

Abstract
Efficient production of ethanol for use as a renewable fuel requires organisms with a high level of ethanol tolerance. However, this trait is complex and increased tolerance therefore requires mutations in multiple genes and pathways. Here, we use experimental evolution for a system-level analysis of adaptation of Escherichia coli to high ethanol stress. As adaptation to extreme stress often results in complex mutational data sets consisting of both causal and noncausal passenger mutations, identifying the true adaptive mutations in these settings is not trivial. Therefore, we developed a novel method named IAMBEE (Identification of Adaptive Mutations in Bacterial Evolution Experiments). IAMBEE exploits the temporal profile of the acquisition of mutations during evolution in combination with the functional implications of each mutation at the protein level. These data are mapped to a genome-wide interaction network to search for adaptive mutations at the level of pathways. The 16 evolved populations in our data set together harbored 2,286 mutated genes with 4,470 unique mutations. Analysis by IAMBEE significantly reduced this number and resulted in identification of 90 mutated genes and 345 unique mutations that are most likely to be adaptive. Moreover, IAMBEE not only enabled the identification of previously known pathways involved in ethanol tolerance, but also identified novel systems such as the AcrAB-TolC efflux pump and fatty acids biosynthesis and even allowed to gain insight into the temporal profile of adaptation to ethanol stress. Furthermore, this method offers a solid framework for identifying the molecular underpinnings of other complex traits as well.
Keywords: experimental evolution, biological networks, ethanol tolerance, bacteria, hypermutation, gene prioritization
Introduction
Experimental evolution offers great potential to gain insights into the molecular mechanisms that contribute to the acquisition of complex traits (Kawecki etal. 2012; Wiser etal. 2013; Anderson etal. 2014). Previously, experimental evolution has been used not only to study the mechanisms underlying clinically (Palmer and Kishony 2013; Van den Bergh etal. 2016; Steenackers etal. 2016) or industrially relevant phenotypes (Winkler and Kao 2014), but also to improve key industrial traits for the production of advanced evolutionary engineered strains (Winkler and Kao 2014). Laboratory evolution experiments usually start from a single clone that is cultivated for prolonged periods of time in predefined conditions. During this period of time, natural selection favors mutations that confer a benefit in the chosen condition leading to improved phenotypes (Dragosits and Mattanovich 2013). Fitness is tracked over time and clones displaying increased fitness are genotyped to identify the underlying mutations (Schlötterer etal. 2015). Although some phenotypes are established by only one or just a few mutations, complex traits often involve mutations in multiple adaptive pathways, severely complicating identification of the causal adaptive mutations (Eyre-Walker 2010).
In this study, we used experimental evolution to study high ethanol tolerance in the bacterium Escherichia coli. Usually, microbial ethanol production capacity is severely limited by the toxic effect of ethanol itself. Therefore, higher ethanol tolerance and increased ethanol production are inherently linked (Huffer etal. 2012; Thammasittirong etal. 2013). Even though understanding and improving this trait is vital for strain engineering, it has been challenging to fully elucidate the underlying mechanisms. Previous studies have identified single genes (Gonzalez etal. 2003; Goodarzi etal. 2010) as well as epistatically interacting genes (Nicolaou etal. 2012) involved in higher ethanol tolerance. However, tolerance to ethanol is clearly a complex trait established by the interaction of multiple genes and pathways (Gonzalez etal. 2003; Goodarzi etal. 2010; Nicolaou etal. 2012; Swinnen etal. 2012; Lam etal. 2014) and a broad understanding of ethanol tolerance in E. coli is currently lacking. Moreover, in a previous study we found that hypermutation drives evolution under severe stress, such as ethanol stress, to enable adaptation of at least some individuals to avoid extinction (Swings etal. 2017). An increased mutation rate was found in all high ethanol tolerant populations and resulted in a higher ratio of passenger versus adaptive mutations, leading to an extremely complex mutational profile. This complexity is reflected in our data set of 16 evolved populations by a total of 2,286 mutated genes, containing 4,470 unique mutations. Consequently, the increased mutation rate combined with the complexity of the phenotype impedes the ability to statistically distinguish between true adaptive mutations and passenger mutations.
In most studies, this distinction between adaptive and passenger mutations is based on identifying mutations or mutated genes that recurrently emerge in independent evolutionary lines (Woods etal. 2006; Dees etal. 2012; Lawrence etal. 2013; Tamborero etal. 2013; Read and Massey 2014; Chen and Shapiro 2015; Hammerstrom etal. 2015). This narrow definition of parallelism assumes that only frequently mutated genes contribute to an adaptive phenotype. However, in populations that evolve independently, there is no guarantee that exactly the same mutation or even the same mutated gene is responsible for the observed adaptive phenotype. Affecting the same pathway through different and not necessarily frequently mutated genes might equally well induce the same adaptive phenotype (Tenaillon etal. 2012; Kvitek etal. 2013; Hong and Gresham 2014). Rather than identifying recurrent mutations, one can search for consistently mutated molecular pathways, assuming that adaptive mutations will hit the same adaptive pathways in independently evolved populations. Approaches that search for consistent changes in molecular pathways are typically network-based and have been applied successfully, mainly in the context of cancer genomics (Leiserson etal. 2015; Babur etal. 2015; Le Van etal. 2016; Pulido-Tamayo etal. 2016) but not yet for the mapping of genotypes to complex traits in clonal micro-organisms such as bacteria.
To cope with the specificities of clonal evolution experiments that aim to study complex traits, we developed a novel network-based method, IAMBEE. The method exploits the information gained from the trajectory of individual mutations along the evolution experiment together with network information to reduce the complexity of identifying adaptive pathways/genes. Our experimental set-up combined with this unique network-based approach resulted in the identification of several adaptive pathways that conferred ethanol resistance in E. coli. The role of the 30S ribosomal subunit pathway (Mars etal. 2015; Suzuki etal. 2015) as well as the osmotic stress response pathway (ompR/envZ) (Cai and Inouye 2002; Quinn etal. 2014) were confirmed. In addition, newly predicted molecular mechanisms such as the multidrug efflux pump AcrAB-tolC and the fatty acid biosynthesis pathway were experimentally validated. These results demonstrate the value of IAMBEE to analyze complex mutational data sets, including even data sets resulting from a hypermutator phenotype, to obtain a comprehensive overview of the pathways and its specific mutated components involved in the establishment of a trait.
Results
Ethanol Tolerant Populations Display a Hypermutator Phenotype
We set up an evolution experiment in which 16 independent E. coli populations were experimentally evolved under increasing ethanol concentrations (fig. 1). Changes in ethanol tolerance due to accumulation of beneficial mutations were tracked over time to obtain a fitness trajectory for each population (supplementary fig. 1, Supplementary Material online). These trajectories for all 16 populations show remarkable selective sweeps between 5% and 6% ethanol tolerance (further referred to as the initial selective sweep) and from 6% to 6.5% ethanol tolerance (further referred to as the second selective sweep). The populations were sampled right before and right after each increase in ethanol tolerance and were subjected to pooled sequencing. Primary analysis of the data showed that each of the ethanol tolerant populations evolved a hypermutation phenotype. In depth study of this observation led to the conclusion that near-lethal conditions require rapid adaptation of at least some individuals to avoid extinction of the population (Swings etal. 2017). Hypermutation considerably facilitates rapid adaptation by increasing the probability to acquire a beneficial mutation (Chao and Cox, 1983; Taddei etal. 1997; Woods etal. 2011; Swings etal. 2017). Although hypermutation enables adaptation, it also leads to complex mutational profiles with multiple mutations in random genes (Woods etal. 2006), further impeding identification of causal mutations. In our data set of 16 evolved populations a total of 2,286 mutated genes, containing 4,470 unique mutations were detected. To identify causal mutation despite this mutational complexity, we developed a new method that overcomes the limitations of only identifying recurrent mutations in evolved populations.
Fig. 1.

Set-up of the experiment, data acquisition and workflow of adaptive pathway identification. A first input consists of all mutations observed in independently evolved populations before and after a selective sweep. As a second input, an interaction network is used which is topology-weighted in order to account for hubs. This network is constructed using publicly available data sets. Subsequently, IAMBEE maps all mutated genes (input 1) to this topology-weighted interaction network (input 2) and calculates a relevance score for each mutation (green genes have higher and red genes lower relevance scores). The details on the calculation of these relevance scores are shown in figure 2. The relevance scores of the genes (nodes in the network) as well as the weights of the edges (interactions between genes), which are derived from the topology-weighting, are used to weight the paths (shown as black lines) between mutated genes from different populations found in the pathfinding step. Thick black lines represent paths that contain genes that have a high probability to be involved in the phenotype while thin black lines depict paths with low probability. Finally, a subnetwork inference step takes place which selects a subset of these paths in such a way that as many as possible paths connecting genes with large relevance scores are selected, but which is forced to select a sparse subnetwork as it minimizes the number of edges included. The result is that overlapping paths tend to be chosen and this leads to the selection of recurrently mutated connected subnetwork components. As a final output IAMBEE shows the inferred subnetwork containing highly prioritized network components that represent the identified adaptive pathways underlying the observed phenotype.
Exploiting Parallel Evolution to Identify Adaptive Pathways
To distinguish between adaptive and passenger mutations and to identify pathways underlying complex traits we have developed IAMBEE, which integrates prior information on gene interactions (i.e., an interaction network) with the specificities of the experimental design.
Key to the concept of IAMBEE is the use of multiple independently evolved populations to search for recurrently mutated molecular pathways. This search is driven by the interaction network and based on a decision theoretic subnetwork inference problem (De Maeyer etal. 2015; De Maeyer etal. 2016). However, given the high mutation rate and the relatively low number of indpedently evolved populations, additional information in the form of functional impact scores of the individual mutations is needed to drive the analysis. We hereby assume a priori that not all mutations are equally likely to be involved in the adaptive phenotype. Mutations that increase in frequency during a selective sweep and/or that have a functional impact on the protein in which they occur, are more likely to be involved in the phenotype.
Figure 1 gives a conceptual overview of IAMBEE. The input consists of called mutations from multiple, independently evolved populations and a genome-wide interaction network of the organism of interest. After topology-weighting the interaction network to downweight the effect of hubs on the final solution (see Materials and Methods), IAMBEE proceeds in three steps (fig. 1): 1) The relevance score is calculated for each mutated gene in each population. The relevance score consists of three components. The first component describes the change in frequency of the mutation during a selective sweep in the population. Mutations that increase in frequency during a selective sweep are more likely to be adaptive than mutations that decrease in frequency. However, not necessarily all adaptive mutations will increase in frequency during a sweep (e.g., potentiating mutations) and conversely passenger mutations that hitchhike with driver mutations will also increase in frequency (Lang etal. 2013). Therefore, the frequency change component is complemented with a second component: the functional impact score. The functional impact score reflects the effect of the mutation on the function of the protein. Mutations that are likely to alter a protein’s function are more likely to be adaptive. A last component of a mutation’s relevance score relates to the mutation rate of the population in which the mutation occurs: we assume that mutations that originate from populations with a significantly higher mutation rate than the other populations should contribute relatively less to the final solution as they contain a larger number of passenger mutations (more noise) and a mutation of such a line should thus exhibit a stronger signal in order to be selected. A detailed overview of the calculation of the relevance scores by IAMBEE is shown in figure 2 and is described in the Materials and Methods. 2) The pathfinding step embodies the search for paths, which are defined as consecutive sets of edges connecting mutated genes from different populations, on the topology-weighted interaction network. These paths are weighted based on the relevance scores of the involved mutations and the weights of the edges involved in the path. The weight of a path reflects the degree of belief that the path is involved in the adaptive phenotype. 3) Subnetwork inference (optimization strategy) is subsequently used to select a subset of the paths found during the pathfinding step. This subset is selected in such a way that a maximum number of mutated genes with high relevance scores are included but a minimal number of edges is selected. This means that overlapping paths are more easily selected as they share edges, which reflects the search for molecular pathways that are consistently mutated throughout independently evolved populations. The resulting subset of paths makes up a subnetwork that consists of multiple connected components which are parts of molecular pathways. For a more detailed explanation of these steps, we refer to the Materials and Methods section.
Fig. 2.

Calculation of the relevance scores for each mutation by IAMBEE. In the data acquisition step, a selective sweep of interest is chosen from an evolution experiment involving multiple parallel evolved populations. Samples taken at time points just before (blue arrows) and just after (orange arrows) this selective sweep are sequenced and mutations are called. For every mutation, a functional impact score and frequency change in the population are determined by the IAMBEE software. The frequency change is derived from the degree to which the mutation changes in frequency before (blue) and after (orange) the selective sweep. Genes with mutations that rise in frequency have higher frequency increase scores (green square) while a low frequency increase score is assigned to genes with mutations that decrease in frequency in the population (red squares). Next, a functional impact score is assigned to each mutation by using SIFT4G (Vaser etal. 2016). Genes with mutations having a high functional impact score are depicted with green triangles and vice versa. In addition, populations with a mutation rate that is significantly higher than the mutation rates of the other populations are detected. The relevance of mutated genes in populations with a significantly higher mutation rate are corrected (red star) to avoid overrepresentation of mutations from these populations. Finally, combining a gene’s frequency score, functional impact score and the correction for mutation rates allows calculating a relevance score for every mutated gene in every population. Mutated genes with a high relevance score (green circles) are more likely to harbor mutations that increase in frequency during the selective sweep, have high functional impact scores and are not involved in a population with significantly higher mutation rate than the rest of the populations.
Validation of IAMBEE Using Synthetic Data
To validate and characterize IAMBEE, we generated 100 synthetic data sets, each with randomly selected adaptive and passenger mutations (supplementary Material, Supplementary Material online). Running IAMBEE on these data sets resulted in one subnetwork containing prioritized mutated genes per combination of data set and parameter setting. Every synthetic data set was run with 50 different parameter settings ranging from settings that result in small subnetworks to settings resulting in larger subnetworks. As in this synthetic setting the true adaptive mutations (true positives) are known, we used PPV (the ratio of true positives to the total number of mutations prioritized by IAMBEE) and sensitivity (the number of true positives to the total number of true positives in the data set) as performance criteria. Results showed that, as expected for a method that makes relevant nonrandom predictions, small subnetworks have a high PPV at the expense of a lower sensitivity and subnetworks (of any size) rarely have both low sensitivity and PPV (supplementary fig. 2, Supplementary Material online). Exploring small solutions allows identifying a restricted number of candidates suitable for experimental validation, whereas exploring larger solutions provides a more complete pathway level view of the adaptive phenotype, but risks identifying false positives. The reliability of the predictions as estimated from the parameter settings at which they were detected is also included in the output of IAMBEE where more opaque edges represent edges which are involved in both small and large solutions (high PPV) whereas less opaque edges are only involved in small solutions (lower PPV) (figs. 3 and 4).
Fig. 3.

Subnetwork consisting of multiple connected components inferred by analyzing the mutation data observed in all 16 populations during the initial selective sweep. Nodes represent genes and edges represent interactions between the genes. Around each node an inner and an outer circle is indicated, which are both divided in 16 equal parts, representing mutations in population HT1 to population HT16 (see legend). A colored part of the inner circle represents a mutation which increases in frequency during an initial selective sweep for that gene in the corresponding population while a colored part of the outer circle represents a mutation which increases in frequency during the second selective sweep in that same population. An overview of all possible mutation patterns can be found at the top of the figure (note that as the outcome of the initial selective sweep is compared with the ancestral strain, it is impossible for a mutation to decrease in frequency during an initial selective sweep). The color of the edges represents their type (see legend). The opacity of the edges represents the maximum edge cost for which those edges were selected (a measure for the degree of belief that the interaction is implicated in the adaptive phenotype). Opaque edges are selected in cases with high edge costs (high degree of belief) while edges with low opacity are only selected in cases with low edge costs (lower degree of belief). The online version of the resulting subnetwork is provided with the article.
Network-Based Analysis Unravels Adaptive Pathways for High Ethanol Tolerance
We pooled the mutation data observed in the 16 different lines for, respectively, the first and second selective sweep and applied IAMBEE to the pooled data of each sweep to unveil pathways that drive increases in ethanol tolerance during each of the sweeps. We identified connected components that were common to both sweeps and components that were unique to each of the sweeps. Identified connected network components representative of adaptive pathways or at least parts of adaptive pathways are shown in figures 3 and 4, respectively, the initial and second selective sweep. During the initial selective sweep 32 connected network components, involving 108 genes harboring 228 mutations, were prioritized by IAMBEE out of a total of 1,646 mutated genes harboring 2,511 mutations in 16 populations. Likewise, in the second selective sweep 22 connected components, involving 90 genes harboring 345 mutations, were prioritized by IAMBEE out of a total 2,286 mutated genes harboring 4,470 mutations in the same 16 populations. 15 of these connected components were partly or entirely selected in both sweeps (supplementary fig. 3, Supplementary Material online). The fact that components were detected that are common to both the initial and second sweep but also specific for each of the sweeps demonstrates fundamental differences between initial adaptation to high ethanol stress and adaptation to prolonged exposure to increasing ethanol concentrations. Below, we describe important identified connected network components and their putative roles in ethanol tolerance. A more detailed overview and description of all identified ethanol tolerance related pathways is given in the supplementary Results, Supplementary Material online.
Fig. 4.

Subnetwork consisting of multiple connected components inferred by analyzing the mutation data observed in all 16 populations during the second selective sweep. Nodes represent genes and edges represent interactions between the genes. Colors of the nodes and the edges are identical to figure 3. The online version of the resulting subnetwork is provided with the article.
The Fatty Acid Biosynthesis Pathway Is Selected Exclusively for Initial Adaptation
An important network component that was exclusively identified for the initial selective sweep corresponds to the fatty acid biosynthesis pathway, encoded by the fab genes. The prioritization of this pathway in the initial, but not in the second selective sweep, means that most mutations in the fab genes were already fixed in the latter step, explaining why they were not selected as being adaptive in the second selective sweep. This early fixation of fab mutations indicates that changing the fatty acid composition in the membrane is an initial adaptation strategy, but does not suffice to confer resistance to higher concentrations of ethanol. Different parallel populations accumulated mutations in both fabA and fabB. The amount of unsaturated fatty acids eventually present in the membrane depends on the competition for intermediates at the FabA branch point in the pathway (Heath and Rock 1996; Loffeld and Keweloh 1996; Zhang and Rock 2008). Mutations in either the fabA gene itself or in genes downstream (e.g., fabB or fabG) can permanently change the ratio of saturated versus unsaturated fatty acids thereby changing the fluidity of the membrane. Changes in membrane composition, such as the ratio of saturated versus unsaturated fatty acids, have previously been reported to affect ethanol tolerance (Buttke and Ingram 1980; Luo etal. 2009) although mutations in the fab genes have not been associated with ethanol resistance in the past.
To validate whether mutations in the fab genes affect membrane composition, we compared fatty acid content of selected strains harboring these mutations with that of the wild type (fig. 5). When exposed to 5% ethanol, the percentage of unsaturated fatty acids in wild-type cells increases 2-fold (fig. 5a). This observation corroborates previous results showing ethanol-induced inhibition of saturated fatty acid synthesis (Buttke and Ingram 1980). The percentage unsaturated fatty acids in the absence of ethanol in the two mutant populations harboring a fabA (HT15) and a fabB (HT12) mutation equals that of the wild-type ancestor. However, in the mutant populations subjected to 5% ethanol the percentage unsaturated fatty acids also increases, but not to the same extent as for the wild type (fig. 5b). This difference suggests a direct effect of the identified fab mutations on the ethanol-induced shift in saturated versus unsaturated fatty acids ratio. Increased proportions of unsaturated fatty acids fluidize the membrane. Mutations in the fab genes possibly counteract this shift to become more tolerant against ethanol by maintaining structural rigidity of the membrane.
Fig. 5.

Effect of fab mutations on the percentage of unsaturated fatty acids in the plasma membrane. (a) The membrane composition of E. coli changes dramatically when grown in the presence of 5% ethanol. Especially the proportion of palmitoleic acid (16_1 w7c) increases considerably, whereas the proportion of palmitic acid (16_0) decreases. Additionally, we can see that the larger fatty acids with chain lengths higher than 18 disappear. A switch to shorter chain length is also part of the response to ethanol stress. The first number in the name of the fatty acids denotes the length (or number of C-atoms) in the chain. The second number denotes the number of double bonds: a zero means a saturated fatty acid and a 1 or 2 means a mono- or di-unsaturated fatty acid. The “w” followed by a number shows the position of the double bond, whereas the “c” means cis instead of trans. (b) The total percentage of unsaturated fatty acids in the wild type doubles upon exposure to ethanol. In the two high ethanol tolerant populations, in which we identified mutations in fabA and fabB the percentage of unsaturated fatty acids still increases, but less pronounced compared with the wild type. These results demonstrate that rewiring of unsaturated fatty acid biosynthesis through involved genes, such as fabA and fabB can confer high tolerance to ethanol.
Pathways Involved in Both Initial and Consecutive Adaptation
Several network components were prioritized by IAMBEE in, respectively, the first and second sweep that corresponded to the same pathway and hence most likely to confer ethanol tolerance. When a putative adaptive pathway is selected in both sweeps, typically few mutated genes were detected in the initial sweep, whereas the remainder of the mutated genes was identified in the second sweep (figs. 3 and4).
Multidrug Efflux Pumps
One network component of particular interest is linked to multidrug efflux complexes. The genes acrA, acrB, and acrD, encoding the AcrAB-TolC and the AcrAD-TolC multidrug efflux pump were found to be frequently mutated during the initial selective sweep. Multidrug efflux pumps usually consist of three parts: an inner-membrane transporter, such as AcrB, a membrane fusion protein such as AcrA and an outer-membrane transport channel such as TolC. AcrD, like AcrB, binds to AcrA and forms a complex with TolC to constitute a multidrug efflux pump. Despite their association with tolerance to organic solvents (White etal. 1997; Atsumi etal. 2010; Bernardi etal. 2016), efflux pumps have to our knowledge not yet been specifically linked to ethanol tolerance. To validate the role of the efflux pump in ethanol tolerance, we constructed a deletion mutation in acrB, one of the subunits of the AcrAB-TolC multidrug efflux pump that accumulated most mutations. Indeed, the ΔacrB deletion mutant has an increased growth rate compared with the wild-type ancestor under 5.5% ethanol stress (fig. 6). Moreover, the relative difference in growth compared with the wild type increases with the ethanol percentage in the medium (supplementary fig. 4, Supplementary Material online). Even though drug resistance mediated by efflux pumps is usually rendered by active efflux of the compound, our results suggest that ethanol tolerance through mutations in acrAB works the other way around. Ethanol might be leaking into the cell through the efflux pump, consequently causing higher ethanol tolerance in an AcrAB deletion mutant. Previously, AcrAB-mediated organic solvent tolerance has been reported to depend on the polarity of the solvent, where deleting acrA causes increased tolerance to the slightly polar isobutanol, suggesting a similar tolerance mechanism for ethanol which is even more polar (White etal. 1997; Atsumi etal. 2010).
Fig. 6.
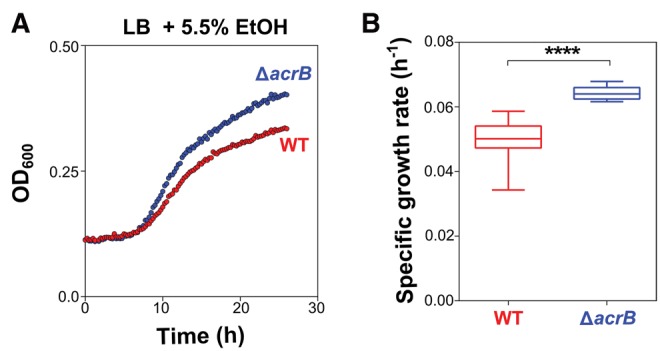
Effect of acrB deletion on growth under high ethanol stress. (a) The graph shows the growth of both the wild-type strain and the acrB deletion mutant under 5.5% ethanol stress. The ΔacrB mutant grows faster and reaches a higher carrying capacity (higher final optical density) under these conditions. (b) The growth curves were fitted using the Gompertz equation (see Materials and Methods) and specific growth rate was extracted. The acrB deletion mutant has a significantly increased growth rate compared with the wild type. Both growth rates were statistically compared using an unpaired two-sided Student’s t test (n = 6, box = median, whiskers = min to max, ****P < 0.0001). These results confirm a selective advantage of the ΔacrB mutant compared with the wild type.
Whereas acrA, acrB, and acrD are mutated in the initial selective sweep, during the second selective sweep additional mutations occurred in mdtA and mdtF. MdtA, like AcrA, is a membrane fusion protein in the MdtABC-TolC multidrug efflux pump (Borges-Walmsley etal. 2002; Nagakubo etal. 2002). MdtF is the counterpart of AcrB and acts as a transporter in the MdtEF-TolC multidrug efflux pump (Nishino and Yamaguchi 2002). The mutations in mdtA and mdtF that rise in frequency during a second selective sweep frequently occurred in populations that already harbored a mutation in the AcrAB-TolC efflux pump originating from the earlier selective sweep (four out of seven). Subsequent mutations in paralogous AcrAB-TolC and MdtABC-TolC multidrug efflux systems thus are likely to enable gradual adaption and further improve fitness under high ethanol concentrations. In addition, even though TolC is the common outer membrane channel for all above mentioned efflux pumps no mutations occurred in the tolC gene. However, TolC is essential for cell functioning, suggesting that mutations in tolC most likely have a high fitness cost compared with the advantage they can provide under ethanol stress.
Other network components were prioritized in both sweeps and have previously been associated with ethanol tolerance. This confirms the ability of IAMBEE to identify true adaptive pathways. Below, we list the most relevant of these pathways. For a more in-depth description we refer to the supplementary Results, Supplementary Material online.
DNA Repair
One highly prioritized network component includes several genes involved in DNA repair mechanisms, such as the methyl-directed mismatch repair pathway (MMR, mutS, mutL, and mutH), the nucleotide excision repair pathway (NER, uvrA, uvrB, and uvrC) and the DNA helicase encoded by uvrD which is involved in both the MMR and NER pathways. Mutations in the aforementioned genes explain the observed higher mutation rates that were observed in the evolution experiment. Prioritization of this network component substantiates our previous work where we demonstrated the crucial role of mutations in the mismatch repair pathway for adaptation to ethanol under high stress conditions (Swings etal. 2017).
Transcription and Translation
Other highly prioritized network components are linked to transcription and translation. We found a frequently mutated connected network component containing several well-known genes involved in transcription termination and antitermination (rho, nusG, and nusA). Additionally, we found a frequently mutated subnetwork containing translation-linked genes encoding for 30 S ribosomal subunit proteins (rpsL, rpsD, rpsB, and rpsH) and the protein chain initiation factor infB. The Selection of genes involved in these pathways further corroborates earlier results that demonstrate early ethanol-induced transcription termination and ethanol-induced translational misreading during protein synthesis. Mutations in genes involved in transcription or translation might confer higher ethanol tolerance through compensation for these toxic effects (Burns and Richardson 1995; Pasman and von Hippel 2000; Haft etal. 2014).
Remarkably, mutations prioritized by IAMBEE in, respectively, (anti) termination factors, such as rho, nusA, and nusG and ribosomal genes, such as rpsB, rpsD, rpsL, and rpsH tend to cooccur. These are consistently observed in the same populations and follow the same trajectories, suggesting that they are dependent on each other in driving the observed tolerance towards ethanol (figs. 3 and4). This observation suggests an epistatic interaction that corresponds to previous findings of Freddolino etal. who showed that mutations in rpsL increase ethanol tolerance in a mutant rho background (Freddolino etal. 2012) and Haft etal. (2014) who demonstrated the role of epistatic interactions between a mutation in rho and rpsQ in ethanol tolerance.
Osmotic Stress Response
The envZ-ompR two-component system that regulates outer membrane porin genes in response to changes in extracellular osmotic pressure was also prioritized (Cai and Inouye 2002; Quinn etal. 2014). Ethanol increases membrane fluidity, thereby causing leakage and osmotic stress as shown by Goodarzi etal. (2010). Mutations in envZ and ompR suggest adaptations to the increased osmotic stress under high ethanol conditions.
Amino Acid Biosynthesis
Finally, several of the prioritized network components and genes are involved in amino acid biosynthesis, such as isoleucine and valine biosynthesis (ilvD, ilvC, ilvB, ilvE, ilvI, and tdcB), alanine and phenylalanine biosynthesis (ilvE and alaA), methionine biosynthesis (metE, metB, and metH), biosynthesis of tetrahydrofolic acid, which is a precursor in the metabolism of amino acids (purH), a gene involved in arginine biosynthesis (argH), and a gene involved in threonine and glycine biosynthesis (itaE) (Keseler etal. 2013). A role of amino acid biosynthesis and transport in tolerance towards ethanol stress has previously been suggested in yeast, because of impaired delivery of amino acids into the cell as a result of membrane functions being disrupted by ethanol (Yoshikawa etal. 2009; Stanley etal. 2010).
Pathways Exclusively Involved in the Second Adaptation Step
One smaller network component that was exclusively, but highly, prioritized in the second selective sweep consists of two genes fadB and gabT. FadB plays a role in fatty acid oxidation and is regulated by FadR (DiRusso etal. 1992). Interestingly, a fadR deletion mutant was also recently found to increase organic solvent tolerance (Oh etal. 2012) pointing to a similar process.
Several additional identified network components correspond to pathways that have never been linked to ethanol tolerance before, but that might influence this trait are discussed in the supplementary Results, Supplementary Material online. In conclusion, we state that IAMBEE is able to detect previously known as well as new adaptive pathways. The identified adaptive pathways in ethanol tolerance might serve as a basis for future strain improvement efforts.
Indications for Epistasis at the Pathway Level
Remarkably, adaptive mutations in the fatty acid pathways tend to occur in a mutual exclusive way: eight populations have mutations in fadB-gabT, nine populations have mutations in the FabA-B system, two populations (HT13 and HT14) have mutations in both pathways and only one population (HT3) has no mutations in either of the respective pathways. Strikingly, this would imply negative epistasis at the pathway level, i.e., a mutation in either pathway increases ethanol resistance but mutations in both mechanisms do not lead to a greater increase in resistance. The incomplete pattern of mutual exclusivity in HT14 can be explained by the fact that both the mutation in fabA (present in 50% of population) and fadB (present in 12% of population) are not fixated in the population. Therefore, it is possible that these mutations exist in different subpopulations. As was the case in the cooccurrence of mutations in the AcrAB-tolC/fab pathway, the incomplete pattern of mutual exclusivity in HT13 could be explained by the fact that the fabA mutation is situated towards the end of the fabA protein (seven amino acids near the end), which makes it likely that this mutation is not functionally relevant. Indeed, by determining the membrane composition we confirmed that the percentage of unsaturated fatty acids in absence and in response to 5% ethanol did not differ from the wild-type strain, suggesting that this particular mutation does not contribute to higher ethanol stress by changing the membrane composition (supplementary fig. 5, Supplementary Material online).
Additionally, mutations in the AcrAB-TolC efflux pump tend to cooccur with mutations in the previously mentioned fab pathway. From the ten populations which have a mutation in acrAB-tolC and the nine populations which have a mutation in the fab pathway, eight populations overlap. According to the mutational trajectories, mutations in both pathways arise during the same selective sweep (5–6%) in seven populations. Only in one population (HT10), the mutation in acrAB-tolC was obtained late during the second selective sweep, following an earlier mutation in the fab pathway. Only population HT13 had a fabA mutation but not a mutation in acrAB-tolC. Again, this specific mutation in fabA is located at seven amino acid residues near the end of the FabA protein (supplementary fig. 5, Supplementary Material online).
Comparison with a per Gene Mutation Frequency Approach
To show the value of the network-based approach of IAMBEE, we compared our results with those obtained by a frequency-based approach which ranks genes based on the number of populations in which they were mutated (fig. 7, supplementary file 1, Supplementary Material online). Although IAMBEE does not explicitly search for genes that are recurrently mutated across populations, results show that IAMBEE is able to also prioritize most of the frequently mutated genes which are associated with ethanol resistance (e.g., rpsL and envZ). Exclusively mapping frequently mutated genes (136 and 153 for, respectively, the first and the second selective sweep) to the genome-wide interaction network showed (supplementary figs. 6 and 7, Supplementary Material online) that adaptive pathways which were identified by IAMBEE, such as the fatty acid biosynthesis pathway, fadB-gabT and AcrAB-TolC, are largely or completely missed (supplementary figs. 6 and 7, Supplementary Material online). This can be explained by the fact that these pathways are composed of genes that are not necessarily frequently mutated (e.g., some connecting genes were only found mutated in one or two populations). By exploiting all mutated genes over the network and using information from mutational trajectories and functional impact scores IAMBEE can, in contrast to the frequency-based approach, extract adaptive pathways consisting of less frequently mutated but highly connected genes (supplementary table 1, Supplementary Material online). However, as IAMBEE is network-based it is possible that some genes are missed because they are not present in the network (e.g., marC and tqsA [Atsumi etal. 2010; Minty etal. 2011]). Nevertheless, this does not outweigh the benefit of IAMBEE to retrieve more complete pathways and to enable reasoning about the temporal aspects of mutation acquisition.
Fig. 7.
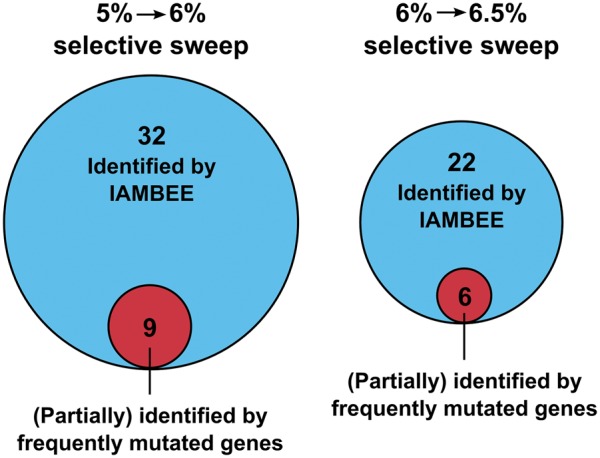
Comparison of the output generated by IAMBEE with the per gene mutation frequency approach. By performing a pooled analysis of the mutation data observed in the 16 populations during the first and second selective sweep IAMBEE identified, respectively, 32 and 22 connected network components. The alternative method using exclusively the number of mutations per gene allowed (partial) identification of 9 and 6 connected network components in the first and second selective sweep, respectively. This result clearly demonstrates that only a fraction of the involved adaptive pathways are identified by using the approach that only takes into account frequently mutated genes. By combining mutation frequency data and functional impact scores, IAMBEE enables identification of the network components underlying an adaptive phenotype. More details on the specific connected network components prioritized by both approaches are given in supplementary table 1 and figures 6 and 7, Supplementary Material online.
Discussion
Evolution experiments have been successfully used to identify the role of specific genes in an adaptive phenotype (Suzuki etal. 2014; Van den Bergh etal. 2016). However, genetic data derived from parallel evolution experiments is usually interpreted by looking at the mutation frequencies of the individual genes (Woods etal. 2006). Especially when dealing with complex traits and hypermutation, these studies do not necessarily yield insight into the complex interactions of the genes that contribute to the adaptive phenotype. Key to unraveling the genetic mechanisms underlying high ethanol tolerance is the development of a dedicated analysis method. IAMBEE is unique in prioritizing adaptive mutations by combining information on each individual mutation inferred from functional impact scores and relative frequency increases during a selective sweep, with information on the interactions between genes (a genome-wide interaction network). Using our newly developed method, we were able to prioritize multiple pathways that were recurrently mutated in different independent high ethanol tolerant populations. Among the highly prioritized pathways, those related to translation, antitermination and amino acid metabolism were previously associated with high ethanol tolerance. Recovering these well-known pathways confirms the ability of IAMBEE to identify true adaptive pathways.
On top of those well-known systems, we identified a yet undescribed role for multidrug efflux pumps in the continuous adaptation to high ethanol stress and the role of fatty acid metabolism in allowing the cell to cope with the toxic effects of ethanol on the membrane (Dombek and Ingram 1984). Related to the latter mechanism, binding and penetration of ethanol into the lipid bilayer increases membrane fluidity (Ramos et al. 2002), thereby inducing secondary effects, such as osmotic stress (Ingram and Buttke 1984; Ingram 1990; Huffer etal. 2011). Response to osmotic stress has previously been shown to induce alterations of the membrane composition including cis-to-trans isomerization of unsaturated fatty-acids as a short-term response (Loffeld and Keweloh 1996) and alteration of the ratio of saturated versus unsaturated fatty acids as a long-term response (Buttke and Ingram 1980; Luo etal. 2009). Both changes to the membrane composition can result in denser packing of the fatty acids thereby increasing the rigidity of the membrane which enables the cell to withstand the toxic effect of ethanol (Ramos etal. 2002; Nicolaou etal. 2012). Although there has been confusion about the effect of unsaturated fatty acids on ethanol tolerance (Buttke and Ingram 1980; Luo etal. 2009; Vanegas etal. 2012), we provide evidence that tempering the shift to higher ratios of unsaturated fatty acids confers higher resistance to ethanol (fig. 5). We could indeed show that mutations in representative genes of the fab pathway resulted in increased ethanol tolerance by affecting the ratio of saturated versus unsaturated fatty acids.
As IAMBEE is designed to be used in combination with a dedicated experimental set-up, which includes sequencing of the evolving populations before and after a selective sweep, it is possible to gain insight in the temporal profile of adaptation. Using this unique feature of the method we found that mutations in the fatty acid biosynthesis pathway occur early in the evolutionary trajectory of ethanol resistance in E. coli whereas mutations in other pathways have less strict temporal constraints. In this context, we also found that mutations in the pathway for fatty acid biosynthesis (fabA, fabG, and fabB) and a pathway involved in fatty acid oxidation (gabT, fadB) were mutually exclusive while having high coverage (15 out of the 16 populations had a mutation in either pathway). This implies that fatty acids play a pivotal role in ethanol tolerance, but that either a mutation in one of the two pathways does not lead to a significant increase in fitness if a mutation in the other pathway is already present (negative epistasis) or that having a mutation simultaneously in both pathways is lethal (synthetic lethality as an extreme form of negative epistasis). In contrast, mutations in the fatty acid biosynthesis pathway (fabA, fabG, and fadB) and the AcrAB-tolC efflux pump (acrA and acrB) significantly co-occur, suggesting positive epistasis between these pathways.
When compared with a naive approach which is based on recurrence of mutations across experiments at the level of individual genes, it is obvious that IAMBEE offers not only the advantage of being able to identify adaptive mutations which are not frequently mutated but also to interpret adaptive mutations and epistasis at the level of pathways. As such, IAMBEE is very useful and meets the need for adequate tools to analyze highly complex mutational data sets.
Conclusions
Experimental evolution can readily yield insight in complex traits assuming low complexity of the resulting mutational profiles. However, in the case of complex traits and especially when hypermutation arises, evolution experiments often lead to complex mutational profiles with high rates of passenger mutations that are difficult to interpret. Traditionally, adaptive genes are identified by counting the number of mutations per gene across independently evolved populations. Although this approach is valid in some data sets, in complex mutational profiles it neglects less frequently mutated genes, resulting in the inability to generate a broad understanding of the adaptive phenotype. Therefore, we developed IAMBEE, a method that exploits the interaction network, combined with information from mutational trajectories and functional impact scores, to identify adaptive genes and pathways from complex mutational data sets. By applying IAMBEE to an evolution experiment consisting of 16 independently evolved E. coli populations subjected to increasing ethanol concentrations, pathways that were previously linked to high ethanol resistance as well as novel pathways, which were experimentally validated, could be identified. In conclusion, IAMBEE is a powerful tool that successfully allows to generate a broader understanding of (complex) traits that could not be fully elucidated so far.
Materials and Methods
Data Acquisition
Experimental Evolution
We used a set of 16 parallel evolved E. coli populations. Of these highly ethanol-tolerant populations, seven originated from our previously conducted evolution experiment (Swings etal. 2017) and we initiated a new experiment using the same workflow to add an additional nine populations to the final data set. All populations acquired a hypermutator phenotype, which is necessary to enable evolution under near-lethal stress conditions (Swings etal. 2017). For ease of reading, we renamed al populations HT1-16 (High Tolerance). In brief, all parallel populations originated from the same ancestral strains SX4 and SX25. We used lysogeny broth (LB) supplemented with 5% (v/v) ethanol as primary stress conditions to initiate the evolution experiment. We maintained growth in exponential phase in each population. As parameters to monitor evolution, we used both the optical density (A595nm) and the time to reach a specific optical density, typical for exponential growth. When a population reached exponential phase (A595nm around 0.2) within 24 h, we transferred it to fresh LB medium that was supplemented with an additional 0.5% (v/v) ethanol. If the population needed >24 h but <14 days to reach exponential growth, we transferred it to fresh medium with the same percentage of ethanol. In case the strain did not grow within 14 days, we revived the sample from the previous time point from the −80 °C stock and used it to restart the evolving population in fresh medium with a 0.5% reduced ethanol concentration. Upon each transfer to fresh medium, a sample was stored in a −80 °C glycerol stock for further analysis. Based on the adaptation trajectories of these populations (supplementary fig. 1, Supplementary Material online) we decided to analyze both the 16 selective sweeps from 5% to 6% ethanol together and the 16 selective sweeps from 6% to 6.5% ethanol together in order to gain insight into the temporal aspects of the adaptive pathways.
Mutation Calling
High-quality genomic DNA from overnight cultures of the ancestor and intermediate points of evolved populations was isolated (DNeasy Blood and Tissue kit, Qiagen). 100 bp paired-end sequencing libraries with an average insert size of 200 bp were prepared at GeneCore (EMBL, Heidelberg) and used for massive parallel sequencing with the Illumina HiSeq2000. We used CLC Genomics Workbench version 7.6 (https://www.qiagenbioinformatics.com) (RRID: SCR_011853) for analysis of the sequences. Following quality assessment of the raw data, reads were trimmed using quality scores of the individual bases (quality limit = 0.01; maximum number of ambiguous bases = 2). Reads shorter than 15 bases were discarded from the set. We used the CLC Assembly Cell 4.0 algorithm to map the trimmed reads to the E. coli MG1655 reference genome (NC_000913.1) yielding a minimal coverage of 150× (mismatch cost = 2; insertion cost = 3; deletion cost = 3; length fraction = 0.8; similarity fraction = 0.8). Mutations were called using the CLC Low Frequency Variant Detector (http://resources.qiagenbioinformatics.com/manuals/clccancerresearchworkbench/200/index.php?manual=Low_Frequency_Variant_Detection.html; last accessed August 3, 2017; required significance = 1%; minimum coverage = 10; minimum frequency = 10%). Finally, the mutations in the SX4 compared with the MG1655 reference genome were discarded. In addition, CLC largely fails in the detection of larger rearrangements, such as large insertions, deletions, inversions or duplications. Therefore, we performed an additional analysis on the sequence data using a dedicated tool, Pindel (Ye etal. 2009), for detection of larger genomic rearrangements. We ran Pindel using default settings.
Mapping of Mutations to Genes
Mutations in coding regions of genes were mapped to those genes, whereas mutations in intergenic regions were only mapped to a gene if they resided in a promoter. Promoter element locations were taken from RegulonDB (RRID: SCR_003499) (Gama-Castro etal. 2016) and PromBase (Kanhere and Bansal 2005). Supplementary table 3, Supplementary Material online, shows the number of mutations retained and used in both analyses.
Functional Impact Scores and Frequency of Mutations
To calculate functional impact scores for each mutation we used SIFT scores which were calculated using the SIFT4G annotator version 2.2 with the E. coli (GCA_000005845.1.21) database (Vaser etal. 2016). Note that while SIFT scores were used in this paper, any functional impact score measure can be inserted in IAMBEE.
As IAMBEE tries to identify the causal molecular pathways which lie at the basis of a selective sweep, mutations which decrease in frequency during a selective sweep are not taken into account. Therefore, we have to determine when a mutation “decreases” in frequency. As the precision of frequency calling of mutations is finite, the naive way of viewing all mutations with a negative frequency increase as “decreasing” is not valid. This would discard too many mutations which remained stable or, most of the time, were fixed previously in the population. As these mutations could be potentiating mutations, we do not want to discard them. Because of this, and the specifications of the CLC variant caller (a required significance of 1%), we viewed all mutations with a decrease in frequency of at least 2% as decreasing. All mutations in both selective sweeps, together with their SIFT scores and their increase in frequency during the selective sweep are given in supplementary file 1, Supplementary Material online.
Genome-Wide Interaction Network
We used a directed genome-wide interaction network of E. coli K-12 MG1655 compiled from (de)methylation, (de)phosphorylation and metabolic interactions from KEGG version 80 (RRID: SCR_012773) (Kanehisa etal. 2014; Kanehisa etal. 2016), protein–DNA, sigma factor binding and sRNA–DNA interactions from regulonDB version 9.2 (Gama-Castro etal. 2016) and protein–protein interactions from STRING version 10 (RRID: SCR_005223) (Szklarczyk etal. 2015). To reduce the number of false positive interactions in the interaction network, only direct (physical) associations with a score of at least 0.8 were retained from STRING. Interactions involving the primary sigma factor RpoD were removed as RpoD regulates over half of the genes in the interaction network. Furthermore, self-edges were deleted. The final genome-wide interaction network contains 2,678 nodes (genes) and 14,702 edges (interactions between genes/sRNAs), representing about 63% of E. coli K-12 genes. This interaction network is supplied together with IAMBEE at http://bioinformatics.intec.ugent.be/IAMBEE.
Construction of the Probabilistic Genome-Wide Interaction Network
IAMBEE is guided by a directed genome-wide interaction network with the nodes representing genes and the edges representing interactions between these genes. A topology-based weighting of the genome-wide interaction network was performed to reduce the effect of hubs in the subsequent analysis steps: a power law distribution (Barabási and Albert 1999) was estimated based on the out-degrees of the nodes in the interaction network. Next, a sigmoidal function was constructed using as inflection point the out-degree that corresponded to the 90th percentile. This leads to following topology-based weighting of each edge between node i and node j (De Maeyer etal. 2015):
This sigmoidal function is utilized to mainly down weight interactions originating from large hubs while avoiding to penalize interactions involving nodes with low out-degrees.
IAMBEE
Calculation of Relevance Scores
Not all mutations are equally likely to be involved in the adaptive phenotype. Therefore, a relevance score was assigned to each mutation based on its estimated functional impact on the coding/promoter sequence and based on its relative increase in frequency in the population during a fitness increase. The functional impact score reflects how likely a mutation causes a functional change in the resulting protein(s). Here it is based on the degree of conservation of amino acid residues in sequence alignments from closely related sequences using the SIFT algorithm (RRID: SCR_012813) (Ng and Henikoff 2001; Kumar etal. 2009; Vaser etal. 2016). To derive frequency increases, for each population the adaptive trajectory (e.g., fitness profile) is used to delineate selective sweeps (sudden jumps in fitness or an increase in adaptation towards the experimental conditions). The frequency increase of a mutation is equal to the difference of its frequency in the population just after and just before the sweep. To assess the relative importance of a mutation’s frequency increase or functional impact score, we first estimate both the distribution functions, based on the frequency increase/impact score of all mutations from all evolved populations. As neither the functional impact score distribution, nor the frequency increase distribution is expected to follow any known mathematical distribution, the distributions are estimated using a nonparametric cumulative distribution function (MathWorks, Inc. 2017). As synonymous mutations would skew the distribution of the functional impact scores towards low functional impact scores, which could result in assigning relatively high relevance scores to mutations with poor functional impact scores, synonymous mutations are removed from the data when estimating the functional impact distribution function. Note that because some synonymous mutations do have relevant functional impact scores they are not discarded but only ignored when estimating the functional impact distribution. Although the functional impact distribution function is estimated using all mutation data from all evolved populations, the frequency increase distribution is estimated on a per-population basis as the population dynamics can differ between populations. This means that one functional impact distribution is estimated, whereas the number of frequency increase distributions is equal to the number of parallel evolved populations are estimated.
Based on these distributions a relevance score is calculated for each mutated gene in each population as follows:
with the value of the cumulative distribution function of the functional impact scores for the mutation in gene S in population n with the most deleterious functional impact score (note the 1- as we used SIFT scores and a low SIFT score corresponds to a high functional impact), the value of the cumulative distribution function in population n of the frequency increases for the mutation in gene in that population with the highest frequency increase. is a value between 0 (gene S is unlikely to be relevant towards adaptation in population n) and 1 (gene S is very likely to be relevant towards adaptation in population n). Genes without mutations are assigned the mean functional impact score and frequency increase when calculating their relevance.
Furthermore, if the data set contains populations with a mutation rate which is significantly higher than the mutation rates of the other populations, the search for paths in the pathfinding step (see following paragraph) would be skewed towards this population (supplementary table 1, Supplementary Material online). To reduce the impact of the population’s mutation rate on the pathfinding without completely discarding these populations, a correction factor for each population is calculated. To detect populations with significantly higher rates we use the modified z-score for outlier detection (Iglewicz and Hoaglin 1993) as follows:
with the number of mutations in population , the median number of mutations in a population, and the mean absolute deviation of population . Note that in the original publication the modified Z score is defined as the absolute value of the measure used in this paper. We intentionally left out the absolute value to avoid down weighting populations with few mutations. Populations with a significantly higher mutation rate are defined as populations having a modified Z score of at least 3.5 (Iglewicz and Hoaglin 1993). From this modified Z score a population specific correction factor is calculated, based on a parameter p which sets the upper limit for the correction factor. In our analysis, we set this to 3 to have an upper limit of 0.85 but based on how a user would like to deal with populations having significantly higher mutation rates, the factor can be anywhere between 0 and 3, 5:
Due to the modified Z score, the correction factor intrinsically assigns a lower value to outlier populations if a larger number of independent populations are available, hereby largely reducing the effects of populations with high mutation rates to reduce noise when a large number of independent populations is present. When only a limited number of independent populations is available, the correction factor will be higher as in that case populations with larger mutation rates are needed to exploit parallelism.
The relevance score and the correction factor are integrated into a single score for every mutated gene in every population. This is implemented as follows:
with a mutated gene in population .
Pathfinding between Mutated Genes
All genes with at least one mutation in any independent population are mapped on the topology-weighted genome-wide interaction network. Subsequently, all possible paths originating from a mutated gene in a population and ending in any other gene which is mutated in another population, are enumerated. A path is defined as a series of consecutive edges in the interaction network. We exclude paths between mutated genes in the same populations, reasoning that because of the clonality a single mutation in a pathway will confer most of its fitness advantage (Pulido-Tamayo etal. 2016) and including paths between mutated genes within one population would not be informative as this does not reflect parallel evolution.
Each path is assigned a probability which reflects the degree of belief that the path is associated with the adaptive phenotype under study. This probability takes into account the weights of the edges which make up the path (calculated based on the network topology in the previous step) and the corrected relevance scores from both the start gene and the terminal gene of the path (calculated based on the frequency increase and the functional impact score of both genes in the data preparation step). Only the relevance scores of the start gene and the terminal gene are considered irrespective of whether or not intermediate genes are mutated. If intermediate genes would be taken into account, even one passenger mutation in the middle of an interesting molecular pathway would severely decrease the probability of every found path in that molecular pathway. This leads to the following equation for the probability of a path:
with the path which starts in gene S, which is mutated in population n and terminates in gene E, which is mutated in population m. is the collection of edges which make up the path and (i, j) is the edge from node i to node j.
Enumerating all possible paths is computationally expensive and leads to a prohibitively large computational cost in the subsequent subnetwork inference step. Therefore, the following heuristics are used: 1) Based on biological considerations (Gitter etal. 2011; Navlakha etal. 2012) the maximum path length is set to four. 2) From all possible paths originating from a mutated gene in a specific population, only the 25 paths with highest probabilities are retained.
Subnetwork Inference and Prioritization of Molecular Pathways
The final step of the analysis is the inference of a subnetwork containing the molecular pathways responsible for the adaptive phenotype. This subnetwork consists of a subset of the paths selected in the previous step. This subset of paths is obtained by optimizing the following function:
Where S(K) is the score of the selected subnetwork and needs to be maximized, is the number of edges selected, is the imposed cost for each edge, R is the collection of populations used in the experiment, Qn is the collection of mutated genes from population n and P(path(mutS,n, mutall)|probabilities) is the probability that there exists a path relevant to the observed phenotype between a mutated gene S in population n and any other mutated gene in any other population, given the degrees of belief (probabilities) of all found paths in the pathfinding step. The calculation of this term is a generalization of the two-terminal reliability problem (Fratta and Montanari 1973; Cook and Ramirez-Marquez 2007). Note that the optimal subnetwork, which is the selected subnetwork with the highest score S(K), is not necessarily a connected graph.
As the complexity of this problem inhibits a deterministic solution, a greedy hill-climbing heuristic is used in which the previously found paths get sampled pseudo-randomly based on the overlap the paths have with each other: Overlapping paths are more likely to be sampled together. As this procedure is stochastic in nature, the procedure is repeated 20 times and the best solution with respect to the optimization score S(K) is used as the solution.
The parameter is an important parameter as it incentivizes IAMBEE to primarily select overlapping paths with high probabilities because doing so a single edge can be used multiple times while the cost for selecting this edge only has to be paid once. This is biologically relevant as molecular mechanisms in which multiple (partly) overlapping paths with high probabilities are found, are likely mechanisms of interest for a specific selective sweep.
Setting the parameter is not trivial as its optimal value is data set specific. If is set too high the subnetwork will be small and multiple causal molecular pathways are likely missed. Conversely if is set too low the subnetwork will be too big and of little practical use as the fraction of false positives in the solution increases (supplementary fig. 2, Supplementary Material online). Therefore, instead of calculating the optimal subnetwork for one specific cost, we perform a parameter sweep over the parameter and summarize the results in the form of a network, which is obtained by taking the union of all found optimal subnetworks and where the edges are prioritized based on the maximum edge cost for which they are still included in an optimal subnetwork. This means that edges with a high priority (visualized in the output as opaque edges) get selected even when the edge cost is high. This is useful as the PPV (positive predictive value) of a subset of opaque edges is higher (Synthetic data in supplementary Material, Supplementary Material online) and thus a good starting point for experimental validation.
Parameter Setting
The parameters of IAMBEE were set as follows for both jumps in ethanol tolerance: The path length was kept at the default value of 4 and the maximum number of paths between every pair of mutated genes was kept at the default value of 25. The sweep over the edge cost parameter was set from 0.1 to 1.5 in steps of 0.025 and the maximum size for an optimal subnetwork to be accepted was set to 80 (in terms of nodes) in order to keep the resulting subnetwork small enough to interpret manually.
Validation of IAMBEE Features
To show the importance of the different features of IAMBEE, which include mutation frequencies, functional scores, a correction factor for populations with extreme mutation rates and the use of an interaction network, the method was adjusted several times to exclude one feature each time. Each of these adjusted versions of IAMBEE was applied to the synthetic data set. PPV and sensitivity plots were constructed (supplementary fig. 2, Supplementary Material online) and demonstrated that each feature led to an increase in performance. The results are discussed in supplementary Material, Supplementary Material online.
Biological Validation Methods
The methods for the biological validation experiments can be found the supplementary Methods section, Supplementary Material online.
Availability of Data and Material
The genome sequencing data set generated and analyzed during the current study are available in the SRA repository of NCBI, PRJNA380734 (https://www.ncbi.nlm.nih.gov/bioproject/380734; last accessed June 15, 2017).
IAMBEE is available at http://bioinformatics.intec.ugent.be/IAMBEE
Supplementary Material
Supplementary data are available at Molecular Biology and Evolution online.
Author Contributions
T.S. and B.W. conceptualized the study, analyzed and interpreted the results and wrote the manuscript. T.S. designed and performed the biological experiments, B.W. designed IAMBEE and used it to analyze the sequence data. T.S. and C.B. helped in performing the biological experiments. N.V., J.M., and K.M. conceptualized the study, designed the experiments, discussed the results, and edited the manuscript.
Supplementary Material
Acknowledgments
We thank S. Xie for providing the E. coli SX4 ancestor strains. T.S. is a fellow of the Agency for innovation by Science and Technology - IWT (121525). The research was supported bythe KU Leuven Research Council (PF/10/010, IDO/13/008), Ghent University Multidisciplinary Research Partnership “Bioinformatics: from nucleotides to networks,” Interuniversity Attraction Poles - Belgian Science Policy Office IAP-BELSPO (IAP P7/28) and Research Foundation Flanders - FWO (G047112N, G055517N, G0A5315N, G0B2515N, FWO15/PRJ/396), IWT/SBO NEMOA. The authors declare no competing financial interests. The funding sources were not involved in study design, data collection and interpretation, or the decision to submit the work for publication.
References
- Anderson J, Wagner M, Rushworth C, Prasad K, Mitchell-Olds T.. 2014. The evolution of quantitative traits in complex environments. Heredity 1121: 4–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atsumi S, Wu T-YY, Machado IM, Huang W-CC, Chen P-YY, Pellegrini M, Liao JC.. 2010. Evolution, genomic analysis, and reconstruction of isobutanol tolerance in Escherichia coli. Mol Syst Biol. 6: 449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babur Ö, Gönen M, Aksoy B, Schultz N, Ciriello G, Sander C, Demir E.. 2015. Systematic identification of cancer driving signaling pathways based on mutual exclusivity of genomic alterations. Genome Biol. 16: 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barabási A-L, Albert R.. 1999. Emergence of scaling in random networks. Science 2865439: 509–512. [DOI] [PubMed] [Google Scholar]
- Bernardi AC, Gai CS, Lu J, Sinskey AJ, Brigham CJ.. 2016. Experimental evolution and gene knockout studies reveal AcrA-mediated isobutanol tolerance in Ralstonia eutropha. J Biosci Bioeng. 1221: 64–69. [DOI] [PubMed] [Google Scholar]
- Borges-Walmsley I, Beauchamp J, Kelly S, Jumel K, Candlish D, Harding S, Price N, Walmsley A.. 2002. Identification of oligomerization and drug-binding domains of the membrane fusion protein EmrA. J Biol Chem. 278: 12903–12912. [DOI] [PubMed] [Google Scholar]
- Burns CM, Richardson JP.. 1995. NusG is required to overcome a kinetic limitation to Rho function at an intragenic terminator. Proc Natl Acad Sci U S A. 92: 4738–4742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buttke TM, Ingram LO.. 1980. Ethanol-induced changes in lipid composition of Escherichia coli: inhibition of saturated fatty acid synthesis invitro. Arch Biochem Biophys. 2032: 565–571. [DOI] [PubMed] [Google Scholar]
- Cai S, Inouye M.. 2002. EnvZ-OmpR interaction and osmoregulation in Escherichia coli. J Biol Chem. 27727: 24155–24161. [DOI] [PubMed] [Google Scholar]
- Chao L, Cox EC.. 1983. Competition between high and low mutating strains of Escherichia coli. Evolution 371: 125–134. [DOI] [PubMed] [Google Scholar]
- Chen P, Shapiro J.. 2015. The advent of genome-wide association studies for bacteria. Curr Opin Microbiol. 25: 17–24. [DOI] [PubMed] [Google Scholar]
- Cook J, Ramirez-Marquez J.. 2007. Two-terminal reliability analyses for a mobile ad hoc wireless network. Reliab Eng Syst Safety 926: 821–829. [Google Scholar]
- Dees N, Zhang Q, Kandoth C, Wendl M, Schierding W, Koboldt D, Mooney T, Callaway M, Dooling D, Mardis E, et al. 2012. MuSiC: Identifying mutational significance in cancer genomes. Genome Res. 228: 1589–1598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Maeyer D, Weytjens B, Renkens J, Raedt L, Marchal K.. 2015. PheNetic: network-based interpretation of molecular profiling data. Nucleic Acids Res. 43: W244–W250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Maeyer D, Weytjens B, Raedt L, Marchal K.. 2016. Network-based analysis of eQTL data to prioritize driver mutations. Genome Biol Evol. 83: 481–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiRusso CC, Heimert TL, Metzger AK.. 1992. Characterization of FadR, a global transcriptional regulator of fatty acid metabolism in Escherichia coli. Interaction with the fadB promoter is prevented by long chain fatty acyl coenzyme A. J Biol Chem. 26712: 8685–8691. [PubMed] [Google Scholar]
- Dombek KM, Ingram LO.. 1984. Effects of ethanol on the Escherichia coli plasma membrane. J Bacteriol. 1571: 233–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dragosits M, Mattanovich D.. 2013. Adaptive laboratory evolution: principles and applications for biotechnology. Microb Cell Fact. 121: 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eyre-Walker A. 2010. Genetic architecture of a complex trait and its implications for fitness and genome-wide association studies. Proc Natl Acad Sci U S A. 107: 1752–1756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fratta L, Montanari UG.. 1973. A Boolean algebra method for computing the terminal reliability in a communication network. IEEE Transact Circ Theory. 203: 203–211. [Google Scholar]
- Freddolino PL, Goodarzi H, Tavazoie S.. 2012. Fitness landscape transformation through a single amino acid change in the rho terminator. PLoS Genet. 85: e1002744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gama-Castro S, Salgado H, Santos-Zavaleta A, Ledezma-Tejeida D, Muñiz-Rascado L, García-Sotelo J, Alquicira-Hernández K, Martínez-Flores I, Pannier L, Castro-Mondragón J, et al. 2016. RegulonDB version 9.0: high-level integration of gene regulation, coexpression, motif clustering and beyond. Nucleic Acids Res. 44(D1): D133–D143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gitter A, Klein-Seetharaman J, Gupta A, Bar-Joseph Z.. 2011. Discovering pathways by orienting edges in protein interaction networks. Nucleic Acids Res. 394: e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez R, Tao H, Purvis JE, York SW, Shanmugam KT, Ingram LO.. 2003. Gene array‐based identification of changes that contribute to ethanol tolerance in ethanologenic Escherichia coli: comparison of KO11 (parent) to LY01 (resistant mutant). Biotechnol Progr. 192: 612–623 [DOI] [PubMed] [Google Scholar]
- Goodarzi H, Bennett BD, Amini S, Reaves ML, Hottes AK, Rabinowitz JD, Tavazoie S.. 2010. Regulatory and metabolic rewiring during laboratory evolution of ethanol tolerance in E. coli. Mol Syst Biol. 6: 378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haft RJ, Keating DH, Schwaegler T, Schwalbach MS, Vinokur J, Tremaine M, Peters JM, Kotlajich MV, Pohlmann EL, Ong IM, et al. 2014. Correcting direct effects of ethanol on translation and transcription machinery confers ethanol tolerance in bacteria. Proc Natl Acad Sci U S A. 11125: E2576–E2585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammerstrom T, Beabout K, Clements T, Saxer G, Shamoo Y.. 2015. Acinetobacter baumannii repeatedly evolves a hypermutator phenotype in response to tigecycline that effectively surveys evolutionary trajectories to resistance. PLoS One 10: e0140489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heath R, Rock C.. 1996. Roles of the FabA and FabZ beta-hydroxyacyl-acyl carrier protein dehydratases in Escherichia coli fatty acid biosynthesis. J Biol Chem. 271: 27795–27801. [DOI] [PubMed] [Google Scholar]
- Hong J, Gresham D.. 2014. Molecular specificity, convergence and constraint shape adaptive evolution in nutrient-poor environments. PLoS Genet. 101: e1004041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huffer S, Clark M, Ning J, Blanch H, Clark D.. 2011. Role of alcohols in growth, lipid composition, and membrane fluidity of yeasts, bacteria, and archaea. Appl Environ Microb. 7718: 6400–6408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huffer S, Roche C, Blanch H, Clark D.. 2012. Escherichia coli for biofuel production: bridging the gap from promise to practice. Trends Biotechnol. 3010: 538–545. [DOI] [PubMed] [Google Scholar]
- Iglewicz B, Hoaglin DC.. 1993. How to detect and handle outliers. Milwaukee: ASQC Quality Press. [Google Scholar]
- Ingram LO. 1990. Ethanol tolerance in bacteria. Crit Rev Biotechnol. 94: 305–319. [DOI] [PubMed] [Google Scholar]
- Ingram LO, Buttke TM.. 1984. Effects of alcohols on micro-organisms. Adv Microb Physiol. 25: 253–300. [DOI] [PubMed] [Google Scholar]
- Kanhere A, Bansal M.. 2005. A novel method for prokaryotic promoter prediction based on DNA stability. BMC Bioinformatics 6: 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanehisa M, Sato Y, Kawashima M, Furumichi M, Tanabe M.. 2016. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 44(D1): D457–D462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanehisa M, Goto S, Sato Y, Kawashima M, Furumichi M, Tanabe M.. 2014. Data, information, knowledge and principle: back to metabolism in KEGG. Nucleic Acids Res. 42(Database issue): D199–D205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawecki T, Lenski R, Ebert D, Hollis B, Olivieri I, Whitlock M.. 2012. Experimental evolution. Trends Ecol Evol 27: 547–560. [DOI] [PubMed] [Google Scholar]
- Keseler I, Mackie A, Peralta-Gil M, Santos-Zavaleta A, Gama-Castro S, Bonavides-Martínez C, Fulcher C, Huerta A, Kothari A, Krummenacker M, et al. 2013. EcoCyc: fusing model organism databases with systems biology. Nucleic Acids Res. 41: D605–D612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar P, Henikoff S, Ng P.. 2009. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc. 47: 1073–1081. [DOI] [PubMed] [Google Scholar]
- Kvitek DJ, Sherlock G, Zhang J.. 2013. Whole genome, whole population sequencing reveals that loss of signaling networks is the major adaptive strategy in a constant environment. PLoS Genet. 911: e1003972.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam FH, Ghaderi A, Fink GR, Stephanopoulos G.. 2014. Biofuels. Engineering alcohol tolerance in yeast. Science 3466205: 71–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang GI, Rice DP, Hickman MJ, Sodergren E, Weinstock GM, Botstein D, Desai MM.. 2013. Pervasive genetic hitchhiking and clonal interference in forty evolving yeast populations. Nature 5007464: 571–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence MS, Stojanov P, Polak P, Kryukov GV, Cibulskis K, Sivachenko A, Carter SL, Stewart C, Mermel CH, Roberts SA, et al. 2013. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 4997457: 214–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leiserson MDM, Vandin F, Wu H-T, Dobson JR, Eldridge JV, Thomas JL, Papoutsaki A, Kim Y, Niu B, McLellan M, et al. 2015. Pan-cancer network analysis identifies combinations of rare somatic mutations across pathways and protein complexes. Nat Genet. 472: 106–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Van T, van Leeuwen M, Fierro CA, De Maeyer D, Van den Eynden J, Verbeke L, De Raedt L, Marchal K, Nijssen S.. 2016. Simultaneous discovery of cancer subtypes and subtype features by molecular data integration. Bioinformatics 3217: i445–i454. [DOI] [PubMed] [Google Scholar]
- Loffeld B, Keweloh H.. 1996. Cis/trans isomerization of unsaturated fatty acids as possible control mechanism of membrane fluidity in Pseudomonas putida P8. Lipids 318: 811–815. [DOI] [PubMed] [Google Scholar]
- Luo LH, Seo P-SS, Seo J-WW, Heo S-YY, Kim D-HH, Kim CH.. 2009. Improved ethanol tolerance in Escherichia coli by changing the cellular fatty acids composition through genetic manipulation. Biotechnol Lett. 3112: 1867–1871. [DOI] [PubMed] [Google Scholar]
- Mars R, Mendonça K, Denham EL, van Dijl J.. 2015. The reduction in small ribosomal subunit abundance in ethanol-stressed cells of Bacillus subtilis is mediated by a SigB-dependent antisense RNA. Biochim Biophys Acta 185310: 2553–2559. [DOI] [PubMed] [Google Scholar]
- MathWorks, Inc. 2017. Nonparametric estimates of cumulative distribution functions and their inverses. Available from: https://tinyurl.com/l4pdsaa [Accessed 13 March 2017].
- Minty JJ, Lesnefsky AA, Lin F, Chen Y, Zaroff TA, Veloso AB, Xie B, McConnell CA, Ward RJ, Schwartz DR, et al. 2011. Evolution combined with genomic study elucidates genetic bases of isobutanol tolerance in Escherichia coli. Microb Cell Fact .101: 1–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagakubo S, Nishino K, Hirata T, Yamaguchi A.. 2002. The putative response regulator BaeR stimulates multidrug resistance of Escherichia coli via a novel multidrug exporter system, MdtABC. J Bacteriol. 18415: 4161–4167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navlakha S, Gitter A, Bar-Joseph Z.. 2012. A network-based approach for predicting missing pathway interactions. PLoS Comput Biol. 88: e1002640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng PC, Henikoff S.. 2001. Predicting deleterious amino acid substitutions. Genome Res. 115: 863–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolaou SA, Gaida SM, Papoutsakis ET.. 2012. Exploring the combinatorial genomic space in Escherichia coli for ethanol tolerance. Biotechnol J. 711: 1337–1345. [DOI] [PubMed] [Google Scholar]
- Nishino K, Yamaguchi A.. 2002. EvgA of the two-component signal transduction system modulates production of the yhiUV multidrug transporter in Escherichia coli. J Bacteriol. 1848: 2319–2323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh H, Lee J, Kim O.. 2012. Increase of organic solvent tolerance of Escherichia coli by the deletion of two regulator genes, fadR and marR. Appl Microbiol Biotechnol. 966: 1619–1627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer A, Kishony R.. 2013. Understanding, predicting and manipulating the genotypic evolution of antibiotic resistance. Nat Rev Genet. 144: 243–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasman Z, von Hippel PH.. 2000. Regulation of rho-dependent transcription termination by NusG is specific to the Escherichia coli elongation complex. Biochemistry 3918: 5573–5585. [DOI] [PubMed] [Google Scholar]
- Pulido-Tamayo S, Weytjens B, De Maeyer D, Marchal K.. 2016. SSA-ME Detection of cancer driver genes using mutual exclusivity by small subnetwork analysis. Sci Rep. 6: 36257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinn HJ, Cameron AD, Dorman CJ.. 2014. Bacterial regulon evolution: Distinct responses and roles for the identical OmpR proteins of Salmonella typhimurium and Escherichia coli in the acid stress response. PLoS Genet. 103: e1004215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramos J, Duque E, Gallegos M-T, Godoy P, Ramos-González M, Rojas A, Terán W, Segura A.. 2002. Mechanisms of solvent tolerance in Gram-negative bacteria. Annu Rev Microbiol. 56: 743–768. [DOI] [PubMed] [Google Scholar]
- Read T, Massey R.. 2014. Characterizing the genetic basis of bacterial phenotypes using genome-wide association studies: a new direction for bacteriology. Genome Med. 611: 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlötterer C, Kofler R, Versace E, Tobler R, Franssen SU.. 2015. Combining experimental evolution with next-generation sequencing: a powerful tool to study adaptation from standing genetic variation. Heredity 1145: 431–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanley D, Bandara A, Fraser S, Chambers PJ, Stanley GA.. 2010. The ethanol stress response and ethanol tolerance of Saccharomyces cerevisiae. J Appl Microbiol. 1091: 13–24. [DOI] [PubMed] [Google Scholar]
- Steenackers HP, Parijs I, Foster KR, Vanderleyden J.. 2016. Experimental evolution in biofilm populations. FEMS Microbiol Rev. 40: 373–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swings T, Van den Bergh B, Wuyts S, Oeyen E, Voordeckers K, Verstrepen KJ, Fauvart M, Verstraeten N, Michiels J.. 2017. Adaptive tuning of mutation rates drives adaptation to high ethanol stress in Escherichia coli. eLife 6: e22939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swinnen S, Schaerlaekens K, Pais T, Claesen J, Hubmann G, Yang Y, Demeke M, Foulquié-Moreno MR, Goovaerts A, Souvereyns K, et al. 2012. Identification of novel causative genes determining the complex trait of high ethanol tolerance in yeast using pooled-segregant whole-genome sequence analysis. Genome Res. 22: 975–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki S, Horinouchi T, Furusawa C.. 2014. Prediction of antibiotic resistance by gene expression profiles. Nat Commun. 5: 5792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki T, Seta K, Nishikawa C, Hara E, Shigeno T, Nakajima-Kambe T.. 2015. Improved ethanol tolerance and ethanol production from glycerol in a streptomycin-resistant Klebsiella variicola mutant obtained by ribosome engineering. Bioresour Technol. 176: 156–162. [DOI] [PubMed] [Google Scholar]
- Szklarczyk D, Franceschini A, Wyder S, Forslund K, Heller D, Huerta-Cepas J, Simonovic M, Roth A, Santos A, Tsafou K, et al. 2015. STRING v10: protein–protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 43(D1): D447–D452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taddei F, Radman M, Maynard-Smith J, Toupance B, Gouyon PH, Godelle B.. 1997. Role of mutator alleles in adaptive evolution. Nature 3876634: 700–702. [DOI] [PubMed] [Google Scholar]
- Tamborero D, Gonzalez-Perez A, Lopez-Bigas N.. 2013. OncodriveCLUST: exploiting the positional clustering of somatic mutations to identify cancer genes. Bioinformatics 2918: 2238–2244. [DOI] [PubMed] [Google Scholar]
- Tenaillon O, Rodríguez-Verdugo A, Gaut RL, McDonald P, Bennett AF, Long AD, Gaut BS.. 2012. The molecular diversity of adaptive convergence. Science 3356067: 457–461. [DOI] [PubMed] [Google Scholar]
- Thammasittirong S, Thirasaktana T, Thammasittirong A, Srisodsuk M.. 2013. Improvement of ethanol production by ethanol-tolerant Saccharomyces cerevisiae UVNR56. Springerplus 21: 1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van den Bergh B, Michiels JE, Wenseleers T, Windels EM, Vanden Boer P, Kestemont D, De Meester L, Verstrepen KJ, Verstraeten N, Fauvart M, Michiels J.. 2016. Frequency of antibiotic application drives rapid evolutionary adaptation of Escherichia coli persistence. Nat Microbiol. 15: 16020. [DOI] [PubMed] [Google Scholar]
- Vanegas J, Contreras M, Faller R, Longo M.. 2012. Role of unsaturated lipid and ergosterol in ethanol tolerance of model yeast biomembranes. Biophys J. 102: 507–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaser R, Adusumalli S, Leng S, Sikic M, Ng P.. 2016. SIFT missense predictions for genomes. Nat Protoc. 111: 1–9. [DOI] [PubMed] [Google Scholar]
- White DG, Goldman JD, Demple B, Levy SB.. 1997. Role of the acrAB locus in organic solvent tolerance mediated by expression of marA, soxS, or robA in Escherichia coli. J Bacteriol. 17919: 6122–6126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winkler J, Kao K.. 2014. Recent advances in the evolutionary engineering of industrial biocatalysts. Genomics 104: 406–411. [DOI] [PubMed] [Google Scholar]
- Wiser MJ, Ribeck N, Lenski RE.. 2013. Long-term dynamics of adaptation in asexual populations. Science 3426164: 1364–1371. [DOI] [PubMed] [Google Scholar]
- Woods RJ, Schneider D, Winkworth C, Riley MA, Lenski RE.. 2006. Tests of parallel molecular evolution in a long-term experiment with Escherichia coli. Proc Natl Acad Sci U S A. 103: 9107–9112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woods RJ, Barrick JE, Cooper TF, Shrestha U, Kauth MR, Lenski RE.. 2011. Second-order selection for evolvability in a large Escherichia coli population. Science 3316023: 1433–1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye K, Schulz MH, Long Q, Apweiler R, Ning Z.. 2009. Pindel: a pattern growth approach to detect break points of large deletions and medium sized insertions from paired-end short reads. Bioinformatics 2521: 2865–2871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshikawa K, Tanaka T, Furusawa C, Nagahisa K, Hirasawa T, Shimizu H.. 2009. Comprehensive phenotypic analysis for identification of genes affecting growth under ethanol stress in Saccharomyces cerevisiae. FEMS Yeast Res. 91: 32–44. [DOI] [PubMed] [Google Scholar]
- Zhang Y-MM, Rock CO.. 2008. Membrane lipid homeostasis in bacteria. Nat Rev Microbiol. 63: 222–233. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.