

Abstract
Background
Genetic causes of dilated cardiomyopathy (DCM) are incompletely understood. LRRC10 (leucine‐rich repeat–containing 10) is a cardiac‐specific protein of unknown function. Heterozygous mutations in LRRC10 have been suggested to cause DCM, and deletion of Lrrc10 in mice results in DCM.
Methods and Results
Whole‐exome sequencing was carried out on a patient who presented at 6 weeks of age with DCM and her unaffected parents, filtering for rare, deleterious, recessive, and de novo variants. Whole‐exome sequencing followed by trio‐based filtering identified a homozygous recessive variant in LRRC10, I195T. Coexpression of I195T LRRC10 with the L‐type Ca2+ channel (Cav1.2, β2CN2, and α2δ subunits) in HEK293 cells resulted in a significant ≈0.5‐fold decrease in ICa,L at 0 mV, in contrast to the ≈1.4‐fold increase in ICa,L by coexpression of LRRC10 (n=9–12, P<0.05). Coexpression of LRRC10 or I195T LRRC10 did not alter the surface membrane expression of Cav1.2. LRRC10 coexpression with Cav1.2 in the absence of auxiliary β2CN2 and α2δ subunits revealed coassociation of Cav1.2 and LRRC10 and a hyperpolarizing shift in the voltage dependence of activation (n=6–9, P<0.05). Ventricular myocytes from Lrrc10 −/− mice had significantly smaller ICa,L, and coimmunoprecipitation experiments confirmed association between LRRC10 and the Cav1.2 subunit in mouse hearts.
Conclusions
Examination of a patient with DCM revealed homozygosity for a previously unreported LRRC10 variant: I195T. Wild‐type and I195T LRRC10 function as cardiac‐specific subunits of L‐type Ca2+ channels and exert dramatically different effects on channel gating, providing a potential link to DCM.
Keywords: cardiomyopathy, genetics, ion channel, pediatrics
Subject Categories: Electrophysiology, Ion Channels/Membrane Transport, Cardiomyopathy, Genetics, Pediatrics
Clinical Perspective
What Is New?
We report a rare, homozygous variant in a cardiac‐specific protein, I195T LRRC10 (leucine‐rich repeat–containing 10), discovered in an infant with a dilated cardiomyopathy (DCM) of unknown cause.
Functional studies demonstrate that wild‐type and I195T LRRC10 act as subunits of L‐type Ca2+ channels exerting opposing effects on Ca2+ currents through the channels.
What Are the Clinical Implications?
Our finding of a homozygous I195T LRRC10 variant in an infant with DCM, other recently identified heterozygous LRRC10 variants associated with adult DCM, and loss of Lrrc10 function studies in mice causing DCM provide evidence that variants in LRRC10 can serve as a genetic cause of DCM.
The newly identified role of LRRC10 as an auxiliary subunit for cardiac L‐type Ca2+ channels provides a potential link to the pathophysiology of DCM.
Dilated cardiomyopathy (DCM), characterized by left ventricular dilation and systolic dysfunction, is a leading cause of heart failure and premature death. Consequently, DCM is the most common indication for cardiac transplantation in both children and adults.1, 2 Seminal work establishing idiopathic DCM as a familial disorder in 20% to 48% of cases3, 4, 5 has led to DCM gene discovery, culminating in the identification of >50 DCM‐associated genes.6 Inheritance of many disease‐causing mutations does not translate into symptomatic heart failure until the fifth decade of life, owing to age‐dependent penetrance. Recently, LRRC10 was selected as a candidate gene for mutation scanning in a cohort of 220 DCM patients, based on its cardiac‐restricted expression and animal knockout studies. Two heterozygous missense mutations were identified (L41V and L163I), associating LRRC10 with adult‐onset, familial DCM segregating as an autosomal dominant trait.7
Leucine‐rich repeats (LRRs) are 20‐ to 29‐residue sequence motifs present in many proteins with diverse biological functions and cellular locations.8 The crystal structure of the porcine ribonuclease inhibitor protein, composed of 15 LRRs, illustrates their solenoid conformation ideal for protein–protein interactions.9 LRR‐containing proteins have been shown to regulate ion channel function by modulating their biophysical properties.10, 11, 12, 13 LRRC10 (LRR‐containing 10) is a cardiac‐specific factor in zebrafish, mice, and humans.14, 15, 16, 17 Knockdown of lrrc10 in zebrafish results in heart failure and embryonic lethality.17 Knockout of Lrrc10 in mice leads to prenatal systolic dysfunction and postnatal DCM.18, 19 These findings indicate that LRRC10 plays a critical role across species in maintaining normal cardiac function. However, the mechanism by which LRRC10 regulates cardiac function and how mutation or knockout of LRRC10 leads to DCM are unknown.
Localization of LRRC10 to the dyad region of adult cardiomyocytes16 suggests it may regulate cardiac ion channels such as L‐type Ca2+ channel (LTCC). We used whole‐exome sequencing (WES) to discover a homozygous recessive variant in the LRR region of LRRC10 in a sporadic case of pediatric DCM. We provide comprehensive characterization of LRRC10 and the DCM‐associated substitution, I195T, revealing its unique impact on the modulation of cardiac LTCCs using both heterologous expression and native cardiomyocyte systems.
Materials and Methods
Because of the sensitive and confidential nature of the clinical and genetic patient data collected for this study, requests for data access will be restricted to deidentified Sanger sequencing data from qualified researchers following approval by the Mayo Clinic institutional review board. Requests may be sent to Dr Timothy M. Olson at the Mayo Clinic. The cDNA constructs used for heterologous expression studies and the mouse model can be provided to qualified researchers upon request to Dr Timothy Kamp at the University of Wisconsin–Madison.
Study Participants
Participants provided written informed consent under a research protocol approved by the Mayo Clinic institutional review board. Diagnostic criteria for DCM were left ventricular diastolic and/or systolic dimensions >95th percentile indexed for body surface area and left ventricular ejection fraction <50% as determined by echocardiography. Genomic DNA was isolated from peripheral blood white cells or saliva. WES and whole‐genome sequencing data from 115 in‐house individuals not affected with DCM were used for subtraction filtering of exome variant calls to identify true positives.
Array Comparative Genomic Hybridization
Array comparative genomic hybridization using a custom 180K oligonucleotide microarray was performed on a DNA sample from the affected patient with a genome‐wide resolution of 100 kb. Clinically reportable results included deletions ≥200 kb and duplications ≥500 kb. Those detected below the size threshold were considered relevant if sufficient evidence supporting pathogenicity was present from Online Mendelian Inheritance in Man (http://ncbi.nlm.nih.gov/omim), PubMed (http://ncbi.nlm.nih.gov/pubmed), GeneReviews (http://ncbi.nlm.nih.gov/books/NBK1116/), or the ClinGen Dosage Sensitivity Map (http://ncbi.nlm.nih.gov/projects/dbvar/clingen/).
WES and Bioinformatics Analysis
WES and variant annotation were initially performed on DNA samples from the family trio, consisting of the affected patient and her clinically unaffected parents, utilizing the Mayo Medical Genome Facility and Bioinformatics Core. The Agilent SureSelect Human All Exon 50 Mb kit (version 2.0) was used for exome capture. Each sample was run in a single lane, and 101‐base‐pair, paired‐end sequencing was performed on Illumina's HiSeq2000 platform. A DNA sample subsequently procured from the unaffected sibling underwent WES utilizing the Agilent SureSelect Human All Exon v5+UTRs for exome capture. Reads were aligned to the hg19 reference genome using Novoalign (http://novocraft.com), followed by sorting and marking of duplicate reads using Picard (http://picard.sourceforge.net). Local realignment of insertions and deletions and base quality score recalibration were then performed using the Genome Analysis Toolkit.20 Single nucleotide variants and insertions and deletions were called across all 4 samples simultaneously using the Genome Analysis Toolkit's UnifiedGenotyper with variant quality score recalibration.21 The resultant variant call format files were analyzed with Qiagen's Ingenuity Variant Analysis software using an iterative filtering process. To determine rarity of variants, minor allele frequencies from 3 publicly available databases were utilized: 1000 Genomes (whole‐genome sequencing data from 1092 individuals),22 the Exome Variant Server (WES data from 6503 individuals; http://evs.gs.washington.edu/EVS/), and the Exome Aggregation Consortium (ExAC; WES data from 60 706 individuals; http://exac.broadinstitute.org). Candidate variants identified through rigorous filtering were further analyzed for their in silico predicted effect on protein function23, 24 and gene‐based expression across tissues.25
Animals
Wild‐type (WT) and Lrrc10 −/− mice in a C57BL/6N genetic background were generated and genotyped, as described previously.18, 26 In this study, age‐matched mice (3–4 months old) were used in biochemistry and whole‐cell patch clamp electrophysiology studies. All procedures were performed in accordance with the Guide for the Care and Use of Laboratory Animals (National Institutes of Health) and approved by the University of Wisconsin–Madison institutional animal care and use committee (protocol M05182).
Isolation of Adult Mouse Ventricular Myocytes
Adult mouse ventricular cardiac myocytes were isolated by enzymatic digestion using 0.1 mg/mL of Blendzyme 3 (Roche) or Liberase TM (Roche), as described previously.27, 28, 29
Plasmids
Human LRRC10 plasmids were constructed by standard methods. Briefly, the full‐length LRRC10 coding sequence was amplified by polymerase chain reaction using LRRC10 in the pCMV‐SPORT6.1 vector (Open Biosystems) as a template and LRRC10 primers. The LRRC10 cDNA was then subcloned into the pcDNA3, pcDNA3.1/myc HisB(−) (Invitrogen), or pEGFP‐N1 vector (Clontech) using appropriate restriction enzyme sites. The LRRC10 point mutation (I195T) was generated in the LRRC10/pCMV‐SPORT6.1 vector using the QuickChange Site‐Directed Mutagenesis kit (Agilent Technologies) with primers containing a nucleotide mutation (T584C). The LRRC10 mutant cDNA was amplified by polymerase chain reaction and subcloned into pcDNA3, pcDNA3.1/myc, or pEGF‐N1. The sequence was verified by diagnostic restriction enzyme digest and nucleotide sequencing.
Cell Culture and Transfections
Human embryonic kidney (HEK293) cells were maintained and transfected, as described previously.30 Briefly, HEK293 cells were transfected with either LTCC subunits (rabbit full length Cav1.2 subunit31 and rabbit skeletal muscle Cavα2δ‐1 cloned into pGW1H; British Biotechnology) with β2CN2 cloned into pcDNA3.1HisTOPO (Invitrogen) and pSV40Tag to increase expression levels, as described previously,30 along with enhanced GFP (green fluorescent protein) only or cotransfected with WT or I195T LRRC10 cloned into pEGF‐N1 using the calcium phosphate transfection method, as described previously.32 For coimmunoprecipitation (co‐IP) and surface biotinylation experiments, plasmids encoding for hemagglutinin‐tagged Cav1.2 (HA‐Cav1.2)33 and Myc‐tagged WT (Myc‐WT) or Myc‐I195T LRRC10 were cotransfected in HEK293 cells.
Co‐IP and Western Blot
Mouse hearts were homogenized using the lysis buffer containing (in mmol/L) 11 Tris‐HCl (pH 8.0), 1.1 EDTA (pH 8.0), 11% glycerol, and 0.2% SDS with protease inhibitors (2 mmol/L phenylmethylsulfonyl fluoride, 12 μg/mL aprotinin, 1 mmol/L benzamidin, 21.5 μmol/L leupeptin, 5 μmol/L pepstatin A, and calpain inhibitors). Transiently transfected HEK293 cells were harvested and sonicated 48 hours after transfection, and the insoluble material was removed by centrifugation. Overall, 1 mg of lysates was used for co‐IP experiments and preincubated with 2 μg of anti‐LRRC10, 0.2 μg antihemagglutinin, 1 μg anti‐Myc, or IgG control for 2 hours in 4°C. Next, 50 µL of Dynabeads Protein G or A/G magnetic beads (ThermoFisher Scientific) were added overnight. Immunoprecipitates were washed in 1% Triton X‐100 and 0.4% deoxycholate or 1% IGEPAL‐CA630 (Sigma‐Aldrich) in Tris‐buffered saline supplemented with protease inhibitors. Immunoprecipitated proteins were analyzed by SDS‐PAGE and Western blotting. Membranes were incubated with anti‐Cav1.2, anti‐LRRC10, antihemagglutinin, anti‐Myc, or the α6F antibody raised against the α1 subunit of the sodium pump, as described previously34 (26174834), and then incubated with HRP‐conjugated secondary antibodies. Western blots performed to detect the expression of Cav1.2 LTCC protein were also incubated with anti‐GAPDH or anti‐LRRC10 polyclonal antibody, as described previously.16
Surface Biotinylation
Representative fractions of cell surface proteins were prepared, as described previously.34 Briefly, HEK293 cells transiently expressing HA‐Cav1.2 (Cav1.2, α2δ, β2CN2 plus T‐antigen) with or without Myc‐LRRC10 (WT or I195T) were treated with 0.5 to 1 mg/mL sulfo‐NHS‐SS‐biotin for 10 minutes at 37°C. Cells were lysed in 1% Triton X‐100 in PBS supplemented with protease inhibitors. Surface membrane proteins were subsequently purified using streptavidin sepharose and used for SDS‐PAGE and Western blotting.
Electrophysiology
LTCC currents (ICa,L) were recorded from isolated ventricular myocytes and transiently transfected HEK293 cells 48 hours after transfection using the whole‐cell configuration. The extracellular recording solution consisted of (in mmol/L) 1 4‐aminopyridine, 1.8 CaCl2, 5 CsCl, 5 Glucose, 10 HEPES, 1 MgCl2, and 145 TEA‐Cl (pH 7.4 using TEA‐OH) for isolated myocytes and 10 CaCl2, 133 CsCl, 10 HEPES, and 5 glucose (pH 7.4 using CsOH) for recordings in HEK293 cells. Borosilicate glass pipettes were pulled to a resistance of 1.2 to 2.7 MΩ when filled with internal solution, which contained (in mmol/L) 114 CsCl, 10 EGTA, 10 HEPES, and 5 Mg‐ATP (pH 7.2 using CsOH). Whole‐cell currents were recorded and analyzed, as described previously.29, 30, 35 The current–voltage protocol described previously was used for recordings in HEK293 cells,30 and a current–voltage protocol that steps to −50 mV from a holding potential of −80 mV to inactivate any sodium current contamination followed by 10‐mV depolarizations from −40 mV to +70 mV was used to record ICa,L in isolated myocytes. Leak and capacitive currents were subtracted using a P/4 protocol and whole‐cell conductance was calculated as described previously.30, 35 In brief, the whole‐cell conductance (shown as G) was calculated from the peak ICa,L obtained in the current–voltage relationship, divided by the driving force (Vtest−Vreversal). The conductance–voltage curve was plotted and fit by a Boltzmann function to the equation G/Gmax=1/[1+e−(V−V1/2)/k] from which the half‐activation potential (V1/2) and slope value (k) are calculated. To determine the voltage dependence of ICa,L inactivation, we used a 2‐pulse protocol with a 500‐ms conditioning pulse from −80 mV to +50 mV, followed by a 25‐ms test pulse to +10 mV. For the steady‐state inactivation data, the peak current during the test pulse was fit to a Boltzmann function using the equation I/Imax=1/[1+e(V−V1/2)k].
Statistical Analyses
Data are expressed as mean±SEM. Using Origin 8.6 software (OriginLab), 1‐way ANOVA followed by post hoc pairwise comparisons with Bonferroni correction was performed to determine statistical significance between multiple experimental groups compared with the control group. The Student t test was used for comparisons between 2 groups. P<0.05 was considered statistically significant.
Results
Presentation of End‐Stage Infantile DCM
The patient presented at 6 weeks of age in cardiogenic shock. Echocardiography demonstrated severe left ventricular enlargement and an ejection fraction of 10% to 15%. Electrocardiography revealed normal sinus rhythm and left ventricular hypertrophy with a strain pattern. Her birth history was unremarkable, and she had no dysmorphic features. Workup for infectious and metabolic etiologies was negative, leading to a diagnosis of idiopathic DCM. She was resuscitated and stabilized with mechanical ventilation and intravenous ionotropic support and eventually transitioned to an outpatient heart failure regimen of carvedilol, enalapril, and furosemide. Because of persistent, severe systolic dysfunction, she was listed for cardiac transplantation, which occurred at 4 months of age. Screening echocardiograms in her nonconsanguineous parents and older brother revealed normal left ventricular size and systolic function (father, aged 35 years; mother, aged 34 years; sibling, aged 8 years).
WES Identifies Homozygous Recessive LRRC10 Variant
Array comparative genomic hybridization excluded chromosomal aneuploidy in the patient. To identify the genetic underpinnings of her DCM, WES was performed on genomic DNA samples from the patient and her unaffected parents (Figure 1A). Each sample yielded >120 million 101‐bp, paired‐end reads and passed quality control standards. More than 95% of the reads mapped to the genome, with at least 87% of the mapped reads demonstrating a read depth of ≥20. Variant call format files from the family trio were loaded into Ingenuity's Variant Analysis, and a comprehensive filtering strategy was used (Figure 1B). Variants with a read depth >5 that mapped to the coding region and passed variant quality score recalibration were filtered to exclude those located in the top 0.5% of the most exonically variable genes and present in ≥5 in‐house, non‐DCM controls. Variants were then filtered for rarity, excluding variants with a minor allele frequency ≥1.0% in the 1000 Genomes, Exome Variant Server, or ExAC databases. Next, all canonical splice‐site, missense, and truncation variants were retained. Variants were then filtered to model all potential modes of inheritance, including de novo, homozygous recessive, compound heterozygous, and uniparental disomy. Comprehensive filtering resulted in only 5 candidate variants in 4 genes.
Figure 1.
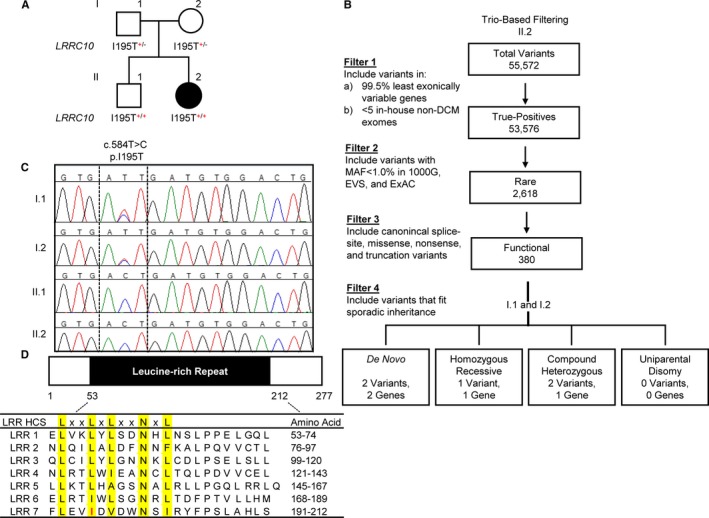
Identification of a homozygous recessive LRRC10 (leucine‐rich repeat–containing 10) variant in sporadic pediatric dilated cardiomyopathy. A, Family pedigree. Square=male; circle=female; solid=affected; open=unaffected. B, Trio‐based filtering scheme for whole‐exome sequencing data. The number of variants identified in the patient are shown. C, Sanger sequencing verification of the I195T variant in the family quartet. D, Gene structure of LRRC10. LRRC10 consists of a single coding exon and contains 7 leucine‐rich repeats, characterized by a highly conserved segment (LxxLxLxxNxL, where L indicates an isoleucine, a leucine, or a valine residue) and a variable segment. Red indicates the location of the I195T variant, within the seventh leucine‐rich repeat, at the second conserved “L” position. 1000G indicates 1000 Genomes; DCM, dilated cardiomyopathy; EVS, Exome Variant Server; ExAC, Exome Aggregation Consortium; HCS, highly conserved segment; LRR, leucine‐rich repeat; MAF, minor allele frequency.
De novo missense variants were identified in TTC28 (tetratricopeptide repeat domain 28) and RIMKLA (ribosomal modification protein rimK‐like family member A; also known as N‐acetylaspartylglutamate synthase II). TTC28 is involved in condensation of spindle midzone microtubules, formation of the midbody, and completion of cytokinesis in COS‐7 and HeLa cells36 and has low expression in the heart (left ventricle: 1.732 RPKM (reads per kilobase of sequence per million reads mapping to transcribed RNAs); highest expression: ovary, 9.205 RPKM).25 The identified c.4062C>A, p.N1354K missense variant (NM_001145418.1) does not fall within a known functional domain, and in silico tools did not predict a deleterious effect (SIFT: 0.12=tolerated, PolyPhen2: 0.223=benign).23, 24 RIMKLA catalyzes the synthesis of the neurotransmitters NAGG (N‐acetylaspartylglutamate)37 and NAGG2 (N‐acetylaspartylglutamylglutamate)38 and is almost exclusively expressed in the central nervous system,37 with low expression in the heart (left ventricle: 1.432 RPKM).25 The identified c.1021A>G, p.T341A missense variant (NM_173642.3) does not fall within a known functional domain and was not predicted to have a deleterious effect (SIFT: 1=tolerated, PolyPhen 2: 0.001=benign).23, 24 Two compound heterozygous candidate variants were identified in SSC5D (scavenger receptor cysteine rich family member with 5 domains) protein, thought to be involved in both adaptive and innate immunity as well as placental function.39 Both variants (c.1802C>T, p.A601V; c.3524T>A, p.F1175Y; NM_001144950.1) are located outside a known functional domain with weak in silico evidence supporting a deleterious effect (A601V SIFT: 0.55=tolerated, PolyPhen 2: 0.005=benign; F1175Y SIFT: 0.05=damaging [low confidence], PolyPhen 2: 0.141=benign).23, 24 SSC5D expression is restricted to monocytes/macrophages and T lymphocytes, being particularly enriched in the placenta,39 with low expression in the heart (left ventricle: 1.304 RPKM).25 The last candidate was a homozygous recessive missense variant in LRRC10, a recently identified DCM‐associated gene (Figure 1A and 1C). The c.584T>C, p.I195T missense variant (rs151080979; NM_201550.2) is located in the sole coding exon of LRRC10, which encodes a 277–amino acid protein exclusively expressed in the heart (left ventricle: 9.96 RPKM).25 The I195T substitution was predicted to be deleterious (SIFT: 0=damaging; PolyPhen2: 0.796=possibly damaging)23, 24 and is located within a highly conserved LxxLxLxxNxL motif40 (Figure 1D).
The heterozygous carrier state in the patient's parents was clinically silent. To further investigate genotype–phenotype correlation in the family, Sanger sequencing of LRRC10 was performed in the patient's unaffected 8‐year‐old sibling. Unexpectedly, he was found to be homozygous for the same I195T variant identified in his affected sister (Figure 1A). In an effort to identify a potential deleterious or beneficial modifier polymorphism that could account for variable penetrance, WES was performed on the sibling. A gene‐targeted filtering scheme was then applied that compared the siblings for variants in genes (1) that are associated with DCM,41 (2) that could regulate LRRC10 expression (GATA4 [GATA binding protein 4], NKX2‐5 [NK2 homeobox 5]), (3) that encode known LRRC10‐interacting partners (ACTC1 [actin, alpha, cardiac muscle 1], ACTN2 [actinin alpha 2]), or (4) that encode LTCC subunits (CACNA1C [calcium voltage‐gated channel subunit alpha1 C], CACNA2D1 [calcium voltage‐gated channel auxiliary subunit alpha2delta 1], CACNB1 [calcium voltage‐gated channel auxiliary subunit beta 1], CACNB2 [calcium voltage‐gated channel auxiliary subunit beta 2], CACNB3 [calcium voltage‐gated channel auxiliary subunit beta 3], CACNB4 [calcium voltage‐gated channel auxiliary subunit beta 4]) or calcium signaling pathway proteins (RYR2 [ryanodine receptor 2], ATP2A1 [ATPase sarcoplasmic/endoplasmic reticulum Ca2+ transporting 1], ATP2A2 [ATPase sarcoplasmic/endoplasmic reticulum Ca2+ transporting 2], ATP2A3 [ATPase sarcoplasmic/endoplasmic reticulum Ca2+ transporting 3], PLN [phospholamban]). No additional variants were identified that would account for the discordant phenotypes. Next, a filter was applied to screen for polymorphisms within microRNAs associated with DCM, revealing a risk allele in MIR499A (microRNA 499A), an MYH7B (myosin heavy chain 7B)–regulating microRNA (rs374644442). Sanger sequencing verified that the patient was heterozygous for the risk‐conferring minor allele, inherited from her mother, whereas her unaffected brother was homozygous for the WT allele. Because the predicted biological impact of the LRRC10 I195T variant on protein function was compelling, in vitro functional characterization of mutant protein and further investigation of LRRC10's role in normal cardiac physiology were carried out.
WT and I195T LRRC10 Differentially Modulate LTCCs Coexpressed in HEK293 Cells
The localization of LRRC10 to T‐tubules where LTCCs localize16 and the central role of LTCCs in regulating cardiac contractility prompted us to investigate the impact of LRRC10 on LTCC function. We determined the effect of coexpression of WT LRRC10 and the DCM‐linked mutation, I195T LRRC10, on LTCC currents. The LTCC complex (Cav1.2, α2δ and β2CN2 subunits) was expressed alone or with WT LRRC10 or I195T LRRC10 in HEK293 cells. ICa,L was measured using whole‐cell patch clamp electrophysiology 48 hours following transfection. Remarkably, WT LRRC10 coexpression with the LTCC complex resulted in larger ICa,L over a range of test potentials in contrast to I195T LRRC10 coexpression, which led to smaller ICa,L (at 0 mV, LTTC alone, −34.1±2.2 pA/pF; coexpression with WT LRRC10, −81±5.3 pA/pF; and coexpression with I195T LRRC10 −18.2±3.3 pA/pF; P<0.05 for all comparisons; see Figure 2A).
Figure 2.
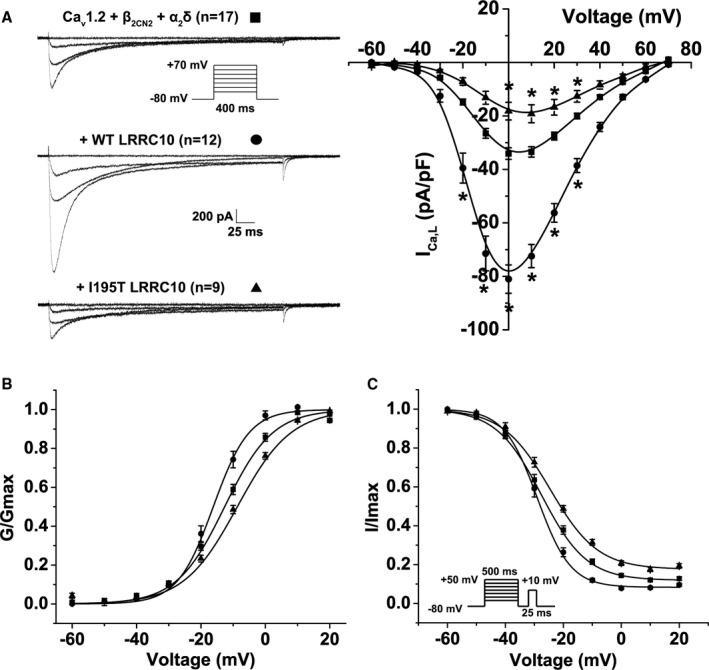
Impact of wild‐type (WT) LRRC10 (leucine‐rich repeat–containing 10) and the dilated cardiomyopathy–linked I195T variant on ICa,L in HEK293 cells. Functional effects of coexpressing WT or I195T LRRC10 with the Cav1.2 complex. A, Representative ICa,L traces recorded at −60, −30, 0, and +30 mV from human embryonic kidney (HEK293) cells transiently expressing either the Cav1.2+β2CN2+α2δ channel complex alone or with WT or I195T LRRC10 using the voltage protocol shown in the inset. Mean current–voltage (I‐V) relationship plotted as mean±SEM. B, Voltage dependence of activation (G/Gmax) and (C) steady‐state inactivation (I/Imax) plotted against voltage (mV) were fitted by Boltzmann distributions, as shown by the solid lines. Data are mean±SEM. *P<0.05.
The voltage dependence of ICa,L activation was determined by calculating the whole‐cell conductance across a range of test potentials (−60 mV to +20 mV) and fitting the data to a Boltzmann distribution. Coexpression of the WT LRRC10 significantly shifted the voltage dependence of activation to more hyperpolarized potentials (V1/2 of −16.2±1.1 mV; P<0.05) compared to the LTCC complex expressed alone (V1/2 of −12.6±0.88 mV), whereas coexpression of the I195T LRRC10 mutation shifted the voltage dependence of activation to more depolarized potentials (V1/2 of −8.7±0.85 mV; P<0.05; Figure 2B). Moreover, the Boltzmann distribution parameter, k, which reflects the slope of the activation curve, was significantly steeper with the coexpression of WT LRRC10 (k=5.6±0.36; P<0.05) compared with the LTCC complex expressed alone (k=7.5±0.26). The k value (k=8.3±0.19) was not different with I195T LRRC10 coexpression. These results show that WT LRRC10 and I195T LRRC10 can affect the activation gating of LTCCs in opposing fashions.
We also analyzed the voltage dependence of steady‐state inactivation of ICa,L (Figure 2C). The V1/2 of ICa,L inactivation was not significantly different comparing LTCC complex alone (V1/2=−27.6±1.1 mV), coexpression with WT LRRC10 (V1/2=−29.0±1.1 mV), or coexpression with I195T LRRC10 (V1/2=−24.6±1.1 mV). However, the slope factor was significantly steeper with WT LRRC10 coexpression (k=5.7±0.28; P<0.05) but not with I195T LRRC10 coexpression (k=8.25±0.50) relative to the LTCC complex expressed alone (k=7.8±0.37). Moreover, we calculated the ratio of ICa,L remaining at +20 mV (slowly inactivating current) to the maximal current (Imax) and found that coexpression of WT LRRC10 resulted in a smaller fraction of slowly inactivating ICa,L (0.08±0.01; P<0.05), whereas coexpression of I195T LRRC10 showed a larger fraction of slowly inactivating ICa,L, (0.18±0.01; P<0.05) compared with the LTCC complex expressed alone (0.12±0.01). These differences in the inactivation of ICa,L may contribute to increased risk of DCM with I195T producing more late ICa,L, which could lead to increased Ca2+ loading.
WT and I195T LRRC10 Do Not Affect the Plasma Membrane Expression of Cav1.2 Channel in Transiently Transfected HEK293 Cells
The changes in the ICa,L density observed with the coexpression of WT or I195T LRRC10 could be due to changes in the surface membrane expression of Cav1.2 channels. To determine whether coexpression of WT or DCM‐linked I195T LRRC10 affects the surface membrane expression of Cav1.2 channels, surface biotinylation experiments were performed using HEK293 cells transiently transfected with the LTCC complex (HA‐Cav1.2+β2CN2+α2δ) alone or with either WT or I195T LRRC10. Lysates were prepared from the biotinylated HEK293 cells and incubated overnight with streptavidin sepharose beads to purify the biotinylated plasma membrane proteins. To specifically detect the plasma membrane expression of the HA‐Cav1.2 channels, immunoblots were incubated with anti‐hemagglutinin antibody. Figure 3 shows a representative immunoblot with the starting material and cell surface fractions for each group of transfected HEK293 cells. The ratio between the cell surface and starting material fractions for the HEK293 cells coexpressing the LTCC complex with the WT or I195T LRRC10 were then normalized relative to the control (LTCC complex). Analysis of the immunoblot from 4 independent surface biotinylation experiments demonstrated no significant differences in the plasma membrane expression of Cav1.2 channels with coexpression of either WT or I195T LRRC10 relative to when the LTCC complex was expressed alone. No significant changes were detected in the protein expression of another membrane protein, Na+/K+ ATPase.
Figure 3.
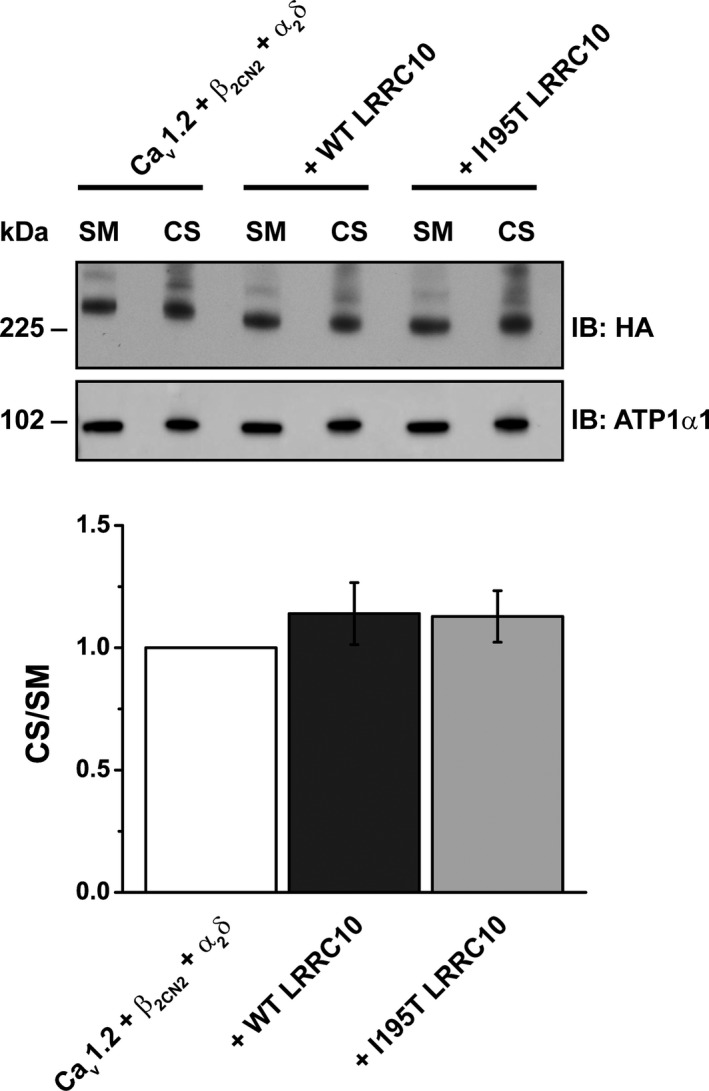
LRRC10 (leucine‐rich repeat–containing 10) coexpression does not affect the expression of L‐type Ca2+ channels on the plasma membrane. The surface membrane proteins of HEK293 cells transiently expressing the Cav1.2 complex alone (HA‐Cav1.2+β2CN2+α2δ) or with Myc‐tagged LRRC10 (wild‐type [WT] or I195T) were biotinylated, purified with streptavidin sepharose and immunoblotted (IB) with anti‐HA antibody to detect the HA‐Cav1.2 protein. Representative immunoblot (n=4) demonstrated no change in surface membrane expression of Cav1.2 complex. Starting material (SM) and cell surface (CS) signals were quantified and normalized to HEK293 cells expressing the Cav1.2 complex alone. Data are mean±SEM with P=0.538. HA indicates hemagglutinin.
LRRC10 Associates With and Directly Modulates Cav1.2 Channel Gating in HEK293 Cells
To determine whether the WT and DCM‐linked I195T LRRC10 mutation associated with the Cav1.2 channels, we coexpressed HA‐Cav1.2 channels with either Myc‐WT or Myc‐I195T LRRC10 in HEK293 cells in the absence of the auxiliary β and α2δ subunits. HEK293 lysates were immunoprecipitated with anti‐hemagglutinin, anti‐Myc, or IgG control antibodies. The immunoprecipitates were then immunoblotted with either antihemagglutinin or anti‐Myc antibodies to detect the HA‐Cav1.2 channel subunit or LRRC10 (WT or I195T) proteins, respectively. Our results show that Cav1.2 associates with both WT and I195T LRRC10 in HEK293 cells (Figure 4A), suggesting that both WT and I195T LRRC10 are able to form a stable complex with the Cav1.2 channel complex.
Figure 4.
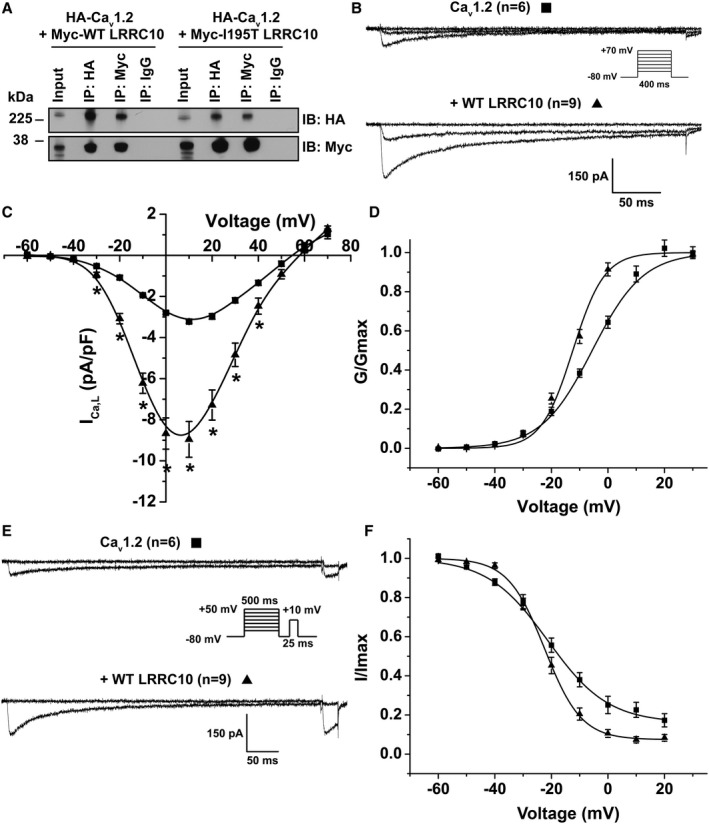
Wild‐type (WT) LRRC10 (leucine‐rich repeat–containing 10) and dilated cardiomyopathy–associated LRRC10–I195T associate with and differentially modulate the Cav1.2 subunit in HEK293 cells. A, Representative immunoblot (n=3) from coimmunoprecipitation experiments of HEK293 cells transiently expressing HA‐Cav1.2 with either Myc‐WT or Myc‐I195T LRRC10 plasmids. Plasmids encoding for the auxiliary β2CN2 or α2δ subunits were not cotransfected. Lysates were immunoprecipitated (IP) with anti‐HA, anti‐Myc, or IgG control antibodies and immunoblotted (IB) with either anti‐HA or anti‐Myc to detect the Cav1.2 or LRRC10 (WT or I195T) proteins, respectively. B, Representative ICa,L traces recorded at −60, −20, and +10 mV from transiently transfected HEK293 cells expressing Cav1.2 alone or with WT LRRC10 showed larger ICa,L compared with cells expressing Cav1.2 alone generated from the voltage protocol shown in the inset. C, Mean current–voltage (I‐V) relationship plotted as mean±SEM, *P<0.05. D, Voltage dependence of activation (G/Gmax) and (E) representative steady‐state inactivation current traces at prepulses −60 and 0 mV with (F) steady‐state inactivation (I/Imax) plotted against voltage (mV). Mean±SEM were fit to Boltzmann distributions. HA indicates hemagglutinin.
Given the biochemical experiments demonstrating an association of LRRC10 and Cav1.2 in the absence of auxiliary β2CN2+α2δ subunits, we tested whether coexpression of LRRC10 with only Cav1.2 in HEK293 cells could alter the ICa,L using whole‐cell patch clamp electrophysiology. WT LRRC10 coexpression with the Cav1.2 channel resulted in significantly larger ICa,L across a range of test potentials compared with Cav1.2 expressed alone (at 10 mV, Cav1.2 alone, −3.22±0.08 pA/pF, n=6; coexpression with WT LRRC10, −8.95±0.87 pA/pF, n=9; P<0.05; Figure 4B and 4C); however, coexpression of I195T LRRC10 did not result in measurable currents (n=7 transfections). Coexpression of WT LRRC10 shifted the normalized whole‐cell conductance–voltage curve in the hyperpolarizing direction (Figure 4D): V1/2=−5.57±1.2 mV for Cav1.2 alone and −12.41±1.1 mV for coexpression of WT LRRC10 (P<0.05, n=6–9). In addition, coexpression of WT LRRC10 increased the steepness of the activation curve, as reflected by fit k values of 9.0±1.2 and 5.57±0.17 for Cav1.2 alone and coexpression with WT LRRC10, respectively (P<0.05, n=6–9). Furthermore, we analyzed the voltage dependence of steady‐state inactivation of ICa,L (Figure 4E and 4F) and found that coexpression of WT LRRC10 did not affect the V1/2 of ICa,L inactivation compared with when Cav1.2 was expressed alone (Cav1.2, −21.27±1.48 mV, n=6; WT LRRC10, −22.36±1.05 mV, n=9). However, the slope factor was significantly steeper with WT LRRC10 coexpression (k=6.48±0.35; P<0.05) relative to when Cav1.2 was expressed alone (k=10.65±0.76). Moreover, calculating the ratio of ICa,L remaining at +20 mV to the maximal current resulted in a smaller fraction of slowly inactivating ICa,L with WT LRRC10 coexpression (0.07±0.02; P<0.05) compared with Cav1.2 alone (0.16±0.03). Together, these results suggest that LRRC10 can act as an auxiliary subunit for Cav1.2 by associating with the channel and modulating its gating.
Reduced ICa,L in Ventricular Myocytes of Lrrc10 −/− Mice
Based on modulation of LTCCs function by LRRC10 observed in the HEK heterologous expression system and our prior finding that LRRC10 −/− mice exhibiting impaired cardiac function and DCM,18 we sought to determine the impact of loss of LRRC10 expression in mouse cardiomyocytes on ICa. In Lrrc10 −/− myocytes, ICa,L was significantly reduced compared with WT (−4.9±0.19 versus −6.95±0.53 pA/pF at 0 mV, P<0.05; Figure 5A). The voltage dependence of current activation was not significantly different between Lrrc10 −/− and WT myocytes; however, the slope factor was significantly different between WT (k=5.2±0.13) and Lrrc10 −/− (k=5.7±0.11; P<0.05; data not shown). To determine whether the sex of the mice differentially affected the effect of knockout of Lrrc10, we separately analyzed the data from male and female mice. A significant reduction in ICa,L was found in cardiomyocytes from both male and female Lrrc10 −/− mice relative to WT mice of the same sex (data at 0 mV; Figure 5A, inset).
Figure 5.
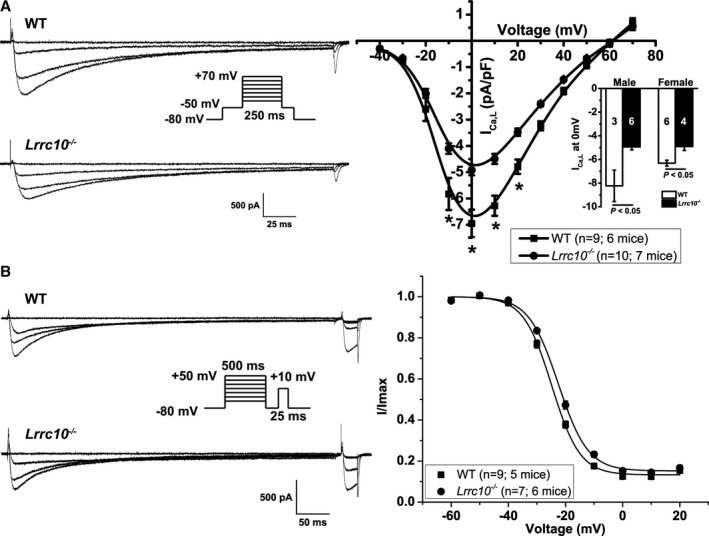
Lrrc10 −/− ventricular myocytes have reduced ICa,L and shifted steady‐state inactivation of ICa,L. A, ICa,L recordings from wild‐type (WT) or Lrrc10 −/− ventricular myocytes at −40, 0, 20, and +40 mV using the voltage protocol in the inset. Mean current–voltage (I‐V) relationship are plotted as mean±SEM *P<0.05. Inset shows mean peak ICa,L at 0 mV separated by sex with sample size shown in bars for 3 male WT mice, 6 female WT mice, 6 for Lrrc10 −/− male mice, and 4 Lrrc10 −/− female mice. B, Representative steady‐state inactivation traces at prepulses −60, −20, 0, and +20 mV. Mean V1/2 of inactivation was shifted to more depolarized potentials in Lrrc10 −/− myocytes compared with WT with data fitted by Boltzmann distributions.
To determine whether loss of Lrrc10 also affected the inactivation of LTCC, we used a steady‐state inactivation protocol that consisted of a 500‐ms prepulse from −60 mV to +50 mV, followed by a 25‐ms test pulse to +10 mV. The peak current elicited by the test pulse normalized to the maximal current (I/Imax) was plotted as a function of voltage and fitted to a Boltzmann distribution. The V1/2 for the Lrrc10 −/− myocytes was more depolarized compared with WT (V1/2 of −22.5±0.44 mV versus −24.8±0.51 mV, P<0.05), with the k value and residual current between the 2 groups unchanged (Figure 5B).
Cav1.2 Protein Expression is Unchanged in Lrrc10 −/− Mouse Hearts
To determine whether the observed decrease in ICa,L of Lrrc10 −/− isolated myocytes is caused by changes in the expression of the Cav1.2 subunit of the LTCC, we performed Western blot analysis using lysates prepared from WT and Lrrc10 −/− mouse hearts. Figure 6A shows a representative immunoblot comparing the protein expression of Cav1.2 LTCC and GAPDH as a loading control in WT and Lrrc10 −/− mouse lysates. Immunoblotting with anti‐LRRC10 antibody confirms the expression of LRRC10 protein in WT but not Lrrc10 −/− hearts. Using the specific Cav1.2 LTCC antibody, we detected the previously described long and short forms of the functional channel, with the short form predominating in native cardiac tissue.43, 44, 45, 46 Although our lysate preparation and Western blot methods allowed the detection of the long form of the channel, the corresponding ≈230‐kDa protein bands were difficult to reproducibly quantitate. Consequently, we performed densitometry analysis using the predominant ≈210‐kDa short form, which showed that the protein expression of Cav1.2 LTCC was not significantly different between WT and Lrrc10 −/− mice.
Figure 6.
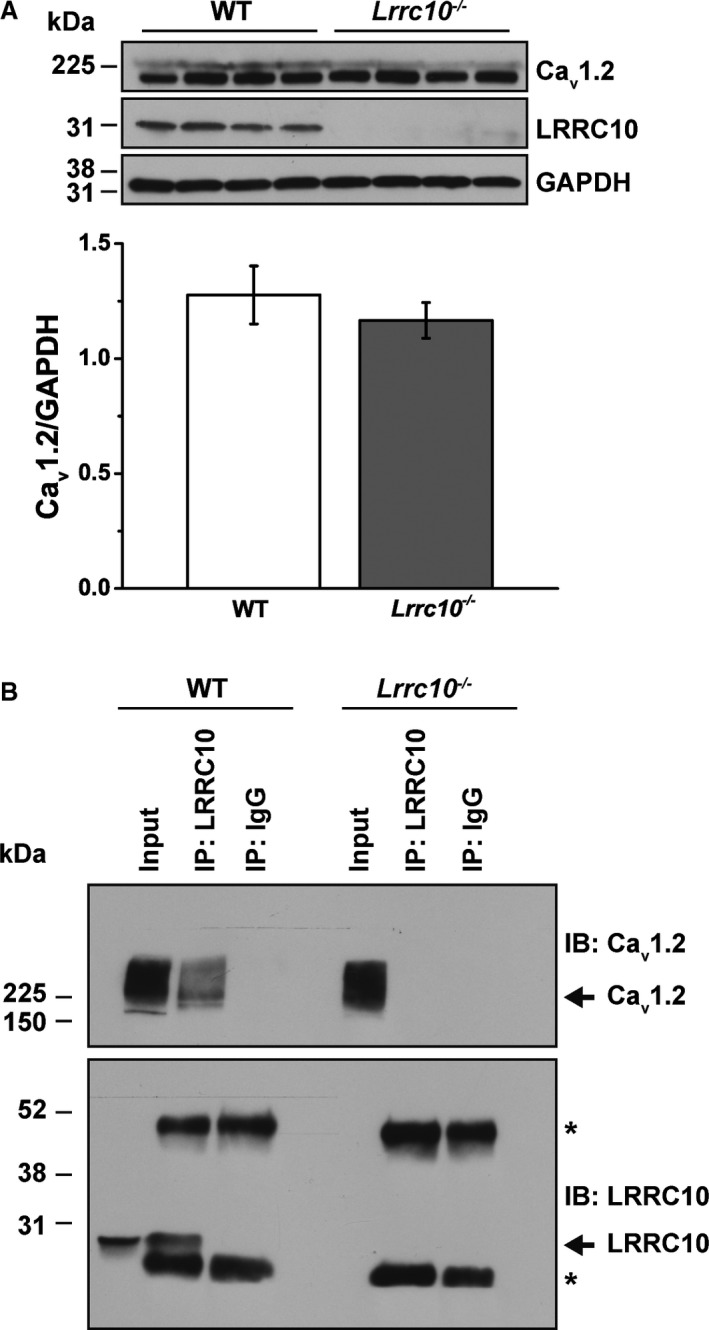
Cav1.2 L‐type Ca2+ channel protein is unchanged in Lrrc10 −/− mice and associates with LRRC10 in wild‐type (WT) mouse hearts. A, Lysates were prepared from WT and Lrrc10 −/− mouse hearts (n=4 each group), and 100 µg of input protein was loaded. Representative immunoblot shows expression of Cav1.2 channel protein and GAPDH loading control in WT and Lrrc10 −/− lysates. Only LRRC10 (leucine‐rich repeat–containing 10) protein is expressed in WT, not Lrrc10 −/− lysates. Normalized Cav1.2 protein signal (Cav1.2/GAPDH) was analyzed by Student t test and found not significantly different between WT and Lrrc10 −/−. B, Representative immunoblot of WT and Lrrc10 −/− mouse heart lysates immunoprecipitated with anti‐LRRC10 or anti‐IgG control antibodies and immunoblotted with anti‐Cav1.2 or anti‐LRRC10 antibodies with the arrows indicating Cav1.2 and LRRC10, respectively. The immunoblot is representative of 3 independent experiments. *IgG heavy and light chains, which are ≈52 and 30 kDa, respectively.
LRRC10 Associates With Cav1.2 Channel Subunit in Mouse Hearts
Mouse heart lysates were used in coimmunoprecipitation experiments to determine whether LRRC10 associates with Cav1.2 in the native cardiomyocytes. WT mouse lysates immunoprecipitated with anti‐LRRC10 antibody and immunoblotted with anti‐Cav1.2 and anti‐LRRC10 antibodies demonstrated coimmunoprecipation of LRRC10 and Cav1.2 (Figure 6B). In contrast, immunoprecipitation with a control IgG failed to demonstrate any immunoreactivity for LRRC10 or Cav1.2 in the immunoprecipitate. As an additional control experiment, immunoprecipitation was performed with anti‐LRRC10 antibody in Lrrc10 −/− mouse heart lysates, and we did not observe LRRC10 in the lysate or immunoprecipitate, consistent with the knockout phenotype, and there was no immunoprecipitation of Cav1.2.
Discussion
We report the use of an unbiased WES approach to uncover a homozygous recessive LRRC10 I195T variant in an infant with DCM and end‐stage systolic heart failure. In addition, we demonstrated that WT LRRC10 and I195T LRRC10 associate with Cav1.2 channels and differentially modulate the gating of heterologously expressed LTCCs. Furthermore, knockout of Lrrc10 in mouse reduces LTCC current. These results suggest that LRRC10 is a cardiac‐specific auxiliary subunit of LTCCs and that variants in LRRC10 can result in distinct differences in LTCC behavior contributing to the genesis of DCM.
LRRC10 was recently selected as a candidate gene for mutation scanning in a cohort of 220 DCM patients based on its cardiac‐restricted expression and animal knockout studies. Two heterozygous missense mutations were identified (L41V and L163I), associating LRRC10 with adult‐onset, familial DCM segregating as an autosomal dominant trait.7 These heterozygous LRRC10 mutations reported by Qu et al are absent in the ExAC database and were identified in individuals ranging in age from 29 to 61 years, all of whom fulfilled diagnostic criteria for DCM.7 By contrast, the heterozygous carrier state of the LRRC10 I195T variant in the patient's parents, both in their mid‐30s, was clinically silent. They may develop heart failure later in life, particularly if they acquire additional risk factors. Indeed, LRRC10 is required to maintain cardiac performance in response to pressure overload,47 implying that mutations in LRRC10 may greatly sensitize human patients to severe cardiomyopathy in the setting of pressure overload, such as in patients with high blood pressure or aortic stenosis.
Heterozygosity for the I195T (rs151080979) variant of LRRC10 is present in 0.56% of ≈60 000 individuals (European descent: 0.77%, n=73 324 alleles; non‐European descent: 0.24%, n=48 016 alleles) of unknown cardiac phenotype in the ExAC database, suggesting it may not be sufficiently pathogenic for DCM but may confer susceptibility to heart failure, given its functional impact on LTCC. Unexpectedly, the identified variant was also present in the homozygous state in the patient's clinically unaffected brother, as well as 2 individuals of unknown age in the ExAC database (frequency: 0.003%), suggesting that even the homozygous state can have variable, age‐dependent penetrance48 and that development of DCM may require additional inherited or acquired risk factors. We considered sex as a possible factor accounting for the variable phenotypes of the female proband and her brother, both of whom were homozygous for the I195T LRRC10 variant, by evaluating for sex‐dependent differences in the Lrrc10 −/− mouse relative to WT. Reanalysis of prior echocardiography data demonstrated that male and female Lrrc10 −/− mice equivalently develop postnatal DCM (data not shown).18 Furthermore, isolated cardiomyocytes from female and male Lrrc10 −/− mice show comparable reductions in ICa,L relative to WT mice. Thus, if the lack of sex effects in the mouse model of loss of LRRC10 function extrapolates to the clinical setting, sex is not a major factor in determining the variable phenotypic presentation of homozygous I195T LRRC10. Post hoc analysis of WES data did not reveal additional variants in DCM‐associated genes or candidate genes relating to LRRC10, LTTC subunits, or cardiomyocyte calcium handling. However, an intriguing risk allele in MIR499A49 was identified that could contribute to the variable penetrance and discordant phenotypes in the patient and her brother. This is of particular interest because MIR499 has been reported to regulate the expression of CACNB2 (calcium voltage‐gated channel auxiliary subunit β2), a Ca2+ channel β subunit gene, providing a potential compensatory effect on ICa,L in the brother.50 Consequently, competing effects on LTCCs by LRRC10 and other Ca2+ channel subunits could contribute to phenotypic differences. Additional unknown modifiers in coding or noncoding regions not covered by WES, epigenetic factors, or environmental differences cannot be excluded. Variable age‐dependent penetrance is known to occur in DCM family members carrying the same mutation, highlighting the possibility that the clinically unaffected sibling may develop DCM as a teenager or adult.41 Analysis of additional participants with the I195T LRRC10 variant in the future is necessary to firmly establish and understand the association between the I195T LRRC10 and DCM.
The results suggest a possible link between the I195T LRRC10 mutation and the DCM phenotype based on the dramatically different effects of WT LRRC10 and I195T LRRC10 on heterologously expressed LTCCs. While WT LRRC10 shifted the voltage dependence of activation in the hyperpolarizing direction, leading to larger currents over the physiological range of the action potential, I195T LRRC10 shifted the activation voltage to more positive potentials and led to smaller currents. Moreover, coexpression of WT LRRC10 with Cav1.2 in the absence of auxiliary β and α2δ subunits resulted in significantly larger ICa,L and a hyperpolarizing shift in the voltage dependence of ICa,L activation, suggesting that although LRRC10 may not affect Cav1.2 channel trafficking, it could still modulate the gating of the channels. Dysregulation of calcium homeostasis by genetic mutations or alteration in the function, localization, and expression of calcium‐handling proteins has been implicated in many cardiac diseases such as pathological cardiac hypertrophy, heart failure, and DCM.19, 51, 52, 53, 54, 55 Although the impact of loss of Lrrc10 in the Lrrc10 −/− mouse on ICa,L is more subtle, there is a consistent decrease in ICa,L in the knockouts. Furthermore, the distinct complement of regulatory proteins, subunits, and posttranslational modifications of LTCCs could contribute to the differences between the effects in heterologously expressed channels and those in native cardiomyocytes.
There is precedence for LRRC proteins acting as modulators of ion channel function, and specifically, a subset of LRRC proteins have been defined as γ subunits for large conductance, calcium, and voltage‐activated potassium (BK) channels. The effects of the LRRC proteins on channel function can be striking, such as LRRC26 causing a ≈140‐mV negative shift in the voltage dependence of activation of BK channels.10 The LRRC26 paralogues, LRRC52, LRRC55, and LRRC38 have also been identified as BK‐γ subunits.11, 12, 56 In addition, LRRC52 has also been demonstrated to be a regulatory protein for Slo3 channels in sperm cells.57 The BK‐γ subunit LRRCs are composed of an intracellular C‐terminus, a transmembrane segment and an extracellular LRRC region.11 However, LRRC10 is an intracellular protein without a predicted transmembrane domain, making it more comparable to cytoplasmic Cavβ auxiliary subunits. Furthermore, LRRC10's localization near the Z‐line in adult myocytes is consistent with it acting as a subunit of dyadic localized LTCCs.
LRR motifs are known to facilitate strong protein–protein interactions. Our co‐IP and Western blot experiments in transiently transfected HEK293 cells demonstrate an association of LRRC10 and the Cav1.2 channel subunit. Co‐IP experiments using WT mouse heart lysates further confirm an association between Cav1.2 and LRRC10 in native tissue. However, the direct binding partner for LRRC10 in the Cav1.2 macromolecular complex in the heart requires further investigation. How does it affect LTCC gating? In addition, it is also possible that LRRC10 has other binding partners and functions besides regulating LTCCs in cardiomyocytes that have yet to be determined. Overall, the role of LRRC10 in cardiac function and DCM is only beginning to be deciphered, and a new link between LRRC10 and LTCCs provides new avenues for understanding.
Sources of Funding
This work was supported by National Institutes of Health grants R01 HL078878 (Kamp), R01 HL071225 (Olson), and predoctoral training program in molecular pharmacology T32GM072474 (Long) and by American Heart Association grants AHA 12GRNT12070021 (Lee), predoctoral AHA‐Midwest affiliate fellowship 14PRE20460194 (Woon), and predoctoral AHA fellowship 14PRE1807007 (Long).
Disclosures
None.
Acknowledgments
We thank the family that participated in this study.
(J Am Heart Assoc. 2018;7:e006428 DOI: 10.1161/JAHA.117.006428.)29431102
Contributor Information
Timothy M. Olson, Email: olson.timothy@mayo.edu.
Timothy J. Kamp, Email: tjk@medicine.wisc.edu.
References
- 1. Dipchand AI, Kirk R, Edwards LB, Kucheryavaya AY, Benden C, Christie JD, Dobbels F, Lund LH, Rahmel AO, Yusen RD, Stehlik J; International Society for Heart and Lung Transplantation . The Registry of the International Society for Heart and Lung Transplantation: sixteenth official pediatric heart transplantation report—2013; focus theme: age. J Heart Lung Transplant. 2013;32:979–988. [DOI] [PubMed] [Google Scholar]
- 2. Lund LH, Edwards LB, Kucheryavaya AY, Dipchand AI, Benden C, Christie JD, Dobbels F, Kirk R, Rahmel AO, Yusen RD, Stehlik J; International Society for Heart and Lung Transplantation . The Registry of the International Society for Heart and Lung Transplantation: thirtieth official adult heart transplant report—2013; focus theme: age. J Heart Lung Transplant. 2013;32:951–964. [DOI] [PubMed] [Google Scholar]
- 3. Michels VV, Moll PP, Miller FA, Tajik AJ, Chu JS, Driscoll DJ, Burnett JC, Rodeheffer RJ, Chesebro JH, Tazelaar HD. The frequency of familial dilated cardiomyopathy in a series of patients with idiopathic dilated cardiomyopathy. N Engl J Med. 1992;326:77–82. [DOI] [PubMed] [Google Scholar]
- 4. Baig MK, Goldman JH, Caforio AL, Coonar AS, Keeling PJ, McKenna WJ. Familial dilated cardiomyopathy: cardiac abnormalities are common in asymptomatic relatives and may represent early disease. J Am Coll Cardiol. 1998;31:195–201. [DOI] [PubMed] [Google Scholar]
- 5. Grünig E, Tasman JA, Kücherer H, Franz W, Kübler W, Katus HA. Frequency and phenotypes of familial dilated cardiomyopathy. J Am Coll Cardiol. 1998;31:186–194. [DOI] [PubMed] [Google Scholar]
- 6. Hershberger RE, Hedges DJ, Morales A. Dilated cardiomyopathy: the complexity of a diverse genetic architecture. Nat Rev Cardiol. 2013;10:531–547. [DOI] [PubMed] [Google Scholar]
- 7. Qu XK, Yuan F, Li RG, Xu L, Jing WF, Liu H, Xu YJ, Zhang M, Liu X, Fang WY, Yang YQ, Qiu XB. Prevalence and spectrum of LRRC10 mutations associated with idiopathic dilated cardiomyopathy. Mol Med Rep. 2015;12:3718–3724. [DOI] [PubMed] [Google Scholar]
- 8. Kobe B, Deisenhofer J. The leucine‐rich repeat: a versatile binding motif. Trends Biochem Sci. 1994;19:415–421. [DOI] [PubMed] [Google Scholar]
- 9. Kajava AV, Kobe B. Assessment of the ability to model proteins with leucine‐rich repeats in light of the latest structural information. Protein Sci. 2002;11:1082–1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Yan J, Aldrich RW. LRRC26 auxiliary protein allows BK channel activation at resting voltage without calcium. Nature. 2010;466:513–516. [DOI] [PubMed] [Google Scholar]
- 11. Zhang J, Yan J. Regulation of BK channels by auxiliary γ subunits. Front Physiol. 2014;5:401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Li Q, Yan J. Modulation of BK channel function by auxiliary beta and gamma subunits. Int Rev Neurobiol. 2016;128:51–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Evanson KW, Bannister JP, Leo MD, Jaggar JH. LRRC26 is a functional BK channel auxiliary γ subunit in arterial smooth muscle cells. Circ Res. 2014;115:423–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Nakane T, Satoh T, Inada Y, Nakayama J, Itoh F, Chiba S. Molecular cloning and expression of HRLRRP, a novel heart‐restricted leucine‐rich repeat protein. Biochem Biophys Res Commun. 2004;314:1086–1092. [DOI] [PubMed] [Google Scholar]
- 15. Adameyko II, Mudry RE, Houston‐Cummings NRM, Veselov AP, Gregorio CC, Tevosian SG. Expression and regulation of mouse SERDIN1, a highly conserved cardiac‐specific leucine‐rich repeat protein. Dev Dyn. 2005;233:540–552. [DOI] [PubMed] [Google Scholar]
- 16. Kim KH, Kim TG, Micales BK, Lyons GE, Lee Y. Dynamic expression patterns of leucine‐rich repeat containing protein 10 in the heart. Dev Dyn. 2007;236:2225–2234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kim KH, Antkiewicz DS, Yan L, Eliceiri KW, Heideman W, Peterson RE, Lee Y. Lrrc10 is required for early heart development and function in zebrafish. Dev Biol. 2007;308:494–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Brody MJ, Hacker TA, Patel JR, Feng L, Sadoshima J, Tevosian SG, Balijepalli RC, Moss RL, Lee Y. Ablation of the cardiac‐specific gene leucine‐rich repeat containing 10 (Lrrc10) results in dilated cardiomyopathy. PLoS One. 2012;7:e51621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Brody MJ, Lee Y. The role of leucine‐rich repeat containing protein 10 (LRRC10) in dilated cardiomyopathy. Front Physiol. 2016;7:337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, DePristo MA. The Genome Analysis Toolkit: a MapReduce framework for analyzing next‐generation DNA sequencing data. Genome Res. 2010;20:1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, Philippakis AA, del Angel G, Rivas MA, Hanna M, McKenna A, Fennell TJ, Kernytsky AM, Sivachenko AY, Cibulskis K, Gabriel SB, Altshuler D, Daly MJ. A framework for variation discovery and genotyping using next‐generation DNA sequencing data. Nat Genet. 2011;43:491–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. 1000 Genomes Project Consortium , Abecasis GR, Auton A, Brooks LD, DePristo MA, Durbin RM, Handsaker RE, Kang HM, Marth GT, McVean GA. An integrated map of genetic variation from 1,092 human genomes. Nature. 2012;491:56–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7:248–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non‐synonymous variants on protein function using the SIFT algorithm. Nat Protoc. 2009;4:1073–1081. [DOI] [PubMed] [Google Scholar]
- 25. GTEx Consortium . The genotype‐tissue expression (GTEx) project. Nat Genet. 2013;45:580–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Manuylov NL, Manuylova E, Avdoshina V, Tevosian S. Serdin1/Lrrc10 is dispensable for mouse development. Genesis. 2008;46:441–446. [DOI] [PubMed] [Google Scholar]
- 27. Benndorf K, Boldt W, Nilius B. Sodium current in single myocardial mouse cells. Pflugers Arch. 1985;404:190–196. [DOI] [PubMed] [Google Scholar]
- 28. Wagoner LE, Zhao L, Bishop DK, Chan S, Xu S, Barry WH. Lysis of adult ventricular myocytes by cells infiltrating rejecting murine cardiac allografts. Circulation. 1996;93:111–119. [DOI] [PubMed] [Google Scholar]
- 29. Balijepalli RC, Foell JD, Hall DD, Hell JW, Kamp TJ. Localization of cardiac L‐type Ca(2+) channels to a caveolar macromolecular signaling complex is required for beta(2)‐adrenergic regulation. Proc Natl Acad Sci USA. 2006;103:7500–7505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Foell JD, Balijepalli RC, Delisle BP, Yunker AMR, Robia SL, Walker JW, McEnery MW, January CT, Kamp TJ. Molecular heterogeneity of calcium channel beta‐subunits in canine and human heart: evidence for differential subcellular localization. Physiol Genomics. 2004;17:183–200. [DOI] [PubMed] [Google Scholar]
- 31. Mikami A, Imoto K, Tanabe T, Niidome T, Mori Y, Takeshima H, Narumiya S, Numa S. Primary structure and functional expression of the cardiac dihydropyridine‐sensitive calcium channel. Nature. 1989;340:230–233. [DOI] [PubMed] [Google Scholar]
- 32. Subramanyam P, Chang DD, Fang K, Xie W, Marks AR, Colecraft HM. Manipulating L‐type calcium channels in cardiomyocytes using split‐intein protein transsplicing. Proc Natl Acad Sci USA. 2013;110:15461–15466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Best JM, Foell JD, Buss CR, Delisle BP, Balijepalli RC, January CT, Kamp TJ. Small GTPase Rab11b regulates degradation of surface membrane L‐type Cav1.2 channels. Am J Physiol Cell Physiol. 2011;300:C1023–C1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Reilly L, Howie J, Wypijewski K, Ashford MLJ, Hilgemann DW, Fuller W. Palmitoylation of the Na/Ca exchanger cytoplasmic loop controls its inactivation and internalization during stress signaling. FASEB J. 2015;29:4532–4543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Cohen RM, Foell JD, Balijepalli RC, Shah V, Hell JW, Kamp TJ. Unique modulation of L‐type Ca2+ channels by short auxiliary beta1d subunit present in cardiac muscle. Am J Physiol Heart Circ Physiol. 2005;288:H2363–H2374. [DOI] [PubMed] [Google Scholar]
- 36. Izumiyama T, Minoshima S, Yoshida T, Shimizu N. A novel big protein TPRBK possessing 25 units of TPR motif is essential for the progress of mitosis and cytokinesis. Gene. 2012;511:202–217. [DOI] [PubMed] [Google Scholar]
- 37. Collard F, Stroobant V, Lamosa P, Kapanda CN, Lambert DM, Muccioli GG, Poupaert JH, Opperdoes F, Van Schaftingen E. Molecular identification of N‐acetylaspartylglutamate synthase and beta‐citrylglutamate synthase. J Biol Chem. 2010;285:29826–29833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lodder‐Gadaczek J, Becker I, Gieselmann V, Wang‐Eckhardt L, Eckhardt M. N‐acetylaspartylglutamate synthetase II synthesizes N‐acetylaspartylglutamylglutamate. J Biol Chem. 2011;286:16693–16706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Gonçalves CM, Castro MAA, Henriques T, Oliveira MI, Pinheiro HC, Oliveira C, Sreenu VB, Evans EJ, Davis SJ, Moreira A, Carmo AM. Molecular cloning and analysis of SSc5D, a new member of the scavenger receptor cysteine‐rich superfamily. Mol Immunol. 2009;46:2585–2596. [DOI] [PubMed] [Google Scholar]
- 40. Matsushima N, Miyashita H. Leucine‐rich repeat (LRR) domains containing intervening motifs in plants. Biomolecules. 2012;2:288–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Long PA, Larsen BT, Evans JM, Olson TM. Exome sequencing identifies pathogenic and modifier mutations in a child with sporadic dilated cardiomyopathy. J Am Heart Assoc. 2015;4:e002443 DOI: 10.1161/JAHA.115.002443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Zhou B, Rao L, Peng Y, Wang Y, Chen Y, Song Y, Zhang L. Common genetic polymorphisms in pre‐microRNAs were associated with increased risk of dilated cardiomyopathy. Clin Chim Acta. 2010;411:1287–1290. [DOI] [PubMed] [Google Scholar]
- 43. Hell JW, Yokoyama CT, Wong ST, Warner C, Snutch TP, Catterall WA. Differential phosphorylation of two size forms of the neuronal class C L‐type calcium channel alpha 1 subunit. J Biol Chem. 1993;268:19451–19457. [PubMed] [Google Scholar]
- 44. Murphy BJ, Rossie S, De Jongh KS, Catterall WA. Identification of the sites of selective phosphorylation and dephosphorylation of the rat brain Na+ channel alpha subunit by cAMP‐dependent protein kinase and phosphoprotein phosphatases. J Biol Chem. 1993;268:27355–27362. [PubMed] [Google Scholar]
- 45. Gao T, Cuadra AE, Ma H, Bunemann M, Gerhardstein BL, Cheng T, Eick RT, Hosey MM. C‐terminal fragments of the alpha 1C (CaV1.2) subunit associate with and regulate L‐type calcium channels containing C‐terminal‐truncated alpha 1C subunits. J Biol Chem. 2001;276:21089–21097. [DOI] [PubMed] [Google Scholar]
- 46. Gerhardstein BL, Gao T, Bünemann M, Puri TS, Adair A, Ma H, Hosey MM. Proteolytic processing of the C terminus of the alpha(1C) subunit of L‐type calcium channels and the role of a proline‐rich domain in membrane tethering of proteolytic fragments. J Biol Chem. 2000;275:8556–8563. [DOI] [PubMed] [Google Scholar]
- 47. Brody MJ, Feng L, Grimes AC, Hacker TA, Olson TM, Kamp TJ, Balijepalli RC, Lee Y. LRRC10 is required to maintain cardiac function in response to pressure overload. Am J Physiol Heart Circ Physiol. 2016;310:H269–H278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Theis JL, Sharpe KM, Matsumoto ME, Chai HS, Nair AA, Theis JD, de Andrade M, Wieben ED, Michels VV, Olson TM. Homozygosity mapping and exome sequencing reveal GATAD1 mutation in autosomal recessive dilated cardiomyopathy. Circ Cardiovasc Genet. 2011;4:585–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Dorn GW, Matkovich SJ, Eschenbacher WH, Zhang Y. A human 3’ miR‐499 mutation alters cardiac mRNA targeting and function. Circ Res. 2012;110:958–967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Ling TY, Wang XL, Chai Q, Lu T, Stulak JM, Joyce LD, Daly RC, Greason KL, Wu LQ, Shen WK, Cha YM, Lee HC. Regulation of cardiac CACNB2 by microRNA‐499: potential role in atrial fibrillation. BBA Clin. 2017;7:78–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Heineke J, Molkentin JD. Regulation of cardiac hypertrophy by intracellular signalling pathways. Nat Rev Mol Cell Biol. 2006;7:589–600. [DOI] [PubMed] [Google Scholar]
- 52. Haase H, Kresse A, Hohaus A, Schulte HD, Maier M, Osterziel KJ, Lange PE, Morano I. Expression of calcium channel subunits in the normal and diseased human myocardium. J Mol Med (Berl). 1996;74:99–104. [DOI] [PubMed] [Google Scholar]
- 53. Bénitah JP, Gómez AM, Fauconnier J, Kerfant BG, Perrier E, Vassort G, Richard S. Voltage‐gated Ca2+ currents in the human pathophysiologic heart: a review. Basic Res Cardiol. 2002;97(suppl 1):I11–I18. [DOI] [PubMed] [Google Scholar]
- 54. Richard S, Leclercq F, Lemaire S, Piot C, Nargeot J. Ca2+ currents in compensated hypertrophy and heart failure. Cardiovasc Res. 1998;37:300–311. [DOI] [PubMed] [Google Scholar]
- 55. McNally EM, Golbus JR, Puckelwartz MJ. Genetic mutations and mechanisms in dilated cardiomyopathy. J Clin Invest. 2013;123:19–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Yan J, Aldrich RW. BK potassium channel modulation by leucine‐rich repeat‐containing proteins. Proc Natl Acad Sci USA. 2012;109:7917–7922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Yang C, Zeng XH, Zhou Y, Xia XM, Lingle CJ. LRRC52 (leucine‐rich‐repeat‐containing protein 52), a testis‐specific auxiliary subunit of the alkalization‐activated Slo3 channel. Proc Natl Acad Sci USA. 2011;108:19419–19424. [DOI] [PMC free article] [PubMed] [Google Scholar]