

Abstract
Background
Transient receptor potential vanilloid 2 is a calcium channel activated by probenecid. Probenecid is a Food and Drug Administration–approved uricosuric drug that has recently been shown to induce positive lusitropic and inotropic effects in animal models through cardiomyocyte transient receptor potential vanilloid 2 activation. The aim of this study was to test the hypothesis that oral probenecid can improve cardiac function and symptomatology in patients with heart failure with reduced ejection fraction and to further elucidate its calcium‐dependent effects on myocyte contractility.
Methods and Results
The clinical trial recruited stable outpatients with heart failure with reduced ejection fraction randomized in a single‐center, double‐blind, crossover design. Clinical data were collected including a dyspnea assessment, physical examination, ECG, echocardiogram to assess systolic and diastolic function, a 6‐minute walk test, and laboratory studies. In vitro force generation studies were performed on cardiomyocytes isolated from murine tissue exposed to probenecid or control treatments. The clinical trial recruited 20 subjects (mean age 57 years, mean baseline fractional shortening of 13.6±1.0%). Probenecid therapy increased fractional shortening by 2.1±1.0% compared with placebo −1.7±1.0% (P=0.007). Additionally, probenecid improved diastolic function compared with placebo by decreasing the E/E′ by −2.95±1.21 versus 1.32±1.21 in comparison to placebo (P=0.03). In vitro probenecid increased myofilament force generation (92.36 versus 80.82 mN/mm2, P<0.05) and calcium sensitivity (pCa 5.67 versus 5.60, P<0.01) compared with control.
Conclusions
Probenecid improves cardiac function with minimal effects on symptomatology and no significant adverse effects after 1 week in patients with heart failure with reduced ejection fraction and increases force development and calcium sensitivity at the cardiomyocyte level.
Clinical Trial Registration
URL: https://www.clinicaltrials.gov. Unique identifier: NCT01814319.
Keywords: echocardiography, Probenecid, TRPV2
Subject Categories: Calcium Cycling/Excitation-Contraction Coupling, Clinical Studies, Contractile function, Ion Channels/Membrane Transport, Translational Studies
Clinical Perspective
What Is New?
Our animal research shows that probenecid has inotropic effects on cardiac function to the bedside by demonstrating that it improves contractility in patients with heart failure with reduced ejection fraction.
We also demonstrate that the effects of probenecid are at least partially caused by increased calcium sensitivity.
What Are the Clinical Implications?
These are the first clinical data to demonstrate that probenecid has inotropic properties in patients with heart failure and is well tolerated in the short term.
Future studies will focus on its long‐term effects as it could potentially become a novel therapeutic option for patients with heart failure with reduced ejection fraction.
Introduction
The current management of heart failure with reduced ejection fraction (HFrEF) is primarily based on afterload reduction, and increasing myocardial contractility in cases of decompensation.1 Available therapies to improve myocardial contractility are limited to digoxin and parenteral inotropes (ie, isoproterenol, dobutamine, and milrinone).2, 3 Most parenteral inotropes share the common end point of increasing cAMP levels in the cardiomyocyte leading to increased calcium influx into the cell that in turn stimulates myocyte contractility.4 Cardiomyocyte loading of calcium increases myocardial contractile force, oxygen consumption, and apoptotic signaling.5, 6, 7 Therefore, despite demonstrated improvements in cardiac function and hemodynamics, the use of inotropes in HFrEF has been demonstrated to have deleterious effects in both the hospital and outpatient setting.8, 9
The TRP family of channels has been studied for many years in the nephrology and neurology literature.10 Several vanilloid subtypes have been shown to be important mediators of vascular tone, cerebral blood flow, neointimal hyperplasia, and pulmonary hypertension.11, 12, 13 Expression of the transient receptor potential vanilloid 2 (TRPV2) channel was noted in abundance in murine myocardial tissue, specifically in the left ventricle.14 Iwata et al initially described that cardiac‐specific overexpression of TRPV2 resulted in chamber dilation of all cavities of the murine heart,15 while our laboratory recently demonstrated increased expression of TRPV2 in diseased hearts.16 Related studies by our laboratory demonstrated that stimulation of the channel with probenecid in isolated cardiomyocytes resulted in increased cytosolic calcium concentrations with improved contractility and relaxation, independently of the β‐adrenergic signaling pathway.17 Further translational studies in healthy and infarcted mice confirmed that probenecid results in improved inotropy and lusitropy in a dose‐dependent manner and does not induce malignant arrhythmias or apoptosis.18, 19
Probenecid is a lipid‐soluble benzoic acid derivative initially developed to decrease the renal tubular excretion of penicillin.20 It has also been used to increase the serum concentration of several other antibiotics and antivirals, and is Food and Drug Administration approved for the treatment of gout.14, 21, 22, 23 It has been in clinical use for >50 years with a limited adverse effect profile.2 Research interest in probenecid has recently increased with the observation that it is an agonist of TRPV2 channels.24 We have previously demonstrated that probenecid increases myocardial contractility via increased calcium cycling on a beat‐to‐beat basis and does not elicit apoptotic or arrhythmic pathways in an animal model.17 However, the effects of probenecid on sarcomeric force generation or its effects on myofilament calcium sensitivity in vitro or its cardiovascular effects in patients with HFrEF have not been tested. Thus, the goal of this study was to test the hypothesis that probenecid affects sarcomeric function distinctly from traditional beta stimulation in vitro and that oral probenecid therapy can improve myocardial contractility in vivo, while improving symptomatology in optimally treated outpatients with New York Heart Association II‐IV HFrEF.
Methods
The data, analytic methods, and study materials will be made available upon request to other researchers for purposes of reproducing the results or replicating the procedure and are available from Dr Rubinstein at the University of Cincinnati.
Clinical Trial Design
This was a randomized, double‐blind, crossover, placebo‐controlled, single‐center trial with each patient serving as his or her own control. The trial protocol was designed and written by the study principal investigator and was approved by the local institutional review board at the University of Cincinnati (IRB#2102‐4048). Upon completion of the study, the trial data were analyzed by independent academic statisticians.
Study Population
Eligible subjects were men and women of any race and ethnic origin 18 years or older with previously established stable HFrEF (left ventricular ejection fraction≤40%) (New York Heart Association II through IV) for at least 3 months on a stable dose of all cardiovascular medications. Subjects enrolled had a systemic blood pressure ≥90 mm Hg systolic and ≥60 mm Hg diastolic. Subjects were identified from the population of patients being treated for heart failure in the practices located within the University of Cincinnati Health system.
Exclusion criteria were pregnant or lactating woman, coadministration of a parenteral positive inotrope (ie, milrinone), history of significant medical noncompliance, symptomatic orthostatic changes, allergy to probenecid, history of gout or renal calculi, significant liver enzyme abnormalities (3 times upper limit of normal), known end‐stage renal disease, worsening renal insufficiency (dialysis dependent or estimated glomerular filtration rate <30), history of gastric ulcerations or significant gastroesophageal reflux, recent history within 3 months before enrollment of unstable coronary artery syndromes including unstable angina, myocardial infarction, or need for any percutaneous or surgical revascularization procedure. Patients with decompensated heart failure were excluded, as were those with history of implant of cardiac resynchronization therapy, transient ischemic attack, stroke, or major surgery in the past 3 months. Those with valvular heart disease (regurgitation or stenosis more than moderate) were excluded as well as patients with hypertrophic obstructive cardiomyopathy, myocarditis, constrictive pericarditis, congenital heart disease, active chemotherapy or malignancy, uncontrolled metabolic disease (hyperthyroidism, Cushing's disease, etc.), current atrial fibrillation with rapid ventricular response, or frequent premature ventricular contractions.
Trial Protocol
Eligible subjects provided written informed consent and were then randomized in lots of 10 with a 1:1 ratio using a preprinted randomization table. The 2 treatment arms were either oral probenecid (1 g twice daily) or placebo.
Dosing
The oral probenecid (Watson Pharmaceuticals, Parsippany, NJ) daily dose was 2 g (1 g twice per day), which based on previous publications corresponds to a mean serum concentration of 149 ng/mL.25 Prior dosing studies in animals revealed an EC50 of 49.33 mg/kg or ≈3 to 4 g daily for an average‐weight individual. The 2 g per day was chosen from previous tolerability studies of probenecid for the treatment of gout as this was the closest dose to the estimated EC50 that was still within the current Food and Drug Administration approved dose. Patients had a total of 5 study visits over 4 weeks. At the initial visit, Investigational Product (IP) (oral probenecid or matching placebo) was dispensed and the subject began the twice daily oral medication that evening. The subject returned for a 1‐week follow‐up study visit. Patients received no IP at the 1‐week follow‐up visit to allow for a 7‐day washout period. At the 2‐week visit they were dispensed the alternate arm of the IP. The subject returned for a 3‐week follow‐up and no IP was dispensed at this visit for another washout period. Finally, the subject returned for 4‐week follow‐up after being off IP for 7 days as a safety visit.
Data Collection
At each visit, subjects who qualified for the study underwent physical examination were evaluated for New York Heart Association status, and completed a current dyspnea assessment using a 5‐point Likert Scale (1=Not Short of Breath to 5=Very Severe Short of Breath). Also, after each treatment arm, patients completed an assessment of their current dyspnea relative to how they felt before beginning that specific IP on a 7‐point Likert scale (−3=Markedly better to +3=Markedly worse).25 All dyspnea assessments were completed after allowing the patient to rest in a room and before a 6‐minute walk test (6MWT). An ECG and echocardiogram were also obtained at each visit. At the end of each visit, blood was drawn for renal function and safety laboratory tests including liver function tests, N‐terminal pro b‐type natriuretic peptide, troponin I, creatinine phosphokinase and complete blood count.
Echocardiography
An echocardiographic study with limited views was performed at each visit in accordance with American Society of Echocardiography guidelines using the Vivid i ultrasound system (GE Vingmed Ultrasound AS, Horten, Norway).26 Specifically, a standardized imaging protocol was used detailing the views and sequences of the images that were to be obtained in the following order: Parasternal long‐axis images were obtained in b‐mode and aligned for mitral and aortic valve opening; a subsequent m‐mode image was obtained immediately below the mitral valve leaflet tips. Left ventricular diameters in diastole and systole were measured from the m‐mode images for calculation of fractional shortening (FS) and for calculation of end diastolic volume and end systolic volume. From the apical 4‐chamber view, the mitral inflow velocities were obtained via pulsed‐wave Doppler and from the septal and lateral walls the myocardial displacement was measured via tissue Doppler. From the pulsed‐wave Doppler the peak early velocity (E wave) was measured while from the tissue Doppler image the early peak velocity was obtained at the level of the mitral annulus of the septal and lateral walls (E′). The ratio of E/E′ was used to estimate diastolic function. All images were obtained by 2 blinded trained cardiovascular fellows and subsequently quantified in a blinded manner by a single American Society of Echocardiography (ASE) certified echocardiographer.
Electrocardiography
A 12‐lead ECG was obtained at each visit upon completion of vital signs using the 1200 Blue Wireless Rest ECG from Norav medical (Delray Beach, FL). The ECGs were blindly reviewed and compared with previous studies by a licensed practitioner on the study team.
6‐Minute Walk Test
Before the test, the subject sat quietly for 5 minutes at the beginning of the course and was asked to walk back and forth during the 6‐minute assessment period at a pace that is brisk but not exertional. After the subject completed the test, a measurement was taken from the starting point to the point where the subject was at when the 6 minutes had expired.
Statistical Analysis
The group size was calculated using power analysis to detect a 15% difference in mean changes in symptomatology (as measured via 6MWT and the Likert Scales), with an estimated SD of 10% (based on the preliminary data) with alpha of 0.05 and a power of 0.8 (with each individual serving as his or her own control).
Statistical analysis was performed using SAS v.9.3 (SAS Institute, Cary, NC). Changes in measurements before and after each treatment arm were compared between probenecid treatment and placebo. Mixed models were used to account for within‐subject correlations between the 2 study arms, and randomization order was tested to ensure lack of carryover effect, with least‐squares means and SE presented. Post hoc subgroup analyses were conducted relative to disease severity and baseline measurements. Two‐sided P<0.05 was considered statistically significant.
Murine Protocol
All animal procedures were performed with the approval of the Institutional Animal Care and Use Committee of the University of Cincinnati and in accordance with the Eighth Edition of the Guide for the Care and Use of Laboratory Animals.27
C57 wildtype mice were divided into 3 groups and treated with 100 mg/kg per day probenecid (Invitrogen, Life Technologies Corp, Eugene, OR) (n=6), 30 mg/kg per day isoproterenol (Sigma Aldrich, St. Louis, MO) (n=6), or saline (n=5) via subcutaneous osmotic pumps for 1 week. Briefly, mice were anesthetized using inhaled isoflurane (Priamal Enterprise, Andhra Pradesh, India) to sedative effect and pre‐emptively given 0.05 mg/kg buprenorphine intraperitoneally for analgesia. The hair was removed and surgical area was cleaned using alternating 70% ethanol and betadine scrubs. A small incision was made between the scapulae, primed pumps were implanted cap down, and the incision was closed using 6–0 Prolene sutures. After 1 week, the mice were euthanized, and hearts were removed and flash frozen for further analysis.
Myocyte Force Generation Analysis
To determine the effect of probenecid on sarcomeric force generation and calcium sensitivity (pCa), we isolated cardiomyocytes from hearts treated with probenecid, saline (negative control), and isoproterenol (positive control) and measured force and pCa dynamics. In brief, permeabilized (skinned) cardiomyocytes were prepared in a relax solution from frozen tissue and analyzed for force‐pCa as previously described.28 Using 1600A Permeabilized Myocyte System (Aurora Scientific Inc, ON, Canada), skinned cells were attached to 2 metal microneedles using UV‐sensitive glue (Norland, Cranbury, NJ) connected to a force transducer (Aurora Scientific Inc, ON, Canada) and a high‐speed piezo translator (Thorlabs, Newton, NJ) (Figure 1A). Myocytes were selected based on ×10 magnification, uniformity of the cell, and regular striation pattern. An 8‐well bath plate of permeabilized myocyte system allowed for seamless transition of the attached myocyte between varying calcium concentrations (pCa 10.0 to pCa 4.5) activating solution. Cells were exposed to maximum calcium saturating activating solution at the beginning and end of the experiment protocol to test the strength of cell attachment and for rundown of force generation. Cells that exhibited >20% rundown in force generation from the first maximal contraction were excluded from the study. Subsequently, isometric force development was recorded at sarcomere length 1.9 μm using activating solution with varying calcium concentrations. At each activating cycle, zero baseline force was subtracted from all force recording to obtained total isometric force. All force measurements were corrected for rundown29 and normalized to cross‐sectional area. Cell cross‐sectional area was calculated by buckling the attached myocyte and measuring dimensions of the 2 sides of the elliptical cell. Sarcomere length was continuously monitored through Aurora's High‐Speed Video Sarcomere Length measurement system. At least 3 cells per heart per group were used for the experiments. Data were acquired using Aurora's 600A real data acquisition and analysis system. Force‐pCa curves were fit using modified Hill equation (Force/Forcemax=[Ca2+]n/(pCa50n+[Ca2+]n), where n is the Hill‐slope.
Figure 1.
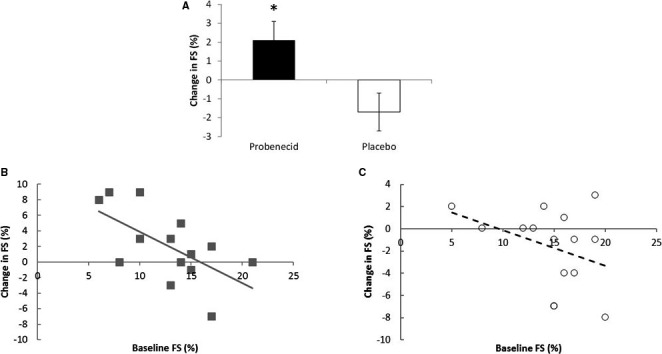
Effect of probenecid on systolic function. A, Change in fractional shortening (FS) after probenecid and placebo. B, Relationship between change in FS and baseline FS after probenecid. C, Relationship between change in FS and baseline FS after placebo. (*P<0.01).
Results
Patient Characteristics
Between June 2013 and April 2015, 20 patients were enrolled. One patient signed informed consent but did not meet inclusion/exclusion criteria and therefore no data were collected. Prior to unblinding of data, 4 additional patients were excluded from the final analysis because of poor echocardiographic image quality. In the remaining 15 patients, data were analyzed and evaluated. Baseline patient characteristics are presented in Table 1. The sample was middle‐aged (mean age 57), 80% male, and predominantly classified as New York Heart Association functional class II (93%). Nine patients had ischemic (ICMP) and 6 had nonischemic cardiomyopathy (NICMP). The mean echocardiographic left ventricular ejection fraction was low upon enrollment (28.1±8.2%). Patients were receiving maintenance therapy with β‐ blockers, angiotensin converting enzyme inhibitors, aldosterone antagonists, digoxin, and diuretics as shown in Table 1.
Table 1.
Baseline Patient Characteristics
| Characteristic | Mean±SD or Median [IQR] | Number (%) |
|---|---|---|
| Age, y | 57.7±6.8 | |
| Sex, n (%) male | 12 (80) | |
| Weight, lb | 211±38 | |
| Body mass index, kg/m2 | 30.8±6.6 | |
| Race, n (%) | ||
| White | 9 (60) | |
| Black | 6 (40) | |
| Systolic blood pressure, mm Hg | 103.7±11.6 | |
| Diastolic blood pressure, mm Hg | 66.5±8.4 | |
| Heart rate, beats/min | 64.1±8.7 | |
| Respiratory rate, breaths/min | 17.5±3.8 | |
| Left ventricular ejection fraction, % | 28.1±8.2 | |
| Left ventricular diastolic dysfunction, E/E′ | 12.2 [6.7, 17.2] | |
| NYHA functional class, n (%) NYHA II | 14 (93.3) | |
| Concomitant medications, n (%) | ||
| ACE inhibitor/ARB | 12 (80) | |
| β‐Blocker | 15 (100) | |
| Aldosterone antagonist | 9 (60) | |
| Digoxin | 4 (27) | |
| Diuretic | 13 (87) | |
| Dose (mg of furosemide or equivalent) | 40 [20, 40] | |
| NT‐proBNP, pg/mL | 1508 [216 to 1709] | |
| eGFR, mL/min per 1.73 m2 | 68 [62 to 75] | |
| Dyspnea score, Likert 5‐point | 1 [1, 1] | |
| 6MWT, feet | 1120 [1027, 1280] | |
Data are presented as n (%), mean±SD or median [interquartile range, IQR]. 6MWT indicates 6‐minute walk test; ACE, angiotensin converting enzyme; ARB, angiotensin receptor blocker; eGFR, estimated glomerular filtration rate; NT‐proBNP, N‐terminal pro‐brain natriuretic peptide; NYHA, New York Heart Association.
Left Ventricular Systolic Function and Chamber Size
On probenecid, FS increased significantly compared with placebo (2.1±1.0 versus −1.7±1.0, P=0.007; Figure 1A and Table 2). On probenecid, end diastolic volume change was significantly lower than on placebo (−14.4±11.8 versus 28.3±11.8; respectively, P=0.02). However, changes in end systolic volume and stroke volume did not significantly differ between probenecid and placebo (P=0.15 and P=0.17, respectively).
Table 2.
Echo, ECG, and Functional Outcome Changes on Probenecid and Placebo
| Probenecid | Placebo | P Value | |||||
|---|---|---|---|---|---|---|---|
| Group Mean Before | Group Mean After | Within‐Person Change | Group Mean Before | Group Mean After | Within‐Person Change | ||
| LV systolic function and chamber size | |||||||
| FS, % | 12.7±4.2 | 14.8±3.8 | 2.1±1.0 | 14.7±4.0 | 13.1±4.3 | −1.7±1.0 | 0.007 |
| Among FS ≤10% | 8.5±1.8 | 13.8±3.7 | 5.3±1.3 | 13.5±5.8 | 10.5±3.4 | −3.0±1.3 | 0.0001 |
| Among FS >10% | 15.4±2.5 | 15.4±3.9 | 0±1.1 | 15.6±2.5 | 14.8±4.0 | −0.8±1.1 | 0.41 |
| EDV, mL | 197.9±57.0 | 183.5±52.2 | −14.4±11.8 | 169.1±40.8 | 197.4±60.3 | 28.3±11.8 | 0.02 |
| ESV, mL | 138.1±57.8 | 128.5±39.0 | −9.6±14.3 | 125.3±35.1 | 146.2±56.7 | 20.9±14.3 | 0.15 |
| Stroke volume, mL | 51.3±18.7 | 56.3±20.8 | 5.00±4.24 | 53.4±18.1 | 50.9±13.0 | −2.47±4.24 | 0.17 |
| LV diastolic function | |||||||
| E/E′ ratio | 16.6±14.9 | 13.6±11.6 | −2.95±1.21 | 14.8±14.6 | 16.2±17.4 | 1.32±1.21 | 0.03 |
| E′ lateral, m/s | 0.06±0.04 | 0.07±0.04 | 0.009±0.005 | 0.07±0.04 | 0.07±0.03 | −0.009±0.005 | 0.009 |
| E′ septal, m/s | 0.05±0.02 | 0.06±0.03 | 0.005±0.003 | 0.06±0.02 | 0.05±0.02 | −0.004±0.003 | 0.07 |
| E wave, m/s | 0.74±0.28 | 0.66±0.32 | −0.03±0.03 | 0.76±0.29 | 0.67±0.34 | −0.05±0.03 | 0.71 |
| ECG assessments | |||||||
| QT interval, ms | 416±34 | 425±46 | 9.3±13.3 | 421±34 | 429±22 | 8.4±6.8 | 0.95 |
| QTc, ms | 443±42 | 464±73 | 21.0±13.2 | 448±49 | 463±57 | 17.5±18.2 | 0.86 |
| QRS width, ms | 112±13 | 120±28 | 8.0±9.3 | 109±15 | 106±20 | −4.6±5.1 | 0.12 |
| PR interval, ms | 201±42 | 195±46 | −6.0±10.1 | 192±35 | 188±23 | −6.5±9.6 | 0.96 |
| Functional assessment | |||||||
| 6MWT (feet) | 1113±353 | 1134±356 | 21.1±24.9 | 1098±341 | 1075±384 | −22.7±24.9 | 0.23 |
Mean within‐person change±SE presented for each treatment arm, from mixed‐effect models including treatment order and treatment order×treatment arm terms, if significant, and random effect of patient included to account for correlated outcomes within patients. P values are presented from these models for treatment arm differences. EDV indicates end diastolic volume; ESV, end systolic volume; FS, fractional shortening; LV, left ventricular; 6MWT, 6‐minute walk test.
Two post hoc analyses were conducted relative to the primary end point of FS by baseline severity and by cause of HFrEF. In the first post hoc analysis, we evaluated whether patients with low baseline FS (≤10%, n=6) versus higher FS (>10%, n=9) would experience different results on probenecid. The patients with a baseline FS ≤10% had an increased FS (5.3±1.3%) in comparison to those with a FS >10% who demonstrated no significant increase in FS on probenecid (0±1.1%; P=0.009). Furthermore, the interaction between baseline FS and treatment arm is highly significant (P=0.0002; Table 2). The change in an individual patient's FS relative to their baseline is shown in Figure 1B (probenecid) and 1C (placebo).
In the second post hoc analysis, the 9 patients with ICMP were compared with the 6 with NICMP. There was no significant interaction of treatment by ischemia group (P interaction=0.67), and, removing the interaction from the model, FS change also did not differ between ICMP and NICMP patients (P=0.65).
Left Ventricular Diastolic Function
The ratio of mitral E velocity to annular E′ velocity (E/E′) was used as the end point for assessment of diastolic function. On probenecid, mean E/E′ dropped significantly compared with placebo (P=0.03; Figure 2A and Table 2). However, when 2 influential individuals with very high baseline E/E′ (>40) were removed, this difference was no longer significant (−1.61±0.96 versus 0.37±0.96, respectively, P=0.11; Figure 2B). The E′ value (m/s) in the lateral wall demonstrated a statistically significant increase on probenecid in comparison to placebo (P=0.01), while the septal wall showed a nonsignificant trend (P=0.14). The change in individual patients' E/E′ relative to their baseline is shown in Figures 2B (probenecid) and 2C (placebo).
Figure 2.
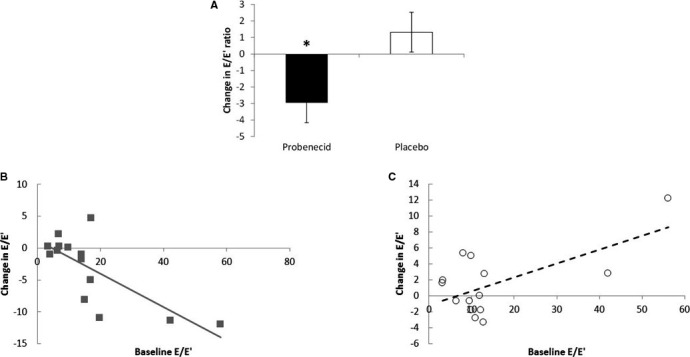
Effect of probenecid on diastolic function. A, Change in E/E′ ratio after probenecid and placebo. B, Relationship between change in E/E′ and baseline E/E′ after probenecid. C, Relationship between change in E/E′ and baseline E/E′ after placebo. (*P=0.03).
ECG Findings
In subjects without pacemakers (n=7), changes in ECG parameters on probenecid and placebo were evaluated. On probenecid, no changes were observed in QRS width (P=0.12), QT interval (P=0.95), QTc interval (P=0.86), or PR interval (P=0.96) when compared with placebo (Table 2).
Clinical Qualitative Assessments
At baseline, the current dyspnea score had a median of 1, corresponding to “Not Short of Breath,” with 2 subjects reporting mild, and 1 reporting severe shortness of breath. On probenecid, 11 of 15 (73%) patients had no change in dyspnea, while 2 experienced slight (1 point) improvement, and 2 experienced slight to moderately (2 point) worse dyspnea. On placebo, 14 of 15 (93%) had no change in dyspnea, while 1 noted slight improvement. Using the comparative scale, 4 subjects felt better after probenecid and none felt worse. Similarly, 4 subjects felt better after taking placebo but 1 felt worse. There were no significant differences in changes in heart rate between probenecid and placebo, either sitting (3.27±2.89 and 4.20±2.89 bpm, respectively, P=0.81) or standing (1.53±2.73 and 1.14±2.82 bpm, respectively; P=0.92). In addition, changes in sitting systolic blood pressure (1.6±3.6 and −2.07±3.60 mm Hg, respectively, P=0.48); standing systolic blood pressure (2.20±3.24 and 0.60±3.24 mm Hg, respectively, P=0.73); sitting diastolic blood pressure (1.29±2.09 and −0.56±2.09 mm Hg, respectively, P=0.54); standing diastolic blood pressure (−2.27±2.41 and −0.47±2.41 mm Hg, respectively, P=0.61); or body weight (0.51±0.84 and −0.49±0.84 kg, respectively, P=0.42) were not significant between probenecid and placebo treatment arms.
6‐Minute Walk Test
The mean 6MWT before probenecid administration was 1113.2±353.2 ft. Changes in distance walked after probenecid treatment (+21.1±24.9 feet) were not statistically different from placebo (−22.7±24.9 feet; P=0.23; Figure 2 and Table 2).
Neurohormones and Biomarkers
In terms of cardiac laboratory values, there were no significant differences in change in N‐terminal pro b‐type natriuretic peptide levels (−37.0±133 and −124±133 pg/mL, respectively; P=0.65) or total creatinine kinase levels (38.0±18.3 and 27.6±18.3 U/L, respectively, P=0.71) after administration of probenecid compared with placebo; troponin I changes also did not differ by treatment (P=0.14). Safety laboratory studies also did not reveal significantly different change in any of the renal (all P≥0.06), hepatic (all P≥0.36), or blood count (all P≥0.14) laboratory values between probenecid and placebo treatment arms, with the exception of a small reduction in total bilirubin during probenecid treatment compared with placebo (−0.06±0.03 and 0.02±0.03, respectively, P=0.04).
Safety
This study was under the oversight of a Data and Safety Monitoring Board. Any potential serious or nonserious adverse events were reported and were promptly adjudicated. No serious adverse reactions occurred in any of the study patients related to the IP.
Force Generation Analysis
Isometric force–pCa analysis was performed in single‐cell cardiomyocytes as sarcomere length (SL) 1.9 μm (Figure 3A) isolated from frozen hearts of mice treated with saline, isoproterenol, and probenecid continuously for 1 week. As expected, skinned cardiomyocytes from isoproterenol (104.30±3.10 mN/mm2) showed significantly increased maximal force development (pCa 4.5) compared with saline controls (80.82±2.56 mN/mm2) (Figure 3B and 3D). Treatment with probenecid also improved maximal force generation (92.36±3.27 mN/mm2) compared with saline (P<0.05); however, not to the same extent as isoproterenol treatment (104.3±3.10 mN/mm2). Similarly, myofilament calcium sensitivity of force development, expressed as pCa50, was also significantly increased in isoproterenol (pCa 5.78±0.01, P<0.0001) and probenecid (5.67±0.02, P<0.01) compared with saline controls (5.60±0.01) (Figure 3C and 3E). Again, the change in calcium sensitivity was less pronounced in probenecid when compared with isoproterenol (P<0.001). Interestingly, the Hill coefficient, which is an index of cooperative activation, was significantly decreased in isoproterenol compared with saline (Figure 3F). Conversely, there was no difference in the Hill coefficient between saline and probenecid (Figure 3F).
Figure 3.
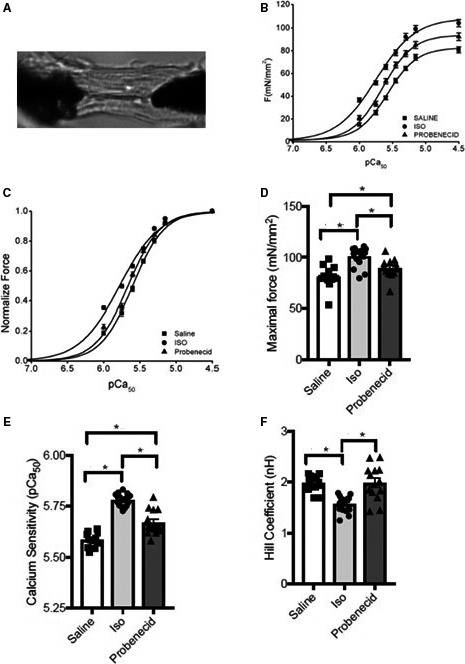
Effect of probenecid on contractility of cardiomyocytes. A, Representative figure of attached cardiomyocytes. B and D, Force‐pCa curves generated from myocytes of saline, isoproterenol (Iso), and probenecid. C and E, Calcium sensitivity of cardiomyocytes from hearts treated with saline, Iso, and probenecid. F, Hill coefficient as an index of cooperative activation of the cardiomyocytes from hearts treated with saline, Iso, and probenecid. (*P<0.05). pCa indicates calcium sensitivity.
Discussion
The development of positive inotropes for treatment of heart failure has been hampered by their adverse effects on heart rate, myocardial energy utilization, and increased risk of arrhythmias in patients with HFrEF.29, 30 The β‐adrenergic stimulation pathway in cardiomyocytes has been well described as activating cAMP, which in turn stimulates protein kinase A to phosphorylate cardiac myosin binding protein C and subsequent downstream phosphorylation of troponin I, troponin C, ryanodine receptor, and phospholamban, all leading to increases in calcium transient and amplitude of force generation.4 In contrast, the mechanism of action of probenecid is via TRPV2 activation that results in transient increases in cytosolic calcium concentrations and myocyte contractility without stimulating the β‐adrenergic pathway, affecting electrical conduction or inducing apoptosis in isolated myocytes, hanging hearts, as well as in vivo mice.17, 19 In this study, we now describe a distinct effect of probenecid in comparison to β‐adrenergic stimulation with regard to maximal isometric force and myofilament calcium sensitivity. Specifically, we demonstrate that while both treatments increase contractility and calcium sensitivity, probenecid is able to accomplish this without affecting the Hill coefficient, thus implying that the thin filaments maintain their cooperation despite increasing workload and higher sensitivity to calcium.
These important molecular findings were then taken to the bedside by administering probenecid orally to patients with HFrEF from ICMP and NICMP causes where improved systolic and diastolic function was observed. Specifically, we found that probenecid increased contractility in most patients after a single week of therapy and that the change was more pronounced in patients with lower baseline function. This finding is consistent with our prior murine studies where probenecid administration increased the EF in healthy mice between 5% and 8% and up to 19% in mice with reduced systolic function at baseline secondary to an induced ischemic event.18 Furthermore, this finding is also consistent with other previous reports that demonstrate that TRPV2 is a stretch‐mediated channel31, 32, 33; thus patients with markedly impaired systolic function and/or significantly dilated left ventricles may be particularly sensitive to its inotropic effects. Specifically, we found that probenecid increased contractility in most patients after a single week of therapy and that the change was more pronounced in patients with lower baseline function, though of course the clinical relevance of this change will require further studies as described below.
Lusitropic Effects
Murine studies from our laboratory previously documented that probenecid also has lusitropic effects.19 In this study we observed that probenecid treatment also resulted in improved diastolic function as measured via the E/E′ ratio (though most of the data were driven by those with very high baseline E/E′). These data are consistent with our basic science findings that demonstrated improved rates of shortening (+dL/dt) and relengthening (−dL/dt) in vitro and also improved rates of contraction (+dP/dt) and relaxation (−dP/dt) in vivo to probenecid.17, 19 Furthermore, in patients with mildly impaired diastolic function (those with an E/E′ between 10 and 20) we found the ratio improving mostly by an increase in the E′; while in those with very high baseline E/E′ (over 30), the ratio was improved mostly by a decrease in the E wave. Therefore, the underlying mechanisms underpinning the improved diastolic function are not immediately clear, but may be secondary to improved contractility without chronotropic effects leading to increased diastolic filling time or to improved volume status.
Safety
The improvement in cardiac function was not associated with any significant adverse events or differences in the safety laboratory tests and ECG measurements including QT interval in this trial. It is important to mention that in previous trials investigating the initial uses of probenecid in gout, there were adverse effects noted. Specifically, gastrointestinal upset was documented at doses of up to 3 g per day, which was relieved by decreasing the dose.34 In our study there were no such symptoms observed. Interestingly, the total bilirubin level decreased with probenecid. The clinical relevance of this finding is unclear, though drug‐specific effects cannot be ruled out and will be monitored closely in subsequent studies.
Symptomatology
The improved cardiac function did not appear to be associated with significant changes in the subjective assessment of symptoms, though a non–statistically significant trend was noted with regard to distance walked on the 6MWT. It is important to consider that these subjects are optimally treated outpatients and 12/15 subjects did not report being short of breath at baseline, thus not allowing for significant improvement. Additionally, it is possible that the length of treatment was too short for these changes to be observed. Future studies will seek to address both issues by exploring longer treatment times and including a more advanced HFrEF cohort of patients.
In summary, the data presented in this article support the basic and translational studies of probenecid, which demonstrated that transient increases in cytosolic calcium concentrations without stimulating the β‐adrenergic pathway are sufficient to increase contractility on a beat‐to‐beat basis without inducing the noxious pathways associated with sympathomimetic drugs.
Historical Background
Probenecid was developed in the 1940s with the explicit goal of administering it as adjuvant therapy for penicillin treatment in order to decrease penicillin excretion and decrease the dosing requirement of the antibiotic for American soldiers during the Second World War.23 Since that time, probenecid has been safely used as adjuvant therapy for other antibiotics,35 antivirals,22 and early antidepressants,36 as well as for the prevention of gout.14 The use of probenecid in patients has decreased in recent years as other therapeutic options for gout have emerged, though it is still actively used in basic science laboratories as a Fura‐2 blocker.37
To the best of our knowledge, before our reports there were no in vitro studies of probenecid on myocyte function, and the only prior report of the use of probenecid in patients with heart failure was reported in 1955 by Bronsky and colleagues. Bronsky et al attempted to measure the uricosuric and diuretic effects of probenecid (known at that time as Benemid) in 13 patients with heart failure38 and found that probenecid produced a significant diuretic effect (up to 3.9 L per day) in patients with heart failure and volume‐overloaded state, as manifested by “diminution of edema, pulmonary rales, hepatomegaly and decrease in venous pressure,” though they also reported that “diuresis with Benemid did not produce the marked urgency and increase in frequency seen with mercurials (diuretics).” It is clear that the authors likely suspected that the mechanism of action of probenecid was as a diuretic and subsequently explained that the diuresis was likely caused by an effect on the proximal renal tubule, whereas a potential cardiac effect was not considered or addressed. However, prior and subsequent studies in patients without HFrEF confirmed a uricosuric effect but have not demonstrated a direct diuretic effect. This result is, in hindsight, and in light of the data presented in this article, open to the interpretation that the diuresis seen in HFrEF patients was caused by a positive inotropic effect. As discussed below, subsequent studies will focus on the use of probenecid for patients with volume overload and acute decompensated failure such as those studied by Bronsky in order to determine whether a positive inotropic response can be elicited acutely to induce diuresis without the adverse effects commonly associated with currently used inotropic drugs.8, 39
Study Limitations
This study was designed to evaluate the effects of the drug on cardiac function, and upon completion demonstrated that after 1 week of therapy both systolic and diastolic function are improved without significant adverse effect, though symptomatology was only minimally improved. Therefore, important limitations of this study include the fact that it was a single‐center study with a short time frame and the lack of harder end points such as rehospitalization rate or mortality. Furthermore, the study used FS as a marker of systolic function, and even though the function improved in those with both NICM as well as ICM, a more precise measurement of change of function in those with a regional wall motion abnormality could not be estimated.
Many drugs have been shown to improve contractility, but have subsequently been found to increase mortality upon further study.2 This risk is reduced as probenecid has been administered safely for decades in patients with multiple comorbidities, though appropriate large‐scale, long‐term, outcome‐driven studies are needed to better establish the risk/benefit profile of the drug in patients with HFrEF.14
Conclusions
We demonstrate that probenecid increased the force generation and calcium sensitivity of single cardiomyocytes that is consistent with improved cardiac function observed in patients with HFrEF. While these increases were not as marked as in isoproterenol‐treated mice, probenecid has shown no pro‐apoptotic signaling or other negative side effects associated with sustained β1‐adrenergic receptor stimulation. Taken together, this suggests that probenecid may modulate myofilament contractility via a pathway independent of β‐adrenergic activation, and likely occurs through stimulation of cardiac TRPV2 channels, which are upregulated in patients with cardiomyopathy and when stimulated induce transient increases in cytosolic calcium concentrations to levels sufficient to increase contractility on a beat‐to‐beat basis without inducing the noxious pathways associated with sympathomimetic drugs. Future long‐term studies will be required in order to evaluate whether probenecid can be administered safely chronically in order to improve symptoms and outcomes such as rehospitalization or mortality.
Sources of Funding
The trial was financially supported by funding from the University of Cincinnati and the American Heart Association (AHA) grant ID 13BGIA17140069.
Disclosures
Dr Rubinstein is a founding member of TRP therapeutics and has not received any fees, salary, or honoraria from this venture. He is also founding member and board member of CinTRYP LLC from which he has received honoraria and royalties. He did not have access to the study data at any point prior to unblinding. All other authors have no disclosures to note.
Acknowledgments
We would like to acknowledge Drs Said Alsidawi and Xu Gao and Michael McDermott for their technical assistance as well as Dr Stephanie Dunlap and the staff from the Advanced Heart Failure Treatment Center at the University of Cincinnati.
(J Am Heart Assoc. 2018;7:e007148 DOI: 10.1161/JAHA.117.007148.)29331959
References
- 1. Gheorghiade M, Zannad F, Sopko G, Klein L, Piña IL, Konstam MA, Massie BM, Roland E, Targum S, Collins SP, Filippatos G, Tavazzi L. Acute heart failure syndromes: current state and framework for future research. Circulation. 2005;112:3958–3968. [DOI] [PubMed] [Google Scholar]
- 2. Abraham WT, Adams KF, Fonarow GC, Costanzo MR, Berkowitz RL, LeJemtel TH, Cheng ML, Wynne J. In‐hospital mortality in patients with acute decompensated heart failure requiring intravenous vasoactive medications: an analysis from the Acute Decompensated Heart Failure National Registry (ADHERE). J Am Coll Cardiol. 2005;46:57–64. [DOI] [PubMed] [Google Scholar]
- 3. Goldhaber JI, Hamilton MA. Role of inotropic agents in the treatment of heart failure. Circulation. 2010;121:1655–1660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Nagy L, Pollesello P, Papp Z. Inotropes and inodilators for acute heart failure: sarcomere active drugs in focus. J Cardiovasc Pharmacol. 2014;64:199–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Endoh M. Ca 2+ in cardiac E‐C coupling. Circ J. 2008;72:1915–1925. Available at: https://www.jstage.jst.go.jp/article/circj/72/12/72_CJ-08-0838/_pdf. Accessed June 28, 2017. [DOI] [PubMed] [Google Scholar]
- 6. Singh K, Xiao L, Remondino A, Sawyer DB, Colucci WS. Adrenergic regulation of cardiac myocyte apoptosis. J Cell Physiol. 2001;189:257–265. [DOI] [PubMed] [Google Scholar]
- 7. Felker GM, O'Connor CM. Inotropic therapy for heart failure: an evidence‐based approach. Am Heart J. 2001;142:393–401. [DOI] [PubMed] [Google Scholar]
- 8. Burger AJ, Elkayam U, Neibaur MT, Haught H, Ghali J, Horton DP, Aronson D. Comparison of the occurrence of ventricular arrhythmias in patients with acutely decompensated congestive heart failure receiving dobutamine versus nesiritide therapy. Am J Cardiol. 2001;88:35–39. [DOI] [PubMed] [Google Scholar]
- 9. Yancy CW, Jessup M, Bozkurt B, Butler J, Casey DE, Drazner MH, Fonarow GC, Geraci SA, Horwich T, Januzzi JL, Johnson MR, Kasper EK, Levy WC, Masoudi FA, McBride PE, McMurray JJV, Mitchell JE, Peterson PN, Riegel B, Sam F, Stevenson LW, Tang WHW, Tsai EJ, Wilkoff BL. 2013 ACCF/AHA guideline for the management of heart failure: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. Circulation. 2013;128:e240–e327. [DOI] [PubMed] [Google Scholar]
- 10. Clapham DE. TRP channels as cellular sensors. Nature. 2003;426:517–524. [DOI] [PubMed] [Google Scholar]
- 11. Muraki K, Iwata Y, Katanosaka Y, Ito T, Ohya S, Shigekawa M, Imaizumi Y. TRPV2 is a component of osmotically sensitive cation channels in murine aortic myocytes. Circ Res. 2003;93:829–838. [DOI] [PubMed] [Google Scholar]
- 12. Makarewich CA, Zhang H, Davis J, Correll RN, Trappanese DM, Hoffman NE, Troupes CD, Berretta RM, Kubo H, Madesh M, Chen X, Gao E, Molkentin JD, Houser SR. Transient receptor potential channels contribute to pathological structural and functional remodeling after myocardial infarction. Circ Res. 2014;115:567–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Yin J, Kuebler WM. Mechanotransduction by TRP channels: general concepts and specific role in the vasculature. Cell Biochem Biophys. 2010;56:1–18. [DOI] [PubMed] [Google Scholar]
- 14. Robbins N, Koch SE, Tranter M, Rubinstein J. The history and future of probenecid. Cardiovasc Toxicol. 2012;12:1–9. [DOI] [PubMed] [Google Scholar]
- 15. Iwata Y, Katanosaka Y, Arai Y, Komamura K, Miyatake K, Shigekawa M. A novel mechanism of myocyte degeneration involving the Ca2+‐permeable growth factor‐regulated channel. J Cell Biol. 2003;161:957–967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Koch SE, Mann A, Jones S, Robbins N, Alkhattabi A, Worley MC, Gao X, Lasko‐Roiniotis VM, Karani R, Fulford L, Jiang M, Nieman M, Lorenz JN, Rubinstein J. Transient receptor potential vanilloid 2 function regulates cardiac hypertrophy via stretch‐induced activation. J Hypertens. 2017;35:602–611. [DOI] [PubMed] [Google Scholar]
- 17. Koch SE, Gao X, Haar L, Jiang M, Lasko VM, Robbins N, Cai W, Brokamp C, Varma P, Tranter M, Liu Y, Ren X, Lorenz JN, Wang H‐S, Jones WK, Rubinstein J. Probenecid: novel use as a non‐injurious positive inotrope acting via cardiac TRPV2 stimulation. J Mol Cell Cardiol. 2012;53:134–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Koch SE, Tranter M, Robbins N, Luther K, Singh U, Jiang M, Ren X, Tee T, Smith L, Varma P, Jones WK, Rubinstein J. Probenecid as a noninjurious positive inotrope in an ischemic heart disease murine model. J Cardiovasc Pharmacol Ther. 2013;18:280–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Rubinstein J, Lasko VM, Koch SE, Singh VP, Carreira V, Robbins N, Patel AR, Jiang M, Bidwell P, Kranias EG, Jones WK, Lorenz JN. Novel role of transient receptor potential vanilloid 2 in the regulation of cardiac performance. Am J Physiol Heart Circ Physiol. 2014;306:H574–H584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Cunningham RF, Israili ZH, Dayton PG. Clinical pharmacokinetics of probenecid. Clin Pharmacokinet. 1981;6:135–151. [DOI] [PubMed] [Google Scholar]
- 21. Craft JC, Feldman WE, Nelson JD. Clinicopharmacological evaluation of amoxicillin and probenecid against bacterial meningitis. Antimicrob Agents Chemother. 1979;16:346–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Laskin OL, de Miranda P, King DH, Page DA, Longstreth JA, Rocco L, Lietman PS. Effects of probenecid on the pharmacokinetics and elimination of acyclovir in humans. Antimicrob Agents Chemother. 1982;21:804–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Boger WP. Probenecid (benemid). AMA Arch Intern Med. 1955;95:83. [DOI] [PubMed] [Google Scholar]
- 24. Bang S, Kim KY, Yoo S, Lee S‐H, Hwang SW. Transient receptor potential V2 expressed in sensory neurons is activated by probenecid. Neurosci Lett. 2007;425:120–125. [DOI] [PubMed] [Google Scholar]
- 25. Pang PS, Collins SP, Sauser K, Andrei A‐C, Storrow AB, Hollander JE, Tavares M, Spinar J, Macarie C, Raev D, Nowak R, Gheorghiade M, Mebazaa A. Assessment of dyspnea early in acute heart failure: patient characteristics and response differences between likert and visual analog scales. Acad Emerg Med. 2014;21:659–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lang RM, Bierig M, Devereux RB, Flachskampf FA, Foster E, Pellikka PA, Picard MH, Roman MJ, Seward J, Shanewise JS, Solomon SD, Spencer KT, Sutton MSJ, Stewart WJ; Chamber Quantification Writing Group, American Society of Echocardiography's Guidelines and Standards Committee, European Association of Echocardiography. . Recommendations for chamber quantification: a report from the American Society of Echocardiography's Guidelines and Standards Committee and the Chamber Quantification Writing Group, developed in conjunction with the European Association of Echocardiography, a branch of the European Society of Cardiology. J Am Soc Echocardiogr. 2005;18:1440–1463. [DOI] [PubMed] [Google Scholar]
- 27. Institutional Animal Care and Use Committee . Guide for the Care and Use of Laboratory Animals: Eighth Edition Guide for the Care and Use of Laboratory Animals. THE NATIONAL ACADEMIES PRESS 500 Fifth Street, NW Washington, DC 20001. 2011:118. [Google Scholar]
- 28. Kumar M, Govindan S, Zhang M, Khairallah RJ, Martin JL, Sadayappan S, de Tombe PP. Cardiac myosin‐binding protein C and troponin‐I phosphorylation independently modulate myofilament length‐dependent activation. J Biol Chem. 2015;290:29241–29249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Butler J, Fonarow GC, Gheorghiade M. Strategies and opportunities for drug development in heart failure. JAMA. 2013;309:1593–1594. [DOI] [PubMed] [Google Scholar]
- 30. Gheorghiade M, Vaduganathan M, Fonarow GC, Bonow RO. Rehospitalization for heart failure: problems and perspectives. J Am Coll Cardiol. 2013;61:391–403. [DOI] [PubMed] [Google Scholar]
- 31. Lorin C, Vögeli I, Niggli E. Dystrophic cardiomyopathy: role of TRPV2 channels in stretch‐induced cell damage. Cardiovasc Res. 2015;106:153–162. [DOI] [PubMed] [Google Scholar]
- 32. Shibasaki K, Murayama N, Ono K, Ishizaki Y, Tominaga M. TRPV2 enhances axon outgrowth through its activation by membrane stretch in developing sensory and motor neurons. J Neurosci. 2010;30:4601–4612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Katanosaka Y, Iwasaki K, Ujihara Y, Takatsu S, Nishitsuji K, Kanagawa M, Sudo A, Toda T, Katanosaka K, Mohri S, Naruse K. TRPV2 is critical for the maintenance of cardiac structure and function in mice. Nat Commun. 2014;5:3932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Selen A, Amidon GL, Welling PG. Pharmacokinetics of probenecid following oral doses to human volunteers. J Pharm Sci. 1982;71:1238–1242. [DOI] [PubMed] [Google Scholar]
- 35. Sowunmi A, Fehintola FA, Adedeji AA, Gbotosho GO, Falade CO, Tambo E, Fateye BA, Happi TC, Oduola AMJ. Open randomized study of pyrimethamine‐sulphadoxine vs. pyrimethamine‐sulphadoxine plus probenecid for the treatment of uncomplicated Plasmodium falciparum malaria in children. Trop Med Int Health. 2004;9:606–614. [DOI] [PubMed] [Google Scholar]
- 36. Shaywitz BA, Cohen DJ, Bowers MB. CSF monoamine metabolites in children with minimal brain dysfunction: evidence for alteration of brain dopamine. J Pediatr. 1977;90:67–71. [DOI] [PubMed] [Google Scholar]
- 37. Di Virgilio F, Steinberg T, Silverstein S. Inhibition of Fura‐2 sequestration and secretion with organic anion transport blockers. Cell Calcium. 1990;11:57–62. [DOI] [PubMed] [Google Scholar]
- 38. Bronsky D, Dubin A, Kushner DS. Diuretic action of benemid. Am J Med. 1955;18:259–266. [DOI] [PubMed] [Google Scholar]
- 39. Communal C, Singh K, Pimentel DR, Colucci WS. Norepinephrine stimulates apoptosis in adult rat ventricular myocytes by activation of the beta‐adrenergic pathway. Circulation. 1998;98:1329–1334. [DOI] [PubMed] [Google Scholar]