 2017;546:498–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Agarwal U, George A, Bhutani S, Ghosh‐Choudhary S, Maxwell JT, Brown ME, Mehta Y, Platt MO, Liang Y, Sahoo S, Davis ME. Experimental, systems, and computational approaches to understanding the microRNA‐mediated reparative potential of cardiac progenitor cell‐derived exosomes from pediatric patients. Circ Res. 2017;120:701–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Liquid Chromatography/Mass Spectrometry Utilized Ascentis Column With 0.1% Formic Acid in Water as Solvent (A) and Acetonitrile as Solvent (B)
Figure S1. Gating strategy to identify cell subpopulations that comprise the innate immune system.
Figure S2. Hydrogel delivery vehicle does not cause reduced innate immune cell infiltration compared with PBS control.
Figure S3. MSCs inhibit neutrophil‐mediated reactive oxygen species formation in vitro. Human neutrophils were cultured with RPMI with and without MSC co‐culture and stimulated with inflammatory TNFα/fMLP for inflammatory stimulation.