

Abstract
The polytopic ligand 1 contains three different metal ion binding subunits forming two substructures that code for the self-assembly of two different coordination structures (helicate and grid type) under metal ion complexation. Reaction with Cu(II) and Zn(II) ions generates the coordinatively unsaturated architectures 8 and 9 resulting from the formation of two double helicate arrangements. Their crystal structure has been determined by x-ray diffraction. These results show that the double helical motif is expressed at the expense of the grid type one, indicating the dominant/recessive behavior of the system. Together with earlier studies on the linear combination and crossover processing schemes, the dominant/recessive generation of 8 and 9 completes the demonstration of principle of the modes of multiple expression of molecular information in a multicode programmed chemical system.
Metal ion-directed self-assembly has been extensively developed as a powerful tool for the spontaneous and efficient generation of complex inorganic architectures (for recent reviews on metal-ion mediated self-assembly, see, for example, refs. 1–13). The goal is to become progressively able to control it in a programmable fashion (3) by means of the information stored in the structure of the ligands and its processing via the coordination algorithms of the metal ions in the course of the complexation process. The information directing the self-assembly is represented by the geometric, steric, and electronic properties of the binding units contained in the structure of the ligand. These subunits are selected so as to encode the ligand for the formation of a given type of superstructure, such as helicate, cage, grid (1–13), etc. As a further step toward more complex processes, we have recently reported (14, 15) the design and properties of ligand systems built from binding subunits of two different types so as to combine within their structure the information required for the processing of two distinct programs corresponding, respectively, to the generation of architectures of helicate-type and grid-type. Such systems offer the possibility of multiple expression of molecular information (16, 17). We have shown in these cases that the subprograms encoded in a particular ligand can be expressed either in an independent way (linear combination) or in a mixed fashion, resulting from the crossing-over of the two subprograms (14, 15). In particular, the outcome of the complexation process could be tuned by making use of different sets of metal ions (representing different reading algorithms) to enforce the self-assembly of different coordination architectures (15). A third possibility is a dominant/recessive behavior where one of the subprograms imposes its own output over the other one(s) (17).
The systems mentioned above operated under conditions in which the ratio ligand/metal ion used was such that all the available coordination sites of the ligands were involved in the binding events. Thus, the information contained in the structure of the ligands was fully exploited by the metal ions. We reasoned that, by an appropriate choice of binding subunits and metal ions, it could be possible, under specific conditions, to trigger the selective expression of one of the encoded coordination programs preferentially to the other(s). This would result in the emergence of the structural motif corresponding to that subprogram, at the expense of alternative, different structures. In the present report, we describe the coordination properties of the multiple subunit ligand 1, which presents such dominant/recessive behavior.
Materials and Methods
Preparation of Cu(II) and Zn(II) Complexes 8 and 9.
The reactions were performed typically on a 20-mg scale of ligand. A suspension of ligand 1 in CH3CN (10 mg/ml) was treated with 2 equivalents of Cu(Tf)2⋅6H2O or Zn(ClO4)2⋅6H2O at reflux for 2 h. The cooled solution (turquoise for Cu and colorless for Zn) was poured over a concentrated aqueous solution of NH4PF6, to quantitatively obtain a precipitate that was centrifuged, and washed several times with water, cold MeOH, and finally ether. All of the subsequent spectral measurements [NMR, electrospray mass spectrometry (ESMS)], as well as the crystallizations for x-ray structure determination, were performed directly on these samples without further purification.
Preparation of Complex [14Co4Cu8(PF6)16], 10.
12.6 mg of ligand 1, 1 equivalent of Co(ClO4)2⋅6H2O, and 2 equivalents of Cu(CH3CN)4ClO4 were heated for 18 h under Ar at 160°C–185°C in 1 ml of ethylene glycol. The resulting solution was cooled and poured dropwise over 4 ml of concentrated aqueous NH4PF6. The precipitate was centrifuged, and washed several times with water, cold iPrOH, and finally ether. Drying under high vacuum afforded a red powder (96% mass recovery). The corresponding complexes with Ni(II) and Fe(II) were prepared in a similar way.
Crystal Structure Data for Complex 9.
[12Zn4]8+⋅8PF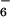 ⋅7 C6H6⋅5 CH3NO2⋅H2O, Fw = 4651.8, crystal size 0.3 × 0.3 × 0.4 mm3, triclinic, P-1, a = 13.7740(2) Å, b = 17.8767(3) Å, c = 22.2208(4) Å, α = 92.340(1)°, β = 104.719(1)°, γ = 97.4800(1)°, V = 5,226.8(1) Å3, Z = 1, ρcalcd = 1.48 g⋅cm−3, 2Θmax = 60.88°, μ = 0.627 mm−1, F(000) = 2,370, 1,285 parameters (full matrix refinement), R1 = 0.1105, wR2 = 0.2762 [for 28,856 refl. I > 2σ(I)], R1 (all data) = 0.2044, wR2 (all data) = 0.3364, S = 1.013, Δρ (min/max) = −0.97/1.55 eÅ−3. The crystallographic data were collected on a Nonius Kappa charge-coupled device (CCD) area-detector diffractometer (Enraf-Nonius, Delft, The Netherlands) by using graphite monochromatized MoKα radiation (λ = 0.71073 Å). Lattice parameters were determined from 10 images recorded with 1Υϕ scans and subsequently refined on all data. The data collections were performed by using ϕ and ω scans with 1Υ steps with an exposure time of 60 s per frame and the crystal-to-detector distance fixed to 35 mm. The data were processed by using denzo-smn v0.93.0 (18). No absorption correction was applied. The structure was solved by direct methods by using shelxs-97 (19) and refined on F2 by using shelxl-97 (20). The hydrogen atoms were calculated to their idealized positions with isotropic temperature factors and refined as riding atoms. The structure is centrosymmetric with a lot of disordered solvent molecules. Two solvent benzene molecules have a disorder over two orientations (0.5 occupancy), one of the remaining benzene molecules is disordered over two positions with occupancy 0.5. Similarly, one of the nitromethane solvent molecule is positionally disordered over two positions (occupancy 0.5), and one rather high residual electron density was treated as positionally (occupancy 0.5) disordered water molecule, H-atoms of which could not be located. The highest residual electron density was found near disordered benzene solvent molecules. Crystallographic data (excluding structure factors) for the structure of [12Zn4(PF6)8], 9 reported in this paper have been deposited with the Cambridge Crystallographic Data Centre as supplementary publication no. CCDC-163255.
⋅7 C6H6⋅5 CH3NO2⋅H2O, Fw = 4651.8, crystal size 0.3 × 0.3 × 0.4 mm3, triclinic, P-1, a = 13.7740(2) Å, b = 17.8767(3) Å, c = 22.2208(4) Å, α = 92.340(1)°, β = 104.719(1)°, γ = 97.4800(1)°, V = 5,226.8(1) Å3, Z = 1, ρcalcd = 1.48 g⋅cm−3, 2Θmax = 60.88°, μ = 0.627 mm−1, F(000) = 2,370, 1,285 parameters (full matrix refinement), R1 = 0.1105, wR2 = 0.2762 [for 28,856 refl. I > 2σ(I)], R1 (all data) = 0.2044, wR2 (all data) = 0.3364, S = 1.013, Δρ (min/max) = −0.97/1.55 eÅ−3. The crystallographic data were collected on a Nonius Kappa charge-coupled device (CCD) area-detector diffractometer (Enraf-Nonius, Delft, The Netherlands) by using graphite monochromatized MoKα radiation (λ = 0.71073 Å). Lattice parameters were determined from 10 images recorded with 1Υϕ scans and subsequently refined on all data. The data collections were performed by using ϕ and ω scans with 1Υ steps with an exposure time of 60 s per frame and the crystal-to-detector distance fixed to 35 mm. The data were processed by using denzo-smn v0.93.0 (18). No absorption correction was applied. The structure was solved by direct methods by using shelxs-97 (19) and refined on F2 by using shelxl-97 (20). The hydrogen atoms were calculated to their idealized positions with isotropic temperature factors and refined as riding atoms. The structure is centrosymmetric with a lot of disordered solvent molecules. Two solvent benzene molecules have a disorder over two orientations (0.5 occupancy), one of the remaining benzene molecules is disordered over two positions with occupancy 0.5. Similarly, one of the nitromethane solvent molecule is positionally disordered over two positions (occupancy 0.5), and one rather high residual electron density was treated as positionally (occupancy 0.5) disordered water molecule, H-atoms of which could not be located. The highest residual electron density was found near disordered benzene solvent molecules. Crystallographic data (excluding structure factors) for the structure of [12Zn4(PF6)8], 9 reported in this paper have been deposited with the Cambridge Crystallographic Data Centre as supplementary publication no. CCDC-163255.
Results and Discussion
Ligand Design and Synthesis.
Ligand 1 contains six metal ion-binding subunits of three different types, two by two distinct in their structural and coordination properties.
The terminal terpyridine (terpy) units ligate metal-ions in a tridentate fashion, thus displaying a strong preference for ions capable of hexa- and penta-coordination over ions presenting tetrahedral binding. The 6,6′-disubstituted bipyridine (bipy) as well as the 6-substituted 2,3′-pyridine-pyridazine (py-pz) sites preferentially bind metal-ions of tetrahedral rather than hexacoordinated coordination mode. Indeed, ligands 2 and 3, analogous to the terminal bipy-terpy motif of ligand 1, form a [2,3,Cu(II)3]6+ double helicate species with three pentacoordinated Cu(II) ions (16). A heterostranded trinuclear double helicate is also generated from a Tris-bipy ligand, a Tris-terpy ligand, and three Cu(II) ions (21). On the other hand, ligand 4a and related ligands are known to bind ions such as Cu(I) or Ag(I) in a tetrahedral fashion, giving grid-type complexes (22, 23), 4a itself forming a tetranuclear [2 × 2] grid (22). The two distinct bidentate sites (bipy and py-pz) are geometrically similar but differ in their electronic properties, bipy being a better σ-donor and poorer π-acceptor than py-pz. Taking advantage of these multiple features of the coordination units incorporated in the structure of 1, we have investigated its self-assembly properties with a variety of metal ions.
Ligand 1 was efficiently synthesized in a manner similar to previous ligands (14, 15). The brominated terpyridine 6 was treated with the monoprotected diol 5b (obtained from the corresponding diol 5a) in the presence of NaH tetrahydrofurane (THF). Quenching of the reaction with water followed by evaporation of the THF produced quantitatively a white precipitate that was recrystallized from chloroform-isopropanol to afford analytically pure 7a in 87% yield. Acid deprotection of 7a quantitatively yielded the alcohol 7b, which was mesylated in CH2Cl2 at −10°C. Vacuum evaporation of the CH2Cl2 followed by washing with dilute NaHCO3, water, MeOH, and finally ether produced 7c in virtually quantitative yield. The ligand 1 was obtained by treating the diol 4b (14) at reflux with NaH in THF followed by 2 equivalents of the mesylate 7c. After one night at reflux, the reaction was quenched with water, the THF evaporated, and the precipitate formed was filtered. Successive washings with hot MeOH, cold CHCl3, and ether afforded ligand 1 in 83% yield. Detailed experimental procedures will be described elsewhere, together with the synthesis of the ligands studied earlier (14, 15).
Formation and Structure of the Cu(II) and Zn(II) Complexes of Ligand 1 in Solution.
Treatment of a suspension of 1 in acetonitrile with 2 equivalents of Cu(CF3SO3)2 or Zn(ClO4)2⋅6H2O at reflux for 2 h (or room temperature for 2 days) resulted in a complete solubilization of the ligand and the appearance of a turquoise color for the Cu(II) complex and a colorless solution for the Zn(II) complex. The initial counteranions could be exchanged by precipitation with an aqueous solution of NH4X, with X = PF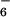 , ClO
, ClO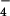 , CF3SO
, CF3SO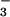 , or SbF
, or SbF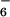 . The ESMS analysis (Fig. 1) of the compounds formed unambiguously shows the formation of molecular entities of stoichiometry [12Cu4(CF3SO3)8], 8 and [12Zn4(PF6)8], 9. The signals observed are due to positively charged species, obtained by successive loss of two, three, four, and five anions from the molecular structures 8 and 9.
. The ESMS analysis (Fig. 1) of the compounds formed unambiguously shows the formation of molecular entities of stoichiometry [12Cu4(CF3SO3)8], 8 and [12Zn4(PF6)8], 9. The signals observed are due to positively charged species, obtained by successive loss of two, three, four, and five anions from the molecular structures 8 and 9.
Figure 1.

Electrospray mass spectra of the complexes [12Cu4(CF3SO3)8], 8 and [12Zn4(PF6)8], 9.
NMR analysis (in CD3CN) of [12Zn4(PF6)8], 9 gives further insight into the structure of the architecture formed. The complex displays a very well resolved and diagnostic spectrum (Fig. 2).
Figure 2.

Five hundred megaherz proton NMR spectrum of the Zn(II) complex 9.
The number of signals corresponding to the aromatic protons clearly shows that each ligand is coordinated in a symmetrical fashion. Each of the four pairs of geminal methylene protons (that were equivalent in the proton NMR spectrum of the free ligand) becomes inequivalent (diastereotopic) in the complex. This behavior is typical of a local double helical environment resulting from the wrapping of two ligand strands around the metal ions. Rotating-frame Overhauser effect spectroscopy (ROESY) experiments (not shown) were used to assign the 1H NMR signals of the complex, as well as for assessing the different spatial relations existing between them. In particular, the observation of a cross-peak at 7.57;5.00 ppm, arising from the interaction between the protons H6C and He strongly supports the simultaneous complexation of a terminal terpyridine unit and of a bipyridine unit to a same metal center. These data, together with the stoichiometry of the complex, support structure 9 (Fig. 3) for this species in solution.
Figure 3.

Self-assembly and structure of the coordination architecture formed by the binding of M(II) = Cu(II) and Zn(II) metal ions by ligand 1 to give complexes 8 and 9, respectively (Upper). Schematic representation of the structure of complexes 10 and 11 (Lower).
Complex 8 presents an electronic d-d transition at λ = 650 nm, ɛ = 400 lm−1⋅cm−1. These values are in agreement with the complexation of Cu(II) by one bipyridine and one terpyridine unit (21, 24) and suggest that 8 has a solution structure analogous to that of 9 (Fig. 3).
Crystal Structures of the Cu(II) and Zn(II) Complexes 8 and 9.
Single crystals of the two complexes were obtained by slow evaporation of a saturated solution of 8 (triflate salt) in a 1:2 mixture of acetonitrile-toluene and by slow diffusion of benzene in a 2 mg/ml solution of 9 (hexafluorophosphate salt) in nitromethane. The solid state structure of both complexes was examined by x-ray crystallography. The present discussion will be limited to complex 9. The data measured for 8 gave a poorer refinement because of twining of the crystals; they were nevertheless sufficient to indicate that the complex cation of 8 had the same structure as that of 9 described here. The structure obtained for 9 confirms the solution state studies (Fig. 4).
Figure 4.

Crystal structure of the Zn(II) complex [12Zn4(PF6)8], 9: front (Left) and side (Right) views; ball-and-stick representations (Upper), space-filling representations (Lower) with the included PF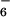 anion shown; all solvent molecules and other counterions excluded.
anion shown; all solvent molecules and other counterions excluded.
The architecture consists of two double-helical domains, linked by the uncomplexed py-pz-py unit. The two double-helical units are dinuclear, homostranded helicates with antiparallel (head-to-tail) orientation of the strands. They are of opposite chirality, the complex being a meso form. Each double-helical domain is formed by the coordination of two Zn(II) ions, each to a terpyridine and to a bipyridine motif. The coordination geometry is a distorted octahedron (Table 1), best described as 5 + 1 coordination with the O-atom being located at the outer limit of a Zn—O bond. The distances between the electronically isolated Zn ions are 8.409 Å (intra-helix) and 13.867 Å (inter-helix).
Table 1.
Bond distances (Å) and angles (°) around the Zn(II) centers
| Zn(1)-N(63) | 2.060(5) | Zn(2)-N(33)#1 | 2.047(5) |
| Zn(1)-N(12)#1 | 2.073(4) | Zn(2)-N(84) | 2.075(4) |
| Zn(1)-N(69) | 2.112(5) | Zn(2)-N(27)#1 | 2.143(5) |
| Zn(1)-N(18)#1 | 2.176(5) | Zn(2)-N(77) | 2.163(4) |
| Zn(1)-N(1)#1 | 2.206(5) | Zn(2)-N(90) | 2.210(4) |
| Zn(1)-O(56) | 2.461(4) | Zn(2)-O(35)#1 | 2.426(4) |
| N(63)-Zn(1)-N(12)#1 | 156.8(2) | N(33)#1-Zn(2)-N(84) | 160.5(2) |
| N(63)-Zn(1)-N(69) | 79.0(2) | N(33)#1-Zn(2)-N(27)#1 | 78.4(2) |
| N(12)#1-Zn(1)-N(69) | 123.8(2) | N(84)-Zn(2)-N(27)#1 | 120.1(2) |
| N(63)-Zn(1)-N(18)#1 | 101.1(2) | N(33)#1-Zn(2)-N(77) | 104.3(2) |
| N(12)#1-Zn(1)-N(18)#1 | 77.0(2) | N(84)-Zn(2)-N(77) | 76.3(2) |
| N(69)-Zn(1)-N(18)#1 | 108.9(2) | N(27)#1-Zn(2)-N(77) | 110.0(2) |
| N(63)-Zn(1)-N(1)#1 | 104.2(2) | N(33)#1-Zn(2)-N(90) | 102.2(2) |
| N(12)#1-Zn(1)-N(1)#1 | 74.9(2) | N(84)-Zn(2)-N(90) | 74.6(2) |
| N(69)-Zn(1)-N(1)#1 | 88.0(2) | N(27)#1-Zn(2)-N(90) | 87.1(2) |
| N(18)#1-Zn(1)-N(1)#1 | 151.8(2) | N(77)-Zn(2)-N(90) | 150.8(2) |
| N(63)-Zn(1)-O(56) | 70.4(2) | N(33)#1-Zn(2)-O(35)#1 | 70.8(2) |
| N(12)#1-Zn(1)-O(56) | 86.4(2) | N(84)-Zn(2)-O(35)#1 | 89.9(2) |
| N(69)-Zn(1)-O(56) | 148.3(2) | N(27)#1-Zn(2)-O(35)#1 | 147.6(2) |
| N(18)#1-Zn(1)-O(56) | 85.6(2) | N(77)-Zn(2)-O(35)#1 | 87.5(2) |
| N(1)#1-Zn(1)-O(56) | 91.5(2) | N(90)-Zn(2)-O(35)#1 | 89.9(2) |
Symmetry operation, #1 −x + 1, −y + 1, −z + 2.
The two double helical motifs are connected through central py-pz-py units, in such a way that the py-pz-py aromatic rings of one ligand are respectively parallel to those of the second ligand. They are positioned with respect to each other in a stair-like fashion. Within the py-pz-py motif, the plane of the central pyridazine forms a dihedral angle of 12.2° with the plane of the adjacent pyridine ring. As may be expected, the central pyridazine is found to be in a transoid conformation with respect to the adjacent pyridines (25).
The cavity created inside the complex 9 is large enough to accommodate one of the PF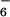 counteranions, which is maintained by strong charge–charge electrostatic binding as well as weak interactions between the aromatic H atoms of the ligand and the fluorides. The corresponding nonbonded (F⋅⋅⋅H) distances vary from 2.46 to 2.57 Å. The other anions and the solvent molecules fill the voids between the complexes 9 in the crystal lattice within Van der Waals contacts.
counteranions, which is maintained by strong charge–charge electrostatic binding as well as weak interactions between the aromatic H atoms of the ligand and the fluorides. The corresponding nonbonded (F⋅⋅⋅H) distances vary from 2.46 to 2.57 Å. The other anions and the solvent molecules fill the voids between the complexes 9 in the crystal lattice within Van der Waals contacts.
Dominant-Recessive Behavior in the Expression of Molecular Information.
The generation of the architectures 8, 9 by coordination of Cu(II) and Zn(II) ions results from the expression of one of the two subprograms encoded in ligand 1, the (bipy-terpy) domains (coding for double helicate formation under pentacoordination), and the repression of the other one, the py-pz-py unit (coding for the formation of a [2 × 2] grid-type structure under tetrahedral coordination). This amounts to a dominant/recessive behavior (17) in the processing of the molecular information contained in this ligand by the Cu(II) and Zn(II) ions.
The directing factors in the formation of complexes 8 and 9 by ligand 1 with Cu(II) and Zn(II) ions, respectively, may be considered to involve structural features, such as relative positioning of the subunits and conformational flexibility; binding properties, pentacoordination of Cu(II) and Zn(II) together with stronger interaction with a pyridine nitrogen site than with a pyridazine one, thus favoring complexation to a (bipy, terpy) pair over complexation to a (py-pz, terpy) one; and contributions from stacking and other interactions (such as charge-dipole).
The presence of free coordination sites in complexes 8 and 9 stresses that, despite the strength of coordination interactions and their tendency to impose maximal site occupation (26), other factors involving internal ligand properties (strain, flexibility, secondary interactions) as well as external effects (medium, counterions, etc.) may interfere with the main assembly directing code and influence the nature of the thermodynamically favored output entity. Such features are also found and have been pointed out in the generation of an equilibrating collection of circular inorganic architectures (27), in the formation of a [4 × 5] grid-type entity containing free coordination sites rather than of the fully coordinated [5 × 5] species (28), and in the medium-dependent adaptive interconversion of two different coordination geometries (24).
All of the contributions, internal as well as external, have to be taken into account to achieve control over the evolution of the system and to gain progressively insight into the requirements for controlling the robustness/adaptability (29) balance in the programming of chemical systems through both storage of suitable information and implementation of specific processing algorithms.
Coordination of Ligand 1 with Other Sets of Metal Ions.
Because ligand 1 contains metal ion binding subunits of three different types, it offers in principle opportunities for generating different coordination architectures by complexation of different sets of metal ions, thus leading to multiple expression of molecular information along the lines presented earlier (15, 17).
Indeed, treatment of ligand 1 in ethylene glycol at 170°C with a mixture of Cu(I) and M(II) = Co(II), Ni(II), or Fe(II) ions in L/M(II)/Cu(I) = 1/1/2 ratio yields a red solution for Co(II)/Cu(I) or Ni(II)/Cu(I) and an intense purple one for Fe(II)/Cu(I). From these solutions the complex formed is precipitated with an aqueous solution of NH4X, X = PF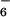 or ClO
or ClO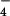 .
.
The proton NMR spectra of the three complexes does not display any sharp signal and is uninformative. However, the ESMS data clearly indicate the almost exclusive formation of the species [14Co4Cu8(PF6)16], 10 and [14Ni4Cu8(PF6)16], 11. The spectra display peaks resulting from the successive loss of PF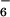 anions from the parent complexes (Fig. 5). The product of the reaction of 1 with a 1/2 Fe(II)/Cu(I) mixture is much less well-defined. It gives an ESMS spectrum displaying the signals corresponding to the species [14Fe4Cu8(PF6)16] together with compounds of lower molecular weight.
anions from the parent complexes (Fig. 5). The product of the reaction of 1 with a 1/2 Fe(II)/Cu(I) mixture is much less well-defined. It gives an ESMS spectrum displaying the signals corresponding to the species [14Fe4Cu8(PF6)16] together with compounds of lower molecular weight.
Figure 5.

Electrospray mass spectra of the complexes [14Co4Cu8(PF6)16], 10 and [14Ni4Cu8(PF6)16], 11.
Despite extensive attempts, it was not possible to obtain crystals of the above complexes suitable for structure determination by x-ray diffraction. On the basis of the ESMS data together with the respective tetrahedral and octahedral coordination geometries of the Cu(I) and M(II) ions and the results obtained earlier with related ligands (14, 15), the architecture schematically represented by 10,11 (Fig. 3) may be suggested for the [14M4Cu8]16+ complex species. A similar structure has been considered earlier (15); however, although likely, it remains tentative in the absence of crystal structure data.
A third type of architecture may be considered for the complexes of 1. Indeed, complexation of 1 with a set of ions presenting penta(trigonalbipyramidal), e.g., Cu(II), and tetrahedral, e.g., Cu(I), coordination in 1/Cu(II)/Cu(I) = 1/2/1 stoichiometry, could be expected to lead to a dodecanuclear circular species of 4/8/4 composition and of the type described earlier (refs. 14 and 15; see structure D, figure 2 in ref. 15). However, the reaction performed in these conditions gave a species [12Cu(II)4]8+ as identified by ESMS, having probably the same structure as 8, 9 above. This observation points to the marked thermodynamic preference for this arrangement even in presence of Cu(I) ions, thus confirming the dominant character of its formation.
Thus, ligand 1 may at least, in principle, lead to three different coordination architectures, depending on the set of metal ions operating on the structural/binding information contained in its molecular strand. Such an ability further stresses the rich behavior of chemical systems capable of multiple expression of molecular information through the operation of different interactional algorithms on the same structural codes (17). It also makes clear that much further work is needed and warranted to fully explore, refine, and exploit all of the consequences of such potential.
Conclusions
The results presented here reveal the dominant/recessive behavior displayed in the generation of architectures 8, 9 by coordination of the multisubunit ligand 1 with Cu(II) or Zn(II) ions. Together with the linear combination and crossover processing schemes described earlier (14, 15), they complete the demonstration of principle for the three ways in which the expression of molecular information may occur in multiple-code programmed chemical systems (17). In fact, as discussed above, ligand 1 could in principle display all three type of behaviors, but such a case remains to be established. Suitable encoding by manipulation of structural subunits and processing through specific interactional algorithms should give access to a variety of other systems. A major facet is the evaluation of the robustness/stability of a given directing code (for instance, of metal coordination nature), i.e., the extent to which it remains unperturbed by interfering interactions (such as secondary metal coordination, hydrogen bonding, van der Waals stacking, etc). On the other hand, sensitivity to perturbations, while limiting the operation range, introduces diversity and adaptability (29) in the self-assembling process. Further exploration along these directions may concern features such as parallel processing, molecular computing, and dynamic information generation (17).
Scheme 1.

Acknowledgments
This publication is dedicated to the memory of Professor Donald J. Cram. We thank Dr. H. Nierengarten and Dr. A. Van Dorsselaer for the ESMS investigations, Dr. A. DeCian and Prof. J. Fischer for crystal structure studies on compound 8, and Dr. R. Graff for the NMR work. K.R. thanks the Academy of Finland for financial support.
Abbreviations
- ESMS
electrospray mass spectrometry
- THF
tetrahydrofurane
Footnotes
Data deposition: The atomic coordinates have been deposited in the Cambridge Structural Database, Cambridge Crystallographic Data Centre, Cambridge CB2 1EZ, United Kingdom (CSD reference no. CCDC-163255).
References
- 1.Baxter P N W. In: Comprehensive Supramolecular Chemistry. Atwood J L, Davies J E D, MacNicol D D, Vögtle F, Lehn J-M, editors. Vol. 9. Oxford: Pergamon; 1996. , Ch. 5. [Google Scholar]
- 2.Constable E C. In: Comprehensive Supramolecular Chemistry. Atwood J L, Davies J E D, MacNicol D D, Vögtle F, Lehn J-M, editors. Vol. 9. Oxford: Pergamon; 1996. , Ch. 6. [Google Scholar]
- 3.Fujita M. In: Comprehensive Supramolecular Chemistry. Atwood J L, Davies J E D, MacNicol D D, Vögtle F, Lehn J-M, editors. Vol. 9. Oxford: Pergamon; 1996. , Ch. 7. [Google Scholar]
- 4.Constable E C. Prog Inorg Chem. 1994;42:67–138. [Google Scholar]
- 5.Lawrence D S, Jiang T, Levett M. Chem Rev. 1995;95:2229–2260. [Google Scholar]
- 6.Fujita M. Chem Soc Rev. 1998;27:417–425. [Google Scholar]
- 7.Saalfrank R F, Demleitner B. In: Transition Metals in Supramolecular Chemistry. Sauvage J-P, editor. New York: Wiley; 1999. pp. 1–51. [Google Scholar]
- 8.Caulder D L, Raymond K N. J Chem Soc Dalton Trans. 1999;8:1185–1200. [Google Scholar]
- 9.Piguet C. J Inclusion Phenom Macrocycl Chem. 1999;34:361–391. [Google Scholar]
- 10.Leininger S, Olenyuk B, Stang P J. Chem Rev. 2000;100:853–908. doi: 10.1021/cr9601324. [DOI] [PubMed] [Google Scholar]
- 11.Swiegers G F, Malefetse T J. Chem Rev. 2000;100:3483–3537. doi: 10.1021/cr000023w. [DOI] [PubMed] [Google Scholar]
- 12.Holliday B J, Mirkin C A. Angew Chem Int Ed Engl. 2001;40:2022–2043. [PubMed] [Google Scholar]
- 13.Lehn J-M. Supramolecular Chemistry: Concepts and Perspectives. New York: VCH; 1995. , Ch. 9. [Google Scholar]
- 14.Funeriu D P, Lehn J-M, Baum G, Fenske D. Chem Eur J. 1997;3:99–104. [Google Scholar]
- 15.Funeriu D P, Fromm K M, Lehn J-M, Fenske D. Chem Eur J. 2000;6:2103–2111. doi: 10.1002/1521-3765(20000616)6:12<2103::aid-chem2103>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- 16.Smith V C M, Lehn J-M. Chem. Commun. 1996. 2733–2734. [Google Scholar]
- 17.Lehn J-M. Chem Eur J. 2000;6:2097–2102. doi: 10.1002/1521-3765(20000616)6:12<2097::aid-chem2097>3.0.co;2-t. [DOI] [PubMed] [Google Scholar]
- 18.Otwinowski Z, Minor W. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 19.Sheldrick G M. SHELXS-97: A Program for Automatic Solution of Crystal Structures. Göttingen, Germany: University of Göttingen; 1997. [Google Scholar]
- 20.Sheldrick G M. SHELXL-97: A Program for Crystal Structure Refinement. Göttingen, Germany: University of Göttingen; 1997. [Google Scholar]
- 21.Hasenknopf B, Lehn J-M, Baum G, Fenske D. Proc Natl Acad Sci USA. 1996;93:1397–1400. doi: 10.1073/pnas.93.4.1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Youinou M T, Rahmouni N, Fischer J, Osborn J A. Angew Chem Int Ed Engl. 1992;31:733–735. [Google Scholar]
- 23.Baxter P N W, Lehn J-M, Fischer J, Youinou M-T. Angew Chem Int Ed Engl. 1994;33:2284–2287. [Google Scholar]
- 24.Baxter P N W, Khoury R G, Lehn J-M, Baum G, Fenske D. Chem Eur J. 2000;6:4140–4148. doi: 10.1002/1521-3765(20001117)6:22<4140::aid-chem4140>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- 25.Cuccia L A, Lehn J-M, Homo J-C, Schmutz M. Angew Chem Int Ed Engl. 2000;39:233–237. doi: 10.1002/(sici)1521-3773(20000103)39:1<233::aid-anie233>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]
- 26.Krämer R, Lehn J-M, Marquis-Rigault A. Proc Natl Acad Sci USA. 1993;90:5394–5398. doi: 10.1073/pnas.90.12.5394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Baxter P N W, Lehn J-M, Rissanen K. Chem. Commun. 1997. 1323–1324. [DOI] [PubMed] [Google Scholar]
- 28.Baxter P N W, Lehn J-M, Baum G, Fenske D. Chem Eur J. 2000;6:4510–4517. doi: 10.1002/1521-3765(20001215)6:24<4510::aid-chem4510>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- 29.Lehn J-M. In: Supramolecular Science: Where It Is and Where it Is Going. Ungaro R, Dalcanale E, editors. Dordrecht, the Netherlands: Kluwer; 1999. pp. 287–304. [Google Scholar]