

Abstract
Background
Breathlessness is the most common symptom in people with pulmonary arterial hypertension and congenital heart disease (CHD‐APAH), previously thought to be caused by worsening PAH, but perhaps also by inflammation and abnormalities of lung function. We studied lung function and airway inflammation in patients with CHD‐APAH and compared the results with controls.
Methods and Results
Sixty people were recruited into the study: 20 CHD‐APAH, 20 CHD controls, and 20 healthy controls. Spirometry, gas transfer, whole body plethysmography and lung clearance index, 6‐minute walk distance, and medical research council dyspnea scoring were performed. Inflammatory markers and endothelin‐1 levels were determined in blood and induced sputum. The CHD‐APAH group had abnormal lung function with lung restriction, airway obstruction, and ventilation heterogeneity. Inverse correlations were shown for CHD‐APAH between medical research council dyspnea score and percent predicted peak expiratory flow (r=−0.5383, P=0.0174), percent predicted forced expiratory flow rate at 50% of forced vital capacity (r=−0.5316, P=0.0192), as well as for percent predicted forced expiratory volume in 1 s (r=−0.6662, P=0.0018) and percent predicted forced vital capacity (r=−0.5536, P=0.0186). The CHD‐APAH patients were more breathless with lower 6‐minute walk distance (360 m versus 558 m versus 622 m, P=0.00001). Endothelin‐1, interleukin (IL)‐β, IL‐6, IL‐8, tumor necrosis factor α, and vascular endothelial growth factor were significantly higher in CHD‐APAH than controls. Serum endothelin‐1 for CHD‐APAH correlated with airflow obstruction with significant negative correlations with percent predicted forced expiratory flow rate at 75% of forced vital capacity (r=−0.5858, P=0.0135).
Conclusions
Raised biomarkers for inflammation were found in CHD‐APAH. Significant abnormalities in airway physiology may contribute to the dyspnea but are not driven by inflammation as assessed by circulating and sputum cytokines. A relationship between increased serum endothelin‐1 and airway dysfunction may relate to its bronchoconstrictive properties.
Keywords: congenital heart disease, inflammation, lung, pulmonary circulation, pulmonary hypertension
Subject Categories: Basic Science Research, Endothelium/Vascular Type/Nitric Oxide, Pathophysiology, Pulmonary Biology, Vascular Biology
Clinical Perspective
What Is New?
Pulmonary arterial hypertension associated with congenital heart disease is associated with restrictive lung function in comparison with a control population of adults with congenital heart disease who do not have pulmonary arterial hypertension.
Airways obstruction is associated with a raised level of endothelin 1.
Measurement of lung clearance index can demonstrate that there is increased ventilator heterogeneity in this population.
Lung clearance index was a more sensitive measure of airway obstruction than standard spirometry but not as sensitive as midexpiratory flows.
Breathlessness in pulmonary arterial hypertension associated with congenital heart disease is associated with older age, restrictive lung function, and raised endothelin 1.
Airways inflammation is not seen in this population and not associated with the systemic inflammation that is present.
What Are the Clinical Implications?
Some of the breathlessness and exercise limitation might be caused by airways disease, rather than pulmonary vascular disease.
As a result, there might be benefit of therapy with endothelin receptor antagonists, which will improve airways mechanics. This effect might be in addition to that of reducing pulmonary vascular resistance.
There is no evidence from this study that the use of inflammatory modulators would alter the airway mechanics in this population.
Introduction
The development of pulmonary arterial hypertension in congenital heart disease (CHD‐APAH) is associated with worsening exercise tolerance with breathlessness being the most common symptom.1, 2 Current drug therapies improve exercise tolerance and reduce the perception of breathlessness on exertion by decreasing pulmonary vascular resistance and increasing cardiac output.3 However, patients usually remain symptomatic despite medication and can expect breathlessness to increase as their disease progresses.3 The prognosis for patients with CHD‐APAH is better than for idiopathic PAH patients such that survival can continue for many years, albeit with very limited exercise activity.4
While abnormalities of pulmonary vasculature in CHD‐APAH have been well described,5 lung function abnormalities are less clear, but could also contribute to symptoms.6 In idiopathic PAH, lung restriction and airflow obstruction have been described.7, 8, 9 Some of these abnormalities correlate with exertion dyspnea and reduction in exercise tolerance.10, 11 While some of these abnormalities have also been described in CHD‐APAH patients,9, 12, 13 a lack of adequate controls makes the significance unclear as abnormal lung function has also been described in CHD patients without APAH.14 These might relate to coexisting scoliosis or previous cardiothoracic surgery that is frequent in this group and may be found in CHD patients both with and without APAH.
We wished therefore to characterize the lung function abnormalities in CHD‐APAH and compare these with CHD patients without APAH as well as with healthy controls. We used standard lung function measurements alongside the novel lung clearance index (LCI), which has been shown to be a more sensitive measure of airway obstruction than spirometry.15 This study also aims to evaluate the potential role of inflammation and endothelin‐1 in airway dysfunction in CHD‐APAH.
Inflammation plays a key role in a number of lung conditions including asthma, chronic obstructive pulmonary disease, and interstitial lung disease.16, 17, 18 Inflammation can result in airflow obstruction through bronchoconstriction, mucus plugging, lung tissue destruction, and airway remodeling.18 In interstitial lung disease, the pattern of chronic inflammation leads to tissue destruction and fibrosis resulting in lung restriction.17 It is not known whether such patterns of lung inflammation occur in CHD‐APAH, and there are only limited reports of airway inflammation in other Group 1 PAH conditions.19
There is, however, growing evidence of a role for inflammation in Group 1 PAH. Lung tissue biopsy samples of patients with idiopathic pulmonary arterial hypertension (IPAH) demonstrate perivascular inflammatory cell infiltrates of T cells, B cells, and macrophages.20, 21 Circulating levels of serum cytokines including interleukin (IL)‐1β, IL‐2, IL‐4, IL‐6, IL‐8, IL‐10, IL‐12p70, and tumor necrosis factor α are significantly elevated when compared with controls.22, 23 IPAH shares a number of features with CHD‐APAH,24 and inflammation could develop in CHD‐APAH because of the high stresses associated with increased pulmonary blood flow and hypoxemia.25 Evidence of inflammation in CHD‐APAH includes inflammatory cell infiltrates around remodeled vessels,26, 27 and elevated circulating levels of IL‐6, tumor necrosis factor α,.MCP‐1, and C‐reactive protein have also been demonstrated.26, 27, 28
While there have been no studies to date looking at airway inflammation or lung inflammation in CHD‐APAH or IPAH, given the proximity of these in the pulmonary vessels, a spillover of such mediators could have effects within the airways. Such protein movement from the systemic circulation into the airways has been shown to occur, and this can increase in the presence of inflammation caused by effects on epithelial permeability.29, 30 If an overspill of these inflammatory mediators and cells into the airways occurs, the resulting airway inflammation could account for the lung function abnormalities seen, as can occur in other respiratory conditions.16, 17, 18
Methods
Because of patient confidentiality, the data, analytic methods, and study materials will not be made available to other researchers for purposes of reproducing the results or replicating the procedure.
Patients with CHD‐APAH were seen in the adult pulmonary hypertension clinic and the data were stored in our hospital database (Heartsuite™) at the Bristol Heart Institute. The clinic is run as part of the UK National Pulmonary hypertension service in conjunction with the Hammersmith Hospital. Patients were invited to participate in our study between 2012 and 2014. Healthy controls and CHD controls without APAH were recruited by internal advertisement in the hospital and were not selected by the research team. All participants provided informed consent and formal ethics approval was obtained. In total, 20 CHD‐APAH, 20 CHD controls, and 20 healthy controls were recruited.
Exclusion criteria were any smoking history, Down syndrome or learning difficulties (because of compliance and consent concerns), pre‐existing lung disease, scoliosis, and left heart dysfunction as assessed by echocardiography. Healthy controls were also excluded if subsequent lung function tests were abnormal with a forced expiratory volume in 1 s (FEV1)/FVC ratio <70% or a TLC of <80% of predicted values.
All participants performed routine spirometry, gas transfer, and whole‐body plethysmography in accordance with guidelines.31 LCI was determined using a multiple‐breath washout technique with a modified Innocor device and 0.2% SF6 as has previously been described.32 Six‐minute walk test was performed enabling maximum distance walked (6MWD), minimum oxygen saturations, and maximum Borg score to be determined.33 MRC dyspnea score and World Health Organization (WHO) performance status were also determined.34 Inflammatory cytokine and endothelin‐1 levels were determined from blood and induced sputum samples. All measurements were obtained at a single visit.
Induced sputum was obtained as previously described.35 Dithiothreitol was used only after sputum supernatant was removed as this has been reported to interfere with detection of endothelin‐1 and several other cytokines of interest.36 Subaliquots of supernatant were removed and stored at −70°C for later analysis.
Sputum was analyzed using trypan blue on a Neubauer hemocytometer to determine the total cell count and using a commercial modified Giemsa stain to determine the cell differential.37 Twenty percent of slides were checked by an independent pathologist to ensure accuracy.
IL‐1β, IL‐6, IL‐8, IL‐10, tumor necrosis factor α, and vascular endothelial growth factor concentrations in sputum and serum samples were determined using Luminex Performance Assay (R&D Systems, Abingdon, UK) and read with a Bio‐Plex Magpix analyzer. Endothelin‐1 levels were determined separately using a commercial ELISA kit according to the manufacturer's instructions (R&D Systems, Abingdon, UK).
Statistical analyses were performed using Stata version 13.1. Comparison between groups was performed using 1‐way ANOVA (parametric data) with post hoc Bonferroni analysis or Kruskal–Wallis (nonparametric data) with post hoc analysis with Dunn's correction where applicable for multiple comparisons. Correlations were assessed using the Spearman's rank correlation coefficient (for nonparametric data). A P value of 0.05 or less was considered significant.
Ethical Approval
The study was sponsored by University Hospitals Bristol NHS Foundation Trust—ME/2009/3142. Ethical approval was granted by the South West Research Ethics Committee—09/H0106/34.
Results
Sixty patients were recruited: 20 patients with CHD‐APAH, 20 CHD, and 20 healthy controls. Standard lung function was performed on all volunteers. Two healthy volunteers were subsequently excluded, as their lung function (FEV1/FVC ratio) was <70%. No healthy volunteers were excluded on the grounds of low total lung capacity (TLC). The demographics of the participants are summarized in Table 1.
Table 1.
Patient Demographics for Patients With CHD‐APAH, CHD Controls, and Healthy Volunteers
| CHD‐APAH (n=20) | CHD (n=20) | Healthy Volunteers (n=18) | P Valuea | |
|---|---|---|---|---|
| Median age, y (IQR) | 42.7 (33.1–57.7) | 40.0 (32.8–50.6) | 31.6 (25.6–38.4) | 0.0373 |
| Sex (M/F) | 8/12 | 10/10 | 9/9 | 0.5266 (χ2) |
| Median height, cm (IQR) | 166 (157–172) | 169 (164–180) | 173 (168–178) | 0.0868 |
| Median weight, kg (IQR) | 64.9 (56.4–77.2) | 72.4 (64.0–81.4) | 69.9 (58.6–85.0) | 0.2575 |
| Median BMI, kg/m2 (IQR) | 23.3 (21.1–27.3) | 24.3 (22.5–26.8) | 22.5 (20.0–29.1) | 0.6511 |
BMI indicates body mass index; CHD, congenital heart disease; CHD‐APAH, congenital heart disease–associated pulmonary artery hypertension; F, female; IQR, interquartile range; M, male.
P values by Kruskal–Wallis unless otherwise stated, comparing 3 patient groups.
There was a slight difference in age among the 3 groups (P=0.0373 Kruskal–Wallis), with CHD‐APAH patients slightly older than healthy controls (42.7 years versus 31.6 years, P=0.0226 Dunn's), but not significantly different from CHD patients (40.0 years, P=0.9595 Dunn's) and the median age of CHD patients was not greater than that of healthy controls (P=0.0725 Dunn's). There was no statistically significant difference in the sex distribution among the 3 groups (P=0.5266 χ2). Height was not significantly different in CHD‐APAH patients, CHD patients, and healthy controls (166 cm versus 169 cm versus 173 cm), nor was body weight (64.9 kg versus 72.4 kg versus 69.9 kg) (P=0.0826 and 0.2526 Kruskal–Wallis, respectively). There was also no statistically significant difference in body mass index (CHD‐APAH 23.3 kg/m2 versus CHD 24.3 kg/m2 versus healthy controls 22.5 kg/m2, P=0.6511 Kruskal–Wallis).
As shown in Table 2, 11 of 20 patients with CHD‐APAH had Eisenmenger syndrome and 9 of 20 patients had APAH following surgical operation.38 All the patients with Eisenmenger syndrome had unrestrictive pulmonary outflow tracts and hence had systemic level pulmonary artery pressures. We do not undertake cardiac catheterization in these patients and therefore have not measured their pulmonary artery pressure directly. The pressures in those patients who had had an operation were either late shunt repairs (the case with atrial septal defect), or had significant residual ventricular septal defect (VSD) (in both those with VSD) that had not been closed. All had undergone cardiac catheterization and the pressures were either systemic or were as described in Table 3. The 3 patients with transposition of the great arteries had had atrial switches and had been fully assessed to ensure there was no evidence of venous pathway obstruction, tricuspid regurgitation, or right ventricle dysfunction. The pulmonary artery pressures were assessed from cardiac catheterization in each case and the pulmonary capillary wedge pressure confirmed the absence of pulmonary venous hypertension. They are classified according to the system proposed by Galie et al.34 There were no patients with CHD‐APAH with a predominant systemic‐to‐pulmonary shunt, or with a small coincidental lesion. The most common congenital heart defect in the CHD‐APAH patient group was a VSD that affected 10 of the 20 patients. Two patients had had an atrial septal defect, and 8 of 20 CHD‐APAH patients had complex congenital heart disease, including transposition of the great arteries, double inlet left ventricle and tricuspid atresia. CHD controls included patients with small atrial septal defect, VSD, and transposition of the great arteries, as well as patients with tetralogy of Fallot, pulmonary stenosis, Ebstein anomaly, and 1 patient with truncus arteriosus. None of the patients had learning difficulties or 22q11 microdeletion.
Table 2.
Clinical Classification and Lesion Type for Patients With CHD‐APAH and CHD Controls38
| Eisenmenger | PAH With Systemic‐Pulmonary Shunt | PAH With Small Defect | PAH After Surgical Operation | CHD Controls Without APAH | |
|---|---|---|---|---|---|
| ASD | 1 | ··· | ··· | 1 | 2 |
| VSD | 8 | ··· | ··· | 2 | 3 |
| TGA | ··· | ··· | ··· | 3 | 5 |
| DILV | 2 | ··· | ··· | 1 | ··· |
| Tricuspid atresia | ··· | ··· | ··· | 2 | ··· |
| Tetralogy of Fallot | ··· | ··· | ··· | ··· | 3 |
| Pulmonary stenosis | ··· | ··· | ··· | ··· | 4 |
| Truncus arteriosus | ··· | ··· | ··· | ··· | 1 |
| Ebstein anomaly | ··· | ··· | ··· | ··· | 2 |
ASD indicates atrial septal defect; CHD‐APAH, congenital heart disease–associated pulmonary artery hypertension; DILV, double inlet left ventricle; TGA, transposition of the great arteries; VSD, ventricular septal defect.
Table 3.
Hemodynamics for Patients With CHD‐APAH
| Eisenmenger | PA Pressure | PAH After Surgical Operation | Mean PA Pressure, mm Hg | SvO2, % | RAp, mm Hg | |
|---|---|---|---|---|---|---|
| ASD | 1 | Systemic | 1 | 62 | 73 | 11 |
| VSD | 8 | Systemic | 2 | 93, 104 | 56, 61 | 8, 6 |
| TGA | ··· | ··· | 3 | 73, 58, 95 | 55, 71, 66 | 13, 12, 14 |
| DILV | 2 | Systemic | ||||
| Tricuspid atresia | ··· | ··· | 2 (Fontan) | 14, 17 |
Both of the patients with Fontan procedure had transpulmonary gradient >8 mm Hg. ASD indicates atrial septal defect; CHD‐APAH, congenital heart disease–associated pulmonary artery hypertension; DILV, double inlet left ventricle; PA, pulmonary artery; RAp, right atrial pressure; SvO2, mixed venous oxygen saturations; TGA, transposition of the great arteries; VSD, ventricular septal defect.
In all patients, echocardiography was undertaken before entry to the study. All the CHD patients and controls were confirmed to have good systemic systolic ventricle function (fractional shortening, S', tricuspid annular plane excursion (TAPSE), mitral annular plane excursion (MAPSE) as appropriate), no or insignificant systemic atrioventricular valve regurgitation and no diastolic dysfunction as judged by inflow Doppler interrogation. There was no evidence of branch pulmonary artery obstruction or other hemodynamic concern that would have altered the interpretation of the data.
Seventeen of 20 CHD control patients had undergone previous sternotomy with none having a prior thoracotomy. The remaining 3 CHD patients included 2 patients with a nonsurgical Amplatzer device closure of atrial septal defect and 1 patient with a small VSD. The 10 CHD‐APAH patients with previous surgery included 6 with prior sternotomy and 4 who had had both a sternotomy and a thoracotomy. No healthy controls had prior cardiothoracic surgery.
The lung function for the 3 groups was as shown in Table 4. These data demonstrate that lung function in patients with CHD‐APAH is abnormal in comparison to CHD patients without associated PAH and also in comparison to healthy controls. Lung volumes including TLC and FVC were reduced in CHD‐APAH in keeping with lung restriction, while residual volume was relatively increased in comparison to TLC, demonstrating relative hyperinflation in these patients. This may be because of gas trapping resulting from airway obstruction that was demonstrated by reductions in FEV1/FVC ratio and reduced midexpiratory flows. This is further supported by LCI measurements that were abnormally high in CHD‐APAH patients and normal in CHD and healthy controls. Gas transfer measurements were also abnormal in both CHD‐APAH patients and CHD patients in comparison to healthy controls, but when accounting for transfer coefficient (KCO), there was no significant difference between CHD patients with and without APAH.
Table 4.
Lung Function by Patient Group
| CHD‐APAH | CHD | Healthy Volunteers | P Valuea | |
|---|---|---|---|---|
| Total number | 20 | 20 | 18 | |
| TLC | 80.0 (77.2–98.0) | 96.3 (86.3–104.6) | 94.2 (87.0–100.2) | 0.0344/0.027/0.046 |
| FVC | 77.8 (73.9–100.0) | 101.8 (90.8–122.2) | 106.1 (96.0–109.5) | 0.0005/0.0011/0.0012 |
| RV | 88.8 (68.8–124.6) | 84.0 (76.9–87.7) | 79.9 (73.8–92.9) | 0.4197 (not significant) |
| RV/TLC | 110.7 (93.6–137.4) | 85.4 (72.8–94.2) | 83.2 (71.1–90.6) | 0.0004/0.0006/0.0018 |
| FEV1 | 70.7 (58.5–81.8) | 95.1 (90.4–112.1) | 99.1 (92.9–105.0) | 0.0001/<0.0001/<0.0001 |
| FEV1/FVC Ratio | 0.719 (0.680–0.778) | 0.790 (0.768–0.818) | 0.815 (0.769–0.869) | 0.0021/0.0272/0.0009 |
| PEFR | 83.7 (65.9–99.9) | 111.0 (88.8–119.6) | 101.5 (90.7–115.0) | 0.0019/0.0014/0.0096 |
| FEF25 | 72.1 (63.2–92.5) | 106.7 (76.8–128.7) | 102.3 (86.6–109.4) | 0.0006/0.0007/0.0027 |
| FEF50 | 44.0 (28.8–63.6) | 82.3 (61.3–103.0) | 85.9 (70.4–94.0) | 0.0001/0.0001/<0.0001 |
| FEF75 | 29.8 (22.0–39.0) | 61.4 (44.9–78.3) | 78.6 (57.3–97.4) | 0.0001/0.0001/<0.0001 |
| FEF25‐75 | 39.6 (27.1–53.5) | 76.3 (61.0–101.2) | 79.0 (70.0–97.1) | 0.0001/<0.0001/<0.0001 |
| TLCO | 64.5 (53.4–74.4) | 89.0 (79.5–100.8) | 95.0 (84.6–104.0) | 0.0001/0.0001/<0.0001 |
| KCO | 84.2 (74.6–94.3) | 89.7 (81.9–100) | 97.2 (86.4–114.8) | 0.0073/0.11/0.0027 |
| LCI | 7.6 (6.99–9.70) | 6.77 (6.56–7.32) | 6.41 (6.14–7.15) | 0.0004/0.0098/0.00024 |
For patients with congenital heart disease–associated pulmonary artery hypertension (CHD‐APAH), CHD controls, and healthy volunteers. Data expressed as median percentage predicted (interquartile range). FEF 25/50/75 indicates forced expiratory flow at 25/50/75% of FVC; FEV1, forced expiratory volume in 1 second; FVC, forced vital capacity; KCO, transfer coefficient; LCI, lung clearance index; PEFR, peak expiratory flow rate; RV, residual volume; TLC, total lung capacity; TLCO, transfer factor for carbon monoxide.
P value: Kruskal–Wallis, and where P<0.05, the following P values are for CHD‐APAH vs CHD and then CHD‐APAH vs healthy controls by post hoc Dunn's correction.
As shown in Table 5, patients with CHD‐APAH were more functionally limited as determined by both 6MWD and WHO‐Functional Class (FC) and more breathless on MRC dyspnea score and by Borg score during the 6‐minute walk test.
Table 5.
Breathlessness and Exercise Tolerance by Group
| CHD‐APAH | CHD | Healthy Volunteers | P Value | |
|---|---|---|---|---|
| Total number | 20 | 20 | 18 | |
| WHO‐FC (I/II/III/IV) | 2/8/10/0 | 13/7/0/0 | 18/0/0/0 |
0.000 Fisher exact |
| WHO, median | 2.5 (2–3) | 1 (1–2) | 1 (1–1) | 0.0001 |
| MRC dyspnea score (1/2/3/4/5) | 2/8/9/1/0 | 13/7/0/0/0 | 18/0/0/0/0 |
0.0001 Fisher exact |
| MRC, median | 2.5 (2–3) | 1 (1–2) | 1 (1–1) | 0.0001 |
| Saturation % at rest | 82 (72–89) | 94 (82–96) | 97 (94–99) | 0.0001 |
| Saturation % at end 6MWD | 68 (45–82) | 89 (69–96) | 96 (94–98) | 0.0001 |
| Mean 6MWD, m (SD) | 360.35 (123.01) | 558.82 (70.59) | 622.0 (80.40) |
0.00001 ANOVA |
For patients with congenital heart disease–associated pulmonary artery hypertension (CHD‐APAH), CHD controls, and healthy volunteers. Kruskal–Wallis for nonparametric data, ANOVA for parametric data, χ2 for proportions, and Fisher exact for proportions where >20% of cells have expected frequencies of <5. 6MWD indicates 6‐minute walk distance; MRC, medical research council; WHO‐FC, World Health Organization Functional Class.
Lung function and its association with breathlessness and exercise tolerance in CHD‐APAH patients were assessed. A weak but significant correlation was shown between percent predicted TLC and 6MWD (r 2=0.2306, P=0.0043) and between percent predicted FVC and 6MWD (r 2=0.3347, P=0.0095), while an inverse correlation was found between residual volume/TLC ratio and 6MWD for these patients (r 2=0.3227, P=0.0139). Significant negative correlations were also shown for CHD‐APAH between MRC dyspnea score and percent predicted peak expiratory flow rate (r=−0.5383, P=0.0174), percent predicted FEF50 (r=−0.5316, P=0.0192), percent predicted FEF75 (r=−0.5927, P=0.0075), and percent predicted FEF25‐75 (r=−0.5536, P=0.0139) as well as for percent predicted FEV1 (r=−0.6662, P=0.0018) and percent predicted FVC (r=−0.5536, P=0.0186), and a positive correlation for percent predicted residual volume/TLC ratio (r=0.573, P=0.0129). There was a significant correlation between age and LCI in CHD‐APAH patients. Multiple regression was therefore performed, and this demonstrated that LCI is significantly determined by presence of APAH (P=0.041) and age (P<0.0001) but CHD‐APAH type did not reach statistical significance (P=0.118). On univariant analysis, there was also no correlation between LCI and body mass index, sex, or type of surgery (thoracotomy or sternotomy).
Figure 1 demonstrates that there were significant differences in serum cytokines levels among the 3 groups, with IL‐β, IL‐6, IL‐8, and vascular endothelial growth factor levels all significantly higher in CHD‐APAH patients than in CHD or healthy controls. Tumor necrosis factor α levels were also greater in CHD‐APAH patients compared with healthy controls but with no significant difference in comparison to CHD controls. There were no differences found in serum IL‐10 levels across the 3 groups. Serum endothelin‐1 levels were also significantly higher in CHD‐APAH patients.
Figure 1.
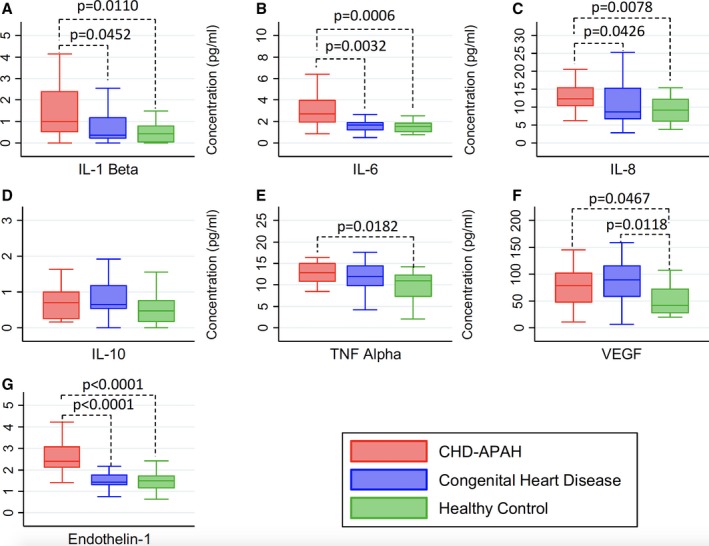
Serum cytokines (pg/mL) by group. Data displayed as box and whisker plots of median, interquartile range, and minimum and maximum values. For cytokine concentrations with a statistically significant difference between groups (P<0.05 Kruskal–Wallis), the P value is indicated (Dunn's). A, IL‐1β (P=0.0214 Kruskal–Wallis), (B) IL‐6 (P=0.0005 Kruskal–Wallis), (C) IL‐8 (P=0.0161 Kruskal–Wallis), (D) IL‐10 (P=0.1119 Kruskal–Wallis), (E) TNFα (P=0.0411 Kruskal–Wallis), (F) VEGF (P=0.0232 Kruskal–Wallis) and (G) endothelin‐1 (P=0.0001 Kruskal–Wallis). IL indicates interleukin; TNFα, tumor necrosis factor α; VEGF, vascular endothelial growth factor.
When sputum samples were analyzed for cell count, no differences were found among the groups (Table 6). There was also no difference in the cytokine levels in sputum, in contrast to the findings in serum (Figure 2).
Table 6.
Total Cell Count for Induced Sputum Samples for Patients With CHD‐APAH, CHD Controls, and Healthy Volunteers
| CHD‐APAH (n=10) | CHD (n=10) | Healthy Control (n=10) | P Value | |
|---|---|---|---|---|
| Total cell count, mean ×106/g (SD) | 1.96 (1.44) | 1.51 (0.84) | 2.58 (1.41) | 0.19 |
| Cell differential count, mean % (SD) | ||||
| Macrophages | 52.1 (28.2) | 53.2 (16.1) | 53.3 (31.3) | 0.9942 |
| Neutrophils | 46.7 (28.3) | 44.0 (15.2) | 45.4 (30.9) | 0.9737 |
| Eosinophils | 0.3 (0.37) | 1.2 (1.8) | 1.0 (1.4) | 0.3509 |
| Lymphocytes | 0.9 (1.1) | 1.6 (2.7) | 0.3 (0.4) | 0.2512 |
Cell differential percentages for induced sputum samples. Data are displayed as mean % (SD). P values ANOVA. CHD‐APAH indicates congenital heart disease–associated pulmonary artery hypertension;
Figure 2.
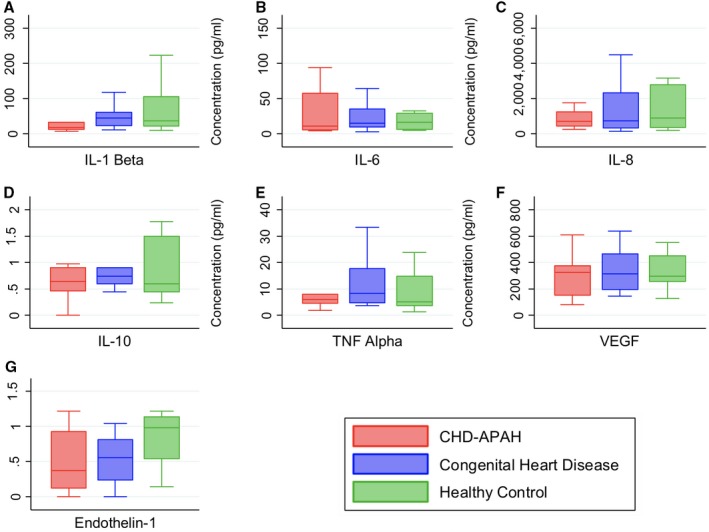
Induced sputum cytokines by group. Data displayed as box and whisker plots demonstrating median, interquartile range, and minimum and maximum values. A, IL‐1β (P=0.2126 Kruskal–Wallis), (B) IL‐6 (P=0.7159 Kruskal–Wallis), (C) IL‐8 (P=0.9351 Kruskal–Wallis), (D) IL‐10 (P=0.5719 Kruskal–Wallis), (E) TNFα (P=0.5719 Kruskal–Wallis), (F) VEGF (P=0.9645 Kruskal–Wallis), and (G) endothelin‐1 (P=0.1810 Kruskal–Wallis). CHD‐APAH indicates congenital heart disease–associated pulmonary artery hypertension; IL, interleukin; TNF Alpha, tumor necrosis factor α; VEGF, vascular endothelial growth factor.
Serum endothelin‐1 levels for CHD‐APAH patients correlated with measures of airflow obstruction with significant negative correlations with percent predicted FEF25‐75 (r=−5417, P=0.0247 Spearman) and percent predicted FEF75 (r=−0.5858, P=0.0135). There were no significant correlations between serum endothelin‐1 levels and FEV1/FVC ratio (r=−0.2770, P=0.2818 Spearman) or LCI (r=0.2170, P=0.4027 Spearman). There were no significant correlations between measures of airflow obstruction and serum endothelin‐1 for CHD and healthy controls. Linear regressions are displayed in Figure 3 showing the endothelin level correlation with restrictive lung function and with LCI.
Figure 3.
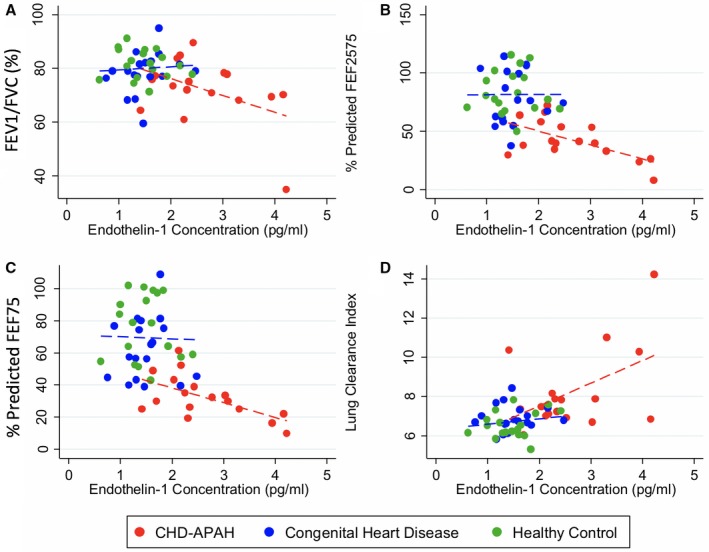
Serum endothelin‐1 and lung function. A, FEV 1/FVC ratio. Linear regression for CHD‐APAH r 2=0.2032, P=0.0693 (red dotted line) and CHD and healthy controls combined r 2=0.0054, P=0.6714 (blue dotted line); (B) Percent predicted FEF25‐75. Linear regression for CHD‐APAH r 2=0.3733, P=0.0092 (red dotted line) and CHD and healthy controls combined r 2=0.0000, P=0.9912 (blue dotted line); (C) Percent predicted FEF75. Linear regression for CHD‐APAH r 2=0.3500, P=0.0124 (red dotted line) and CHD and healthy controls combined r 2=0.0008, P=0.8733 (blue dotted line); (D) Lung clearance index. Linear regression for CHD‐APAH r 2=0.2307, P=0.0510 (red dotted line) and CHD and healthy controls combined r 2=0.0308, P=0.3062 (blue dotted line). CHD‐APAH indicates congenital heart disease–associated pulmonary artery hypertension; FEF 25/50/75, forced expiratory flow at 25/50/75% of FVC; FEV 1, forced expiratory volume in 1 second; FVC, forced vital capacity.
Discussion
Breathlessness is the most common symptom experienced by patients with CHD‐APAH. It is the presenting symptom in up to 70% of patients and affects almost all with advanced disease.39, 40 While breathlessness in CHD‐APAH patients has been attributed to abnormal pulmonary hemodynamics and reduced cardiac output, this study demonstrates that these patients also have abnormal lung function in comparison to both healthy controls and CHD patients without APAH. These abnormalities include lung restriction with reduced TLC and FVC, and airway obstruction with reductions in FEV1/FVC ratio, midexpiratory flows, and increased LCI compared with CHD and healthy controls. It was also demonstrated that both CHD‐APAH and CHD patients had reduced gas transfer measurements in comparison to healthy controls.
Reductions in TLC and FVC described in the present study were in keeping with other studies that have also described lung restriction.12, 13, 41 However, these studies lacked CHD controls for comparison. The findings of the present study, however, are in conflict with a study that demonstrated that Eisenmenger patients have near normal lung volumes, but that study had no controls—either healthy or CHD patients without PAH.42 In addition, unselected CHD patients have been reported as having reduced FVC, which may be related to features such as scoliosis (which was excluded from the present study) and previous cardiothoracic surgery.14, 43 The present study demonstrates that lung restriction occurs in CHD‐APAH compared with CHD and healthy controls without scoliosis. While thoracotomy was associated with lung restriction, sternotomy was not, which is in keeping with previous studies.14 However, patients who had not had previous cardiothoracic surgery also had lower lung volumes than predicted values, suggesting that the presence of APAH may have a separate effect on lung restriction in keeping with lung restriction that has been described in IPAH patients.8, 9, 13, 44, 45
All measurements of airflow were reduced in CHD‐APAH patients including FEV1/FVC ratio, FEV1, peak expiratory flow rate, FEF25, FEF50, FEF75, and FEF25‐75, indicating the presence of airway obstruction. While this has been suggested by previous studies, such studies have lacked adequate controls for comparison.9, 41, 42 The FEV1/FVC ratio was >70% in this present study and could be interpreted as an absence of airway obstruction in keeping with other reports; however, this would be an oversimplification.12, 13 This study confirms that the FEV1/FVC ratio is significantly reduced in comparison to both control groups, and in addition all other markers of airway obstruction were significantly less. This therefore confirms that airway obstruction is found in CHD‐APAH patients compared with both healthy controls and CHD controls without APAH.
LCI in CHD‐APAH has not been previously described, but the elevation further supports the presence of airway obstruction in CHD‐APAH. This is a measurement of ventilation heterogeneity that has been shown in cystic fibrosis to be a more sensitive measure of airway obstruction than spirometry.15 Although more sensitive than routine spirometry, it was not as sensitive as midexpiratory flow measures such as FEF25‐75 and FEF75. However, the clinical utility of midexpiratory flows is limited by its reproducibility and lack of agreed limits of normality.46 LCI may therefore be a useful adjunct, which has a clear abnormal upper limit and thus is much more straightforward to interpret.15 In addition, use of LCI may also have a role in obtaining lung function measures in CHD‐APAH patients with learning difficulties, which are common in this patient group and where reliable measures of lung function are difficult to obtain.47 For LCI, patients are required to breathe through a mouthpiece with normal tidal breathing throughout, without the need for any complex maneuvers.
Airway obstruction would account for gas trapping demonstrated by the raised residual volume/TLC ratio found. This is in keeping with studies in which a raised residual volume/TLC ratio has been described in both CHD‐APAH patients42 and IPAH patients.7 The degree of gas trapping may be related to the severity of pulmonary vascular disease in IPAH, and may therefore result in an increase in TLC in more advanced disease. This could also explain some of the conflicting reports regarding lung volumes in CHD‐APAH.12, 13, 41, 42
Gas transfer measurements for CHD‐APAH patients were reduced as in previous studies.9, 13, 41, 42 We showed a significant difference in TLCO between CHD patients with and without APAH, but no significant difference was demonstrated in TLCO or KCO between CHD patients without APAH and healthy controls. Potential causes for abnormal gas transfer measure in CHD‐APAH patients include reduced pulmonary capillary blood volume and pulmonary membrane diffusion capacity as a consequence of increased pulmonary vascular resistance, reduced cardiac output, and endothelial cell proliferation. While pulmonary edema could affect gas transfer measurements through widening of the alveolar‐capillary membrane, patients with left heart dysfunction on echocardiogram were excluded from this study and therefore would not explain the results of this present study.
It is of note that there were 2 patients with Fontan procedures in our study population. There is much discussion about the definition of pulmonary hypertension, but both patients had transpulmonary gradients of >8 mm Hg and thus would seem reasonable to include in a study on pulmonary vascular disease. Neither patient had impaired systemic ventricular function, or systemic atrioventricular valve regurgitation, so it must be presumed that there was increased pulmonary vascular stiffness if not pulmonary arterial hypertension by the formal definition.
CHD‐APAH patients were more breathless with higher MRC dyspnea scores and higher Borg scores during the 6‐minute walk test, and these patients were confirmed to be more functionally limited with higher WHO‐FC and reduced 6MWD.48 This present study demonstrates that lung restriction correlates with 6MWD and Borg score during the 6‐minute walk test, which is in conflict with a previous study that did not demonstrate such a relationship.42 However, as discussed previously, this prior study did not find any reduction in lung volumes, which would account for this lack of association described. However, our study did not find a correlation between lung volumes and MRC dyspnea score or WHO‐FC. FEV1/FVC ratio was not associated with any measures of breathlessness or exercise tolerance. While LCI was shown to be a more sensitive measure of airway obstruction than spirometry, this also failed to demonstrate any significant correlation with symptoms or functional limitation. FEF25‐75 and FEF75 were more sensitive than both FEV1/FVC ratio and LCI, and correlated with MRC dyspnea scores and WHO‐FC, but there were no significant associations with 6MWD or Borg score. No significant relationship between KCO and measures of breathlessness of functional limitation were demonstrated. While lung restriction appeared to be related to some symptom and exercise tolerance measures, this could reflect a relationship with previous thoracotomy or more complex CHD rather than reflecting an association between APAH and lung volumes.
It may also be possible that the measures of breathlessness and exercise tolerance were not sensitive enough to demonstrate such a relationship. The WHO‐FC and MRC dyspnea score are 4‐ and 5‐point scales, respectively, and while the Borg score has a wider 10‐point scale, this was used to measure breathlessness in response to a specific task rather than an assessment of symptoms during usual activities. The use of cardiopulmonary exercise testing, which is a maximal exercise test, may have achieved a clearer relationship with lung function abnormalities. Lung function abnormalities could be an innocent bystander, a result of another problem, outweighed by abnormalities in pulmonary vasculature and cardiac output. A larger study would be required to investigate this area further.
It is known that patients with CHD‐APAH have evidence of systemic inflammation,26, 27, 28 and elevated serum endothelin‐1 levels.49 While airway inflammation plays a key role in a number of respiratory diseases such as asthma, chronic obstructive pulmonary disease, and interstitial lung disease,16, 17, 18 endothelin‐1 can cause potent bronchoconstriction in vitro,50 and has been found in elevated levels in induced sputum and bronchoalveolar lavage samples in cystic fibrosis and chronic obstructive pulmonary disease,51, 52 but airway inflammation has not been studied in the present group. It is thus possible that inflammation and endothelin‐1 could play a role in the pathogenesis of lung function abnormalities demonstrated in this patient group. This present study demonstrated evidence of systemic inflammation in keeping with these previous studies, but this was not confirmed in induced sputum across the 3 patient groups. While it is recognized that a limited number of inflammatory cytokines were assessed, the findings of this study do not support a role of inflammation in the pathophysiology of abnormal lung function in CHD‐APAH.
Endothelin‐1 is a vasoconstrictor that is found in elevated levels in CHD‐APAH and contributes to the development of PAH49 and is a potent bronchoconstrictor. Thus, it was hypothesized that endothelin‐1 could play a key role in airway obstruction in CHD‐APAH patients.50 Serum endothelin‐1 levels were increased in CHD‐APAH patients in comparison to CHD and healthy controls in keeping with other studies. While induced sputum levels were not elevated in CHD‐APAH patients, it was shown that endothelin‐1 levels correlated with measures of airway obstruction in CHD‐APAH. While a proposed role for endothelin‐1 in airway obstruction in CHD‐APAH has biological plausibility, a key limitation is that causation was not shown and would require further investigation.
Conclusion
Lung function is abnormal in CHD‐APAH with evidence of lung restriction and airway obstruction in comparison to CHD and healthy controls. Evidence of airway obstruction was also supported by the demonstration of increased ventilation heterogeneity with abnormally elevated LCI in this patient group. Lung restriction may in part be caused by previous cardiothoracic surgery, while airway obstruction may be related to elevated serum endothelin‐1 levels. While systemic inflammation is present in these patients, airway inflammation was not demonstrated and was not shown to be associated with airway obstruction. LCI was a more sensitive measure of airway obstruction than standard spirometry, but not as sensitive as midexpiratory flows. LCI may be a more reproducible measure than midexpiratory flows and may be easier to perform in patients with learning disabilities, which are a large cohort of CHD‐APAH patients, though this requires further investigation. Correlations between these lung function abnormalities, breathlessness, and exercise limitation were demonstrated, which may indicate they contribute to patients' symptoms. However, further work is required in this area to elucidate the pathophysiology of these abnormalities and determine whether treatment may benefit CHD‐APAH patients.
Sources of Funding
The study was funded by an Investigator Initiated study from Pfizer (WS775311) and the David Telling Trust (2014/360). This study was supported by the NIHR Biomedical Research Centre at the University Hospitals Bristol NHS Foundation Trust and the University of Bristol. The views expressed in this publication are those of the author(s) and not necessarily those of the NHS, the National Institute for Health Research, or the Department of Health. There was no involvement from Pfizer in the conduct of this study or in the publication.
Disclosures
Tulloh and Howard have received honoraria and lecture fees from Encysive, Pfizer, Bayer, GSK, and Actelion. The remaining authors have no disclosures to report.
(J Am Heart Assoc. 2018;7:e007249 DOI: 10.1161/JAHA.117.007249.)29444773
References
- 1. Diller G‐P, Dimopoulos K, Broberg CS, Kaya MG, Naghotra US, Uebing A, Harries C, Goktekin O, Gibbs JSR, Gatzoulis MA. Presentation, survival prospects, and predictors of death in Eisenmenger syndrome: a combined retrospective and case‐control study. Eur Heart J. 2006;27:1737–1742. [DOI] [PubMed] [Google Scholar]
- 2. Engelfriet PM, Duffels MGJ, Moller T, Boersma E, Tijssen JGP, Thaulow E, Gatzoulis MA, Mulder BJM. Pulmonary arterial hypertension in adults born with a heart septal defect: the Euro Heart Survey on adult congenital heart disease. Heart. 2007;93:682–687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Galie N, Manes A, Palazzini M, Negro L, Marinelli A, Gambetti S, Mariucci E, Donti A, Branzi A, Picchio FM. Management of pulmonary arterial hypertension associated with congenital systemic‐to‐pulmonary shunts and Eisenmenger's syndrome. Drugs. 2008;68:1049–1066. [DOI] [PubMed] [Google Scholar]
- 4. McLaughlin VV, Presberg KW, Doyle RL, Abman SH, McCrory DC, Fortin T, Ahearn G. Prognosis of pulmonary arterial hypertension—ACCP evidence‐based clinical practice guidelines. Chest. 2004;126:78S–92S. [DOI] [PubMed] [Google Scholar]
- 5. D'Alto M, Mahadevan VS. Pulmonary arterial hypertension associated with congenital heart disease. Eur Respir Rev. 2012;21:328–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Low AT, Medford ARL, Millar AB, Tulloh RMR. Lung function in pulmonary hypertension. Respir Med. 2015;109:1244–1249. [DOI] [PubMed] [Google Scholar]
- 7. Meyer FJ, Ewert R, Hoeper MM, Olschewski H, Behr J, Winkler J, Wilkens H, Breuer C, Kubler W, Borst MM; German PPHSG . Peripheral airway obstruction in primary pulmonary hypertension. Thorax. 2002;57:473–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sun X‐G, Hansen JE, Oudiz RJ, Wasserman K. Pulmonary function in primary pulmonary hypertension. J Am Coll Cardiol. 2003;41:1028–1035. [DOI] [PubMed] [Google Scholar]
- 9. Jing Z‐C, Xu X‐Q, Badesch DB, Jiang X, Wu Y, Liu J‐M, Wang Y, Pan L, Li H‐P, Pu J‐L, Zhang Z‐L, Yang Y‐J. Pulmonary function testing in patients with pulmonary arterial hypertension. Respir Med. 2009;103:1136–1142. [DOI] [PubMed] [Google Scholar]
- 10. Richter MJ, Voswinckel R, Tiede H, Schulz R, Tanislav C, Feustel A, Morty RE, Ghofrani HA, Seeger W, Reichenberger F. Dynamic hyperinflation during exercise in patients with precapillary pulmonary hypertension. Respir Med. 2012;106:308–313. [DOI] [PubMed] [Google Scholar]
- 11. Laveneziana P, Garcia G, Joureau B, Nicolas‐Jilwan F, Brahimi T, Laviolette L, Sitbon O, Simonneau G, Humbert M, Similowski T. Dynamic respiratory mechanics and exertional dyspnoea in pulmonary arterial hypertension. Eur Respir J. 2013;41:578–587. [DOI] [PubMed] [Google Scholar]
- 12. Burke CM, Glanville AR, Morris AJR, Rubin D, Harvey JA, Theodore J, Robin ED. Pulmonary‐function in advanced pulmonary‐hypertension. Thorax. 1987;42:131–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Romano AM, Tomaselli S, Gualtieri G, Zoia MC, Fanfulla F, Berrayah L, Cerveri I. Respiratory function in precapillary pulmonary hypertension. Monaldi Arch Chest Dis. 1993;48:201–204. [PubMed] [Google Scholar]
- 14. Alonso‐Gonzalez R, Borgia F, Diller G‐P, Inuzuka R, Kempny A, Martinez‐Naharro A, Tutarel O, Marino P, Wustmann K, Charalambides M, Silva M, Swan L, Dimopoulos K, Gatzoulis MA. Abnormal lung function in adults with congenital heart disease: prevalence, relation to cardiac anatomy, and association with survival. Circulation. 2013;127:882–890. [DOI] [PubMed] [Google Scholar]
- 15. Horsley AR, Gustafsson PM, Macleod KA, Saunders C, Greening AP, Porteous DJ, Davies JC, Cunningham S, Alton EW, Innes JA. Lung clearance index is a sensitive, repeatable and practical measure of airways disease in adults with cystic fibrosis. Thorax. 2008;63:135–140. [DOI] [PubMed] [Google Scholar]
- 16. Bousquet J, Jeffery PK, Busse WW, Johnson M, Vignola AM. Asthma. From bronchoconstriction to airways inflammation and remodeling. Am J Respir Crit Care Med. 2000;161:1720–1745. [DOI] [PubMed] [Google Scholar]
- 17. Selman M, King TE, Pardo A. Idiopathic pulmonary fibrosis: prevailing and evolving hypotheses about its pathogenesis and implications for therapy. Ann Intern Med. 2001;134:136–151. [DOI] [PubMed] [Google Scholar]
- 18. Hogg JC, Chu F, Utokaparch S, Woods R, Elliott WM, Buzatu L, Cherniack RM, Rogers RM, Sciurba FC, Coxson HO, Pare PD. The nature of small‐airway obstruction in chronic obstructive pulmonary disease. N Engl J Med. 2004;350:2645–2653. [DOI] [PubMed] [Google Scholar]
- 19. Fernandezbonetti P, Lupiherrera E, Martinezguerra ML, Barrios R, Seoane M, Sandoval J. Peripheral airways obstruction in idiopathic pulmonary‐artery hypertension (primary). Chest. 1983;83:732–738. [DOI] [PubMed] [Google Scholar]
- 20. Tuder RM, Groves B, Badesch DB, Voelkel NF. Exuberant endothelial‐cell growth and elements of inflammation are present in plexiform lesions of pulmonary‐hypertension. Am J Pathol. 1994;144:275–285. [PMC free article] [PubMed] [Google Scholar]
- 21. Dorfmuller P, Zarka V, Durand‐Gasselin I, Monti G, Balabanian K, Garcia G, Capron F, Coulomb‐Lhermine A, Marfaing‐Koka A, Simonneau G, Emilie D, Humbert M. Chemokine RANTES in severe pulmonary arterial hypertension. Am J Respir Crit Care Med. 2002;165:534–539. [DOI] [PubMed] [Google Scholar]
- 22. Humbert M, Monti G, Brenot F, Sitbon O, Portier A, Grangeotkeros L, Duroux P, Galanaud P, Simonneau G, Emilie D. Increased interleukin‐1 and interleukin‐6 serum concentrations in severe primary pulmonary‐hypertension. Am J Respir Crit Care Med. 1995;151:1628–1631. [DOI] [PubMed] [Google Scholar]
- 23. Soon E, Holmes AM, Treacy CM, Doughty NJ, Southgate L, Machado RD, Trembath RC, Jennings S, Barker L, Nicklin P, Walker C, Budd DC, Pepke‐Zaba J, Morrell NW. Elevated levels of inflammatory cytokines predict survival in idiopathic and familial pulmonary arterial hypertension. Circulation. 2010;122:920–927. [DOI] [PubMed] [Google Scholar]
- 24. Stacher E, Graham BB, Hunt JM, Gandjeva A, Groshong SD, McLaughlin VV, Jessup M, Grizzle WE, Aldred MA, Cool CD, Tuder RM. Modern age pathology of pulmonary arterial hypertension. Am J Respir Crit Care Med. 2012;186:261–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bradford R, Tulloh R. Eisenmenger's syndrome: a review of the pathophysiology and therapeutic options. Br J Card Nurs. 2008;3:138–145. [Google Scholar]
- 26. Hamada H, Terai M, Kimura H, Hirano K, Oana S, Niimi H. Increased expression of mast cell chymase in the lungs of patients with congenital heart disease associated with early pulmonary vascular disease. Am J Respir Crit Care Med. 1999;160:1303–1308. [DOI] [PubMed] [Google Scholar]
- 27. Pinto RFA, Higuchi MD, Aiello VD. Decreased numbers of T‐lymphocytes and predominance of recently recruited macrophages in the walls of peripheral pulmonary arteries from 26 patients with pulmonary hypertension secondary to congenital cardiac shunts. Cardiovasc Pathol. 2004;13:268–275. [DOI] [PubMed] [Google Scholar]
- 28. Diller G‐P, van Eijl S, Okonko DO, Howard LS, Ali O, Thum T, Wort SJ, Bedard E, Gibbs JSR, Bauersachs J, Hobbs AJ, Wilkins MR, Gatzoulis MA, Wharton J. Circulating endothelial progenitor cells in patients with Eisenmenger syndrome and idiopathic pulmonary arterial hypertension. Circulation. 2008;117:3020–3030. [DOI] [PubMed] [Google Scholar]
- 29. Stockley RA, Mistry M, Bradwell AR, Burnett D. Study of plasma‐proteins in the sol phase of sputum from patients with chronic‐bronchitis. Thorax. 1979;34:777–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Gadek JE, Klein HG, Holland PV, Crystal RG. Replacement therapy of alpha‐1‐antitrypsin deficiency—reversal of protease‐antiprotease imbalance within the alveolar structures of PiZ subjects. J Clin Invest. 1981;68:1158–1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Miller MR, Hankinson J, Brusasco V, Burgos F, Casaburi R, Coates A, Crapo R, Enright P, van der Grinten CPM, Gustafsson P, Jensen R, Johnson DC, MacIntyre N, McKay R, Navajas D, Pedersen OF, Pellegrino R, Viegi G, Wanger J. Standardisation of spirometry. Eur Respir J. 2005;26:319–338. [DOI] [PubMed] [Google Scholar]
- 32. Horsley A, Macleod K, Gupta R, Goddard N, Bell N. Enhanced photoacoustic gas analyser response time and impact on accuracy at fast ventilation rates during multiple breath washout. PLoS One. 2014;9:e98487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. ATS statement: guidelines for the six‐minute walk test. Am J Respir Crit Care Med. 2002;166:111–117. [DOI] [PubMed] [Google Scholar]
- 34. Galie N, Humbert M, Vachiery JL, Gibbs S, Lang I, Torbicki A, Simonneau G, Peacock A, Vonk Noordegraaf A, Beghetti M, Ghofrani A, Gomez Sanchez MA, Hansmann G, Klepetko W, Lancellotti P, Matucci M, McDonagh T, Pierard LA, Trindade PT, Zompatori M, Hoeper M. 2015 ESC/ERS guidelines for the diagnosis and treatment of pulmonary hypertension: the Joint Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS): endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC), International Society for Heart and Lung Transplantation (ISHLT). Eur Heart J. 2016;37:67–119.26320113 [Google Scholar]
- 35. Paggiaro PL, Chanez P, Holz O, Ind PW, Djukanovic R, Maestrelli P, Sterk PJ. Sputum induction. Eur Respir J. 2002;20:3S–8S. [DOI] [PubMed] [Google Scholar]
- 36. Chalmers GW, Thomson L, Macleod KJ, Dagg KD, McGinn BJ, McSharry C, Patel KR, Thomson NC. Endothelin‐1 levels in induced sputum samples from asthmatic and normal subjects. Thorax. 1997;52:625–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Efthimiadis A, Spanevello A, Hamid Q, Kelly MM, Linden M, Louis R, Pizzichini MMM, Pizzichini E, Ronchi C, Van Overveld F, Djukanovic R. Methods of sputum processing for cell counts, immunocytochemistry and in situ hybridisation. Eur Respir J. 2002;20:19S–23S. [DOI] [PubMed] [Google Scholar]
- 38. Simonneau G, Gatzoulis MA, Adatia I, Celermajer D, Denton C, Ghofrani A, Gomez Sanchez MA, Kumar RK, Landzberg M, Machado RF, Olschewski H, Robbins IM, Souza R. Updated clinical classification of pulmonary hypertension. J Am Coll Cardiol. 2013;62:D34–D41. [DOI] [PubMed] [Google Scholar]
- 39. Rubin LJ. Primary pulmonary hypertension. Chest. 1993;104:236–250. [DOI] [PubMed] [Google Scholar]
- 40. Barst RJ, Ivy DD, Foreman AJ, McGoon MD, Rosenzweig EB. Four‐ and seven‐year outcomes of patients with congenital heart disease‐associated pulmonary arterial hypertension (from the REVEAL Registry). Am J Cardiol. 2014;113:147–155. [DOI] [PubMed] [Google Scholar]
- 41. Macarthur CGC, Hunter D, Gibson GJ. Ventilatory function in the Eisenmenger syndrome. Thorax. 1979;34:348–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Broberg CS, Van Woerkom RC, Swallow E, Dimopoulos K, Diller G‐P, Allada G, Gatzoulis MA. Lung function and gas exchange in Eisenmenger syndrome and their impact on exercise capacity and survival. Int J Cardiol. 2014;171:73–77. [DOI] [PubMed] [Google Scholar]
- 43. Iozzo A, Cosentino P, Ghai PC, Garbagni R. Alveolar‐arterial gradients and small airways in kyphoscoliosis. Respiration. 1983;44:314–320. [DOI] [PubMed] [Google Scholar]
- 44. Horn M, Ries A, Neveu C, Moser K. Restrictive ventilatory pattern in precapillary pulmonary‐hypertension. Am Rev Respir Dis. 1983;128:163–165. [DOI] [PubMed] [Google Scholar]
- 45. Dalonzo GE, Bower JS, Dantzker DR. Differentiation of patients with primary and thromboembolic pulmonary‐hypertension. Chest. 1984;85:457–461. [DOI] [PubMed] [Google Scholar]
- 46. Quanjer PH, Weiner DJ, Pretto JJ, Brazzale DJ, Boros PW. Measurement of FEF25‐75% and FEF75% does not contribute to clinical decision making. Eur Respir J. 2014;43:1051–1058. [DOI] [PubMed] [Google Scholar]
- 47. Low AT, Giesen L, Gin‐Sing W, Howard L, Tulloh RMR. The advantage of a shared care service in improved access to pulmonary hypertension therapy for adult congenital heart disease associated pulmonary hypertension. Cardiol Young. 2014;24:110–111. [Google Scholar]
- 48. Galie N, Hoeper MM, Humbert M, Torbicki A, Vachiery JL, Barbera JA, Beghetti M, Corris P, Gaine S, Gibbs JS, Gomez‐Sanchez MA, Jondeau G, Klepetko W, Opitz C, Peacock A, Rubin L, Zellweger M, Simonneau G. Guidelines for the diagnosis and treatment of pulmonary hypertension. Eur Respir J. 2009;34:1219–1263. [DOI] [PubMed] [Google Scholar]
- 49. Yoshibayashi M, Nishioka K, Nakao K, Saito Y, Matsumura M, Ueda T, Temma S, Shirakami G, Imura H, Mikawa H. Plasma endothelin concentrations in patients with pulmonary‐hypertension associated with congenital heart‐defects—evidence for increased production of endothelin in pulmonary circulation. Circulation. 1991;84:2280–2285. [DOI] [PubMed] [Google Scholar]
- 50. Uchida Y, Ninomiya H, Saotome M, Nomura A, Ohtsuka M, Yanagisawa M, Goto K, Masaki T, Hasegawa S. Endothelin, a novel vasoconstrictor peptide, as potent bronchoconstrictor. Eur J Pharmacol. 1988;154:227–228. [DOI] [PubMed] [Google Scholar]
- 51. Chalmers GW, Macleod KJ, Sriram S, Thomson LJ, McSharry C, Stack BHR, Thomson NC. Sputum endothelin‐1 is increased in cystic fibrosis and chronic obstructive pulmonary disease. Eur Respir J. 1999;13:1288–1292. [DOI] [PubMed] [Google Scholar]
- 52. Bacakoglu F, Atasever A, Ozhan MH, Gurgun C, Ozkilic H, Guzelant A. Plasma and bronchoalveolar lavage fluid levels of endothelin‐1 in patients with chronic obstructive pulmonary disease and pulmonary hypertension. Respiration. 2003;70:594–599. [DOI] [PubMed] [Google Scholar]