

Abstract
Background
A disintegrin‐like metalloproteinase with thrombospondin motif type 1 member 13 (ADAMTS13), the von Willebrand factor–cleaving enzyme, decreases leukocyte and platelet recruitment and, thus, reduces thrombosis and inflammation. Recombinant human ADAMTS13 (rhADAMTS13) is a novel drug candidate for ischemia/reperfusion injury and has shown short‐term benefits in mouse models of myocardial injury, but long‐term outcome has not been investigated.
Methods and Results
We evaluated the impact of rhADAMTS13 on cardiac remodeling, scarring, and contractile function, under chronic left ventricular pressure overload. The role of von Willebrand factor and the effect of rhADAMTS13 treatment were studied. This model of heart failure, based on ascending aortic constriction, produces a coronary inflammatory response and microvascular dysfunction, resulting in fibrotic remodeling and cardiac failure. Mice were treated with either rhADAMTS13 or vehicle and assessed for coronary vascular inflammation and ventricular function at several postsurgical time points, as well as for cardiac fibrosis after 4 weeks. Early upon induction of pressure overload under rhADAMTS13 treatment, we detected less endothelial‐lumen–associated von Willebrand factor, fewer platelet aggregates, and decreased activated transforming growth factor‐β1 levels than in vehicle‐treated mice. We observed significant preservation of cardiac function and decrease in fibrotic remodeling as a result of rhADAMTS13 administration.
Conclusions
Herein, we show that rhADAMTS13 decreases coronary vascular dysfunction and improves cardiac remodeling after left ventricular pressure overload in mice. We propose that this effect may, at least in part, be the result of decreased von Willebrand factor–mediated recruitment of platelets, a major source of the activated profibrotic cytokine transforming growth factor‐β1. Our study further supports the therapeutic potential of rhADAMTS13 for conditions characterized by inflammatory cardiac damage that results in fibrosis.
Keywords: ADAMTS13, cardiac remodeling, fibrosis, heart failure, von Willebrand factor
Subject Categories: Heart Failure, Fibrosis, Remodeling, Animal Models of Human Disease, Basic Science Research
Clinical Perspective
What Is New?
Recombinant human a disintegrin‐like metalloproteinase with thrombospondin motif type 1 member 13 (ADAMTS13) treatment, by preventing inflammation, platelet recruitment, and microvessel obstruction, improves cardiac functionality and reduces fibrotic remodeling after pressure overload injury in mice.
What Are the Clinical Implications?
ADAMTS13 should be investigated as a treatment option for conditions characterized by coronary microvessel dysfunction.
Introduction
During a lifetime, the heart may be exposed to acute and chronic injuries that result in myocardial cell death, fibrotic remodeling, and subsequent heart failure, which bears substantial morbidity and mortality.1 Myocardial infarction is a common acute event causing ischemic damage of the affected heart muscle. The urgent need for interventional restoration of blood flow to the affected myocardial region leads to reperfusion injury. This is characterized by cardiomyocyte swelling, platelet microthrombi, release of soluble thrombogenic substances, and neutrophil plugging of the microvasculature.2 Thus, myocardial ischemia/reperfusion injury contributes substantially to the overall damage after myocardial infarction.2 Other injuries leading to unfavorable cardiac remodeling are more chronic, yet they present a significant health problem in the elderly population. Pressure overload to the left ventricle attributable to arterial hypertension or aortic valve stenosis over time results in myocardial hypertrophy, fibrotic remodeling, ventricular stiffening, and subsequent congestive heart failure.3 The role of the von Willebrand factor (VWF)–a disintegrin‐like metalloproteinase with thrombospondin motif type 1 member 13 (ADAMTS13) axis in myocardial hypertrophy has not been investigated. In mouse models, myocardial ischemia/reperfusion injury and increased left ventricular (LV) pressure overload are both associated with coronary vessel inflammation, myocardial leukocyte infiltration,4, 5, 6 and, eventually, fibrotic remodeling.7, 8
Inflammatory or ischemic endothelial activation results in release of ultralarge VWF from Weibel‐Palade bodies.9 The presence of ultralarge VWF at the release site is controlled by the endogenous metalloproteinase ADAMTS13, which specifically cleaves VWF and, thus, decreases its activity. Deficiency of this enzyme is associated with thrombotic thrombocytopenic purpura (TTP),10 and low activity likely contributes to other microangiopathies. Moreover, in past studies, our group and others have shown that recombinant human ADAMTS13 (rhADAMTS13) reduces infarct size and improves heart function in mice 24 hours after myocardial ischemia/reperfusion injury. This was VWF mediated11 and at least partially attributed to a decrease in myocardial leukocyte infiltration.12, 13 This is further supported by a study that showed enhanced general leukocyte recruitment in ADAMTS13−/− mice.14 ADAMTS13 was also shown to reduce VWF‐mediated acute inflammation and postischemic hypoperfusion after stroke.15, 16, 17 Given the evidence of the anti‐inflammatory and protective role of rhADAMTS13, we aimed to assess the role of VWF and that of ADAMTS13 on long‐term cardiac remodeling and function after chronic cardiac injury. For the latter, we chose a mouse model of ascending aortic constriction (AAC) leading to LV pressure overload, coronary vessel inflammation, and severe fibrotic remodeling.
Methods
The data, analytic methods, and study materials, with the exception of rhADAMTS13, provided as a gift by Baxalta, will be/have been made available to other researchers for purposes of reproducing the results or replicating the procedure on request. rhADAMTS13 is commercially available from other sources.
Mice
Wild‐type (WT) C57Bl/6J mice (purchased from the Jackson Laboratory) were 9 to 10 weeks old at the time of AAC or 3 to 4 weeks old when used for intravital microscopy. ADAMTS13−/− mice were bred in‐house at Children's Hospital and routinely backcrossed to a C57BL/6J background. For angiotensin II (AngII) infusion experiments, C57BL/6, VWF+/+, and VWF−/− mice were bred in‐house at KU Leuven and were 8 to 17 weeks old when implanted with osmotic pumps. Mice for AngII infusion experiments were always weight matched across groups in each experiment. All experimental procedures were reviewed and approved by the Institutional Animal Care and Use Committee of Boston Children's Hospital (protocol no. 14‐02‐2609R) or KU Leuven (protocol no. P185/2013). All surgically operated mice received 0.1 mg/kg of buprenorphine before surgery and every 12 hours subsequently for the first 72 hours.
rhADAMTS13 Treatment
Treatment with rhADAMTS13 (3460 U/kg) or vehicle (0.9% NaCl) was performed daily for 1 week in mice after AAC, or after osmotic pump implantation, via retro‐orbital intravenous administration. Preparation of rhADAMTS13 has been previously described.12
Intravital Microscopy
ADAMTS13−/− male mice, 3 to 4 weeks old, were anesthetized by an intraperitoneal injection of 2.5% (vol/vol) tribromoethanol (300 mg/kg). Rhodamine 6G was given via retro‐orbital injection to fluorescently label platelets and leukocytes. A midline incision was performed, and the intestines were externalized to reveal mesenteric vessels. A single venule between 200 and 300 μm in diameter was visualized for each mouse and assessed for platelet string formation in venules without treatment (5 minutes) and after retro‐orbital injection of 3460 U/kg rhADAMTS13 (10 minutes). WT mice, 3 to 4 weeks old, were injected with vehicle or rhADAMTS13 and assessed for baseline leukocyte rolling, as previously described.18
VWF Antigen Assay
Medisorp microtiter plates (Nunc) were coated with a rabbit polyclonal anti–VWF‐Ig solution (1:1000; Dako) overnight at 4°C. The plates were washed 3 times with PBS containing 0.5% Tween‐20. The wells were blocked using 3% milk in PBS for 2 hours at room temperature (RT). Serial dilutions of citrate‐anticoagulated plasma samples and a normal plasma pool from at least 10 WT mice were incubated for 1.5 hours at 37°C, followed by 9 washes. A pool from at least 10 VWF−/− mice was used as a negative control. HRP‐conjugated anti–VWF‐Ig (1:3000; Dako) was added and incubated for 1 hour at RT, followed by 9 washes. The substrate 3,3,5,5‐tetramethylbenzidine (Sigma‐Aldrich) was applied, and the coloring reaction was stopped after 15 to 20 minutes with 0.5 mol/L H2SO4. Absorbance was determined at 450 nm.
ADAMTS13 Activity Assay
Fluorescence resonance energy transfer substrate (FRETS)‐VWF73 (VWF residues 1596–1668) was used as a proteolytic substrate to determine the activity of ADAMTS13 in mouse plasma samples. Cleavage was performed according to the manufacturer's instructions. In brief, 4 μL of mouse plasma was diluted in reaction buffer (5 mmol/L bis‐tris, 25 mmol/L CaCl2, and 0.005% Tween‐20, pH 6) in a 96‐well plate. The reaction was initiated by the addition of an equal volume of 4 μmol/L VWF‐73 substrate solution (Peptanova). Fluorescence was measured at 30°C at 1‐minute intervals for 1 hour using the Synergy Neo microplate reader (Biotex). Results were compared with normal pooled mouse plasma (plasma from 20 WT mice) and expressed as the average of activity curves.
Immunohistochemistry
Formalin‐fixed hearts were embedded in paraffin and sectioned at 10‐μm thickness using a microtome. Sections were deparaffinized and washed, permeabilized using 0.05% Triton X‐100, and incubated with Bloxall Endogenous Peroxidase and Alkaline Phosphatase Blocking solution (Vector Laboratories) for 10 minutes at RT. After blocking with 10% normal rabbit or goat serum for 30 minutes at RT, sections were incubated with anti‐VWF antibody (0.3 g/mL; Dako) or anti‐mouse intercellular adhesion molecule‐1 (ICAM‐1) antibody (YN1/1.7.4; 2.5 μg/mL; Biolegend) overnight. The VectaStain ABC‐AP kit, followed by Vector Red Alkaline Phosphatase Substrate (Vector Laboratories), was used according to manufacturer's instructions. Nuclei were counterstained using Methyl Green (Vector Laboratories), and then sections were washed and cleared for mounting with Permount. Fields of view (5–10) were captured using a Zeiss Axioplan 2 bright‐field microscope. VWF was quantified as the percentage positive staining of total tissue area using ImageJ software (National Institutes of Health). ICAM quantification was performed by an investigator (T.W.) blinded to the identity of the samples using a scoring system of 0 to 4, with 0 defined as no ICAM‐1 staining; 1, minimal ICAM‐1 staining in few vessels; 2, moderate ICAM‐1 staining in fewer than half of vessels; 3, intense ICAM‐1 staining in more than half of vessels; and 4, intense ICAM‐1 staining in all vessels.
Immunofluorescence
Perfused hearts were snap frozen in optimal cutting temperature medium and cryosectioned at 8‐ to 10‐μm thickness. After fixation with 2% paraformaldehyde, sections were blocked in 3% bovine serum albumin, and then incubated with anti‐CD41 (clone MWReg30; 0.5 μg/mL; Biolegend) or anti‐myeloperoxidase (0.32 μg/mL; Dako) antibodies overnight at 4°C. After washing, samples were incubated with Alexa555‐conjugated anti‐rat IgG or Alexa488‐conjugated anti‐rabbit IgG (1.5 μg/mL) for 2 hours at RT. Sections were then washed, and DNA was counterstained with Hoechst 33342 (1:10 000) for 10 minutes before mounting with antifade Fluorogel mounting medium (Electron Microscopy Sciences). Images were acquired using a Zeiss Axiovert 200 inverted wide‐field fluorescence microscope coupled to an Axiocam MRm monochromatic charge‐coupled device camera. Channels were acquired in grayscale and pseudocolored with Zeiss Axiovision software.
Induction of AAC
Mice were anesthetized with 3.5% isoflurane, and anesthesia was maintained at 1.5% to 2% in 100% oxygen. Mice underwent either sham operation or AAC, as described previously.19 Briefly, the chest was opened at the second to third intercostal space, and the aortic arch was visualized. A 26G needle was placed adjacent to the aorta, and a suture was tied around both the needle and aorta. The needle was then removed, leaving a 26G opening in the aorta and a resultant significant stenosis. Finally, the chest was closed and the animal was weaned off the respirator as soon as spontaneous breathing had stabilized.
Echocardiography
Cardiac function and heart dimensions were measured in male WT mice that underwent sham surgery (control) and in mice subjected to AAC, followed by treatment. Images were obtained 3 days, 1 week, and 4 weeks after surgery using the Vevo 2100 (Visual Sonics) ultrasound at the Small Animal Imaging Laboratory at Boston Children's Hospital. The mice were anesthetized using 1.5% isoflurane, and 4 limbs were attached to the corresponding ECG electrodes. M mode of the parasternal short‐axis view was used to evaluate LV internal dimension, LV interventricular septum, and LV posterior wall at end diastole and end systole. Echocardiograms were stored digitally, and ejection fraction (percentage) was calculated using Vevostrain software. An investigator (T.W.) blinded to the study groups performed echocardiography and data analysis.
Masson Trichrome Staining
At euthanasia (4 weeks after surgery or after osmotic pump implantation), heart tissue was prepared for histology. To assess collagen content in heart tissue, the Masson trichrome stain kit (Sigma) was used, according to the manufacturer's protocol. Nuclei were not stained with hematoxylin to avoid interference with the quantification of collagen content. For quantification, pictures of 3 whole cross sections were taken by bright‐field microscopy by an investigator (T.W. and K.M.) blinded to the identity of the samples, using either a Zeiss Axioplan microscope coupled with a Zeiss AxioCam HRc camera or a Hamamatsu Nanozoomer RS. The area of blue fibers (collagen) per heart tissue was calculated using ImageJ software.
AngII Infusion Model
Osmotic pumps (Alzet minipumps; model 2004) were loaded with AngII (Sigma), implanted subcutaneously in the intrascapular area, and left implanted for 28 days. These pumps released 0.25 μL/h for 28 days, resulting in a dose of 1000 ng/kg per minute of AngII. Control mice were implanted with saline‐filled pumps.
Statistical Analysis
Data are expressed as mean±SEM or medians with interquartile range (IQR), as indicated. Data were analyzed using a 2‐sided Student t test or by Mann‐Whitney U test, where indicated. A 1‐way ANOVA with a Holm‐Sidak multiple‐comparisons test with multiplicity‐adjusted P values was used when multiple measurements were averaged per mouse, as indicated. Statistical analysis was performed using Prism, version 5.0b or 6.0 (GraphPad Software Inc). P<0.05 was considered statistically significant.
Results
AAC Leads to Increased VWF Secretion and Platelet and Leukocyte Abundance in the Myocardium
We found increased VWF staining in hearts of mice 3 days after AAC compared with sham–operated mice (Figure 1A). Small‐vessel surroundings were heavily stained, suggesting a strong Weibel‐Palade body release response. Even small‐vessel thrombosis was observed (black arrows). We also observed significantly elevated VWF plasma levels (elevated by 81%) by ELISA (data not shown) in mice that underwent AAC compared with sham 3 days after surgery. As platelets bind to VWF,20 we performed immunofluorescent staining of the myocardium for platelets using anti‐CD41 (integrin α IIb) antibody. We found significantly more platelet aggregates in hearts that underwent AAC compared with sham–operated mice (Figure 1B). Within vessels, we observed leukocytes, mostly neutrophils, and platelets aligning along the vessel wall as proof of endothelial activation by AAC (Figure 1C). We also observed significantly increased myocardial neutrophil infiltration caused by afterload stress, as quantified by fluorescence‐activated cell sorting analysis of dissociated heart tissue at postsurgical day 3 (Figure 1C). Single cells that were positive for Ly6G, CD11b, and CD45 were identified as neutrophils. The extensive release of VWF and cellular recruitment confirmed our interest in the effect of rhADAMTS13 treatment on hearts exposed to increased afterload stress.
Figure 1.
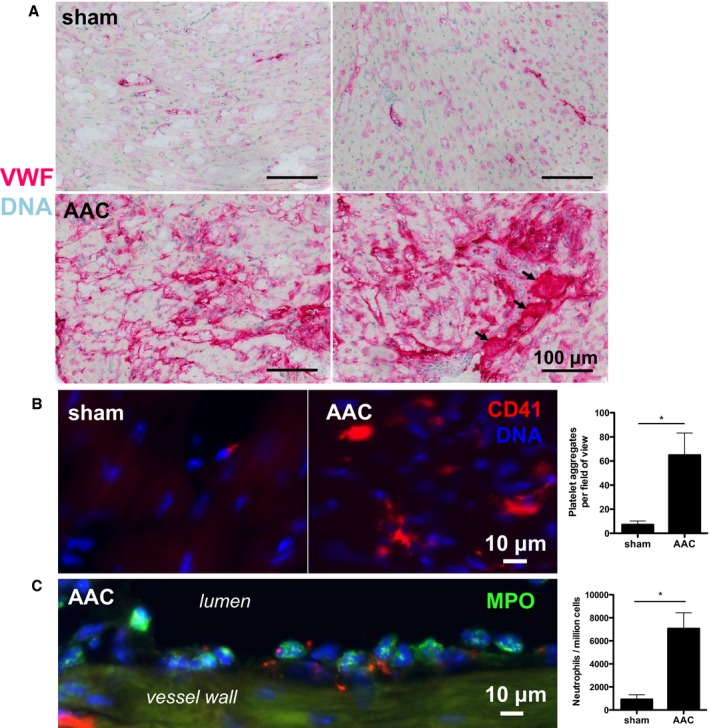
Ascending aortic constriction (AAC) increases endothelial von Willebrand factor (VWF) secretion and inflammatory cell recruitment in coronary vessels. A, Immunohistochemical staining of VWF in the myocardium in mice that underwent AAC or sham surgery 3 days earlier. Each image depicts a representative myocardial left ventricular portion of 1 mouse. VWF staining was clearly stronger in myocardium exposed to afterload stress by AAC. The black arrows point to vessels that are filled with VWF‐rich material, indicating possible microvessel thrombosis. B, The myocardium of these mice was stained for platelets using immunofluorescent anti‐CD41 antibodies. Platelet aggregates in the tissue were quantified by an investigator blinded to treatment in 5 fields of view per mouse. Afterload‐stressed hearts (n=5) had significantly more myocardial platelet accumulation than control hearts (n=4). C, Representative image showing myeloperoxidase‐positive (MPO+) cells (green) and CD41+ platelets (red) accumulating along the myocardial vessel wall 3 days after AAC, showing the inflammatory response induced by the model. Myocardial neutrophil (Ly6G+, CD11b+, and CD45+ cells) infiltration was significantly increased in hearts that underwent AAC (n=4) compared with sham–operated hearts (n=3), as assessed by flow cytometric analysis of single‐cell suspensions prepared from digested heart tissue. Significance was assessed by Mann‐Whitney U tests. *P<0.05.
rhADAMTS13 Works in Vivo in a Dose‐Dependent Manner
We used intravital microscopy of mesenteric venules in ADAMTS13−/− mice to test the efficacy of different doses of rhADAMTS13 in mice (Figure 2A and 2B). In ADAMTS13−/− mice, platelet‐string formation along ULVWF occurs on mesenteric exteriorization. A blinded investigator (T.W.) quantified the formation of new strings per minute before and after rhADAMTS13 treatment. The treatment led to a reduction in platelet‐string formation in a dose‐dependent manner. Reduction was highly significant at the highest dose of 3460 U/kg, whereas the lowest dose of 40 U/kg had no effect (Figure 2B).
Figure 2.
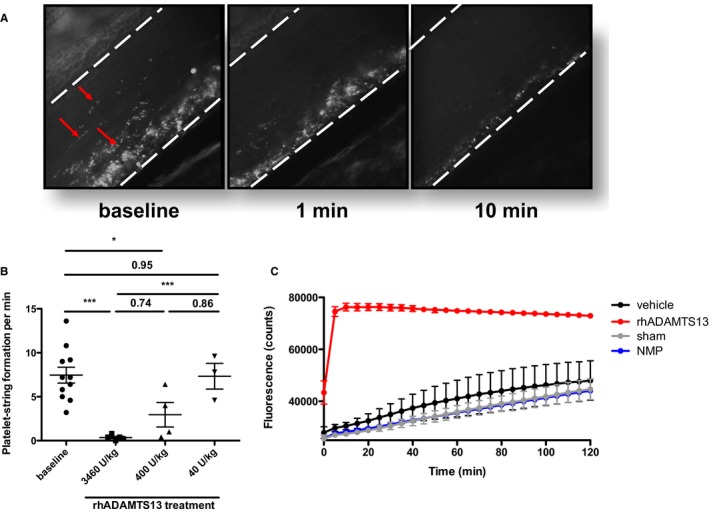
Infusion of recombinant human a disintegrin‐like metalloproteinase with thrombospondin motif type 1 member 13 (rhADAMTS13) has an immediate and dose‐dependent effect on platelet‐string formation in vivo. A, Mice deficient in ADAMTS13 (ADAMTS13−/−) were prepared for intravital microscopy of the mesenteric venules. Rhodamine 6G was infused to visualize platelets. Representative images of ADAMTS13−/− venules, forming visible platelet/von Willebrand factor (VWF) strings (left panel, strings indicated by red arrows). One minute after infusion of 3460 U/kg rhADAMTS13 in the same mouse, there were fewer platelet/VWF strings (middle panel); by 10 minutes, these were nearly entirely released from the vessel wall (right panel). B, Platelet‐strings observed per minute were quantified by a blinded investigator (T.W.) in ADAMTS13−/− mice before (5 minutes) and after (10 minutes) treatment with indicated doses of rhADAMTS13. A highly significant effect on platelet/VWF string formation could only be observed after treatment with 3460 U/kg rhADAMTS13 (n=5). Treatment with 400 U/kg (n=4) had a smaller, but still significant, effect, whereas 40 U/kg (n=3) had no effect. C, Plasma ADAMTS13 activity was measured by fluorescence resonance energy transfer substrate (FRETS)‐VWF73 assay in mice that underwent ascending aortic constriction after treatment with either rhADAMTS13 (red) or vehicle (black), or in sham–operated mice (gray), 24 hours after injection and operation. A strong increase in plasma ADAMTS13 activity, even 24 hours after retro‐orbital injection of rhADAMTS13, demonstrated the enhanced and systemic effect of the treatment with a high dose of rhADAMTS13. No decrease in ADAMTS13 activity was observed in vehicle‐treated mice that underwent AAC compared with sham–operated mice. Both vehicle‐treated and sham–operated mice had similar activity to a pool of normal mouse plasma (NMP; blue). *P<0.05, ***P<0.001.
FRETS assay was used to measure ADAMTS13 activity in plasma of mice that underwent sham operation or that underwent AAC and were treated with the highest dose of rhADAMTS13 or vehicle, 24 hours after surgery (Figure 2C). There was no decrease of ADAMTS13 activity in mice that underwent AAC compared with sham–operated mice, indicating that there was no relative ADAMTS13 deficiency induced by the model. ADAMTS13 activity was substantially increased in rhADAMTS13‐treated mice that underwent AAC compared with vehicle‐treated mice that underwent AAC or sham–operated mice, even 24 hours after injection of rhADAMTS13. The observational time was limited to 2 hours because FRETS‐VWF73 substrate is a human VWF sequence fragment. Therefore, endogenous mouse ADAMTS13 activity may be underestimated in our assay.
rhADAMTS13 Treatment Reduces Coronary VWF, Platelet‐Aggregate Formation, and Active Transforming Growth Factor‐β1 Release
To study possible effects of rhADAMTS13 administration on cell infiltration into the myocardium after AAC, we chose postsurgical day 1. At this early time point, we had previously observed that inflammatory cell recruitment was highest.21 Although we found no difference in leukocyte recruitment (data not shown), we saw a significant decrease of myocardial platelet‐aggregate abundance in mice that underwent AAC with subsequent rhADAMTS13 treatment compared with vehicle treatment (Figure 3A). We observed a decrease of VWF staining in rhADAMTS13‐treated hearts that underwent AAC to a level comparable to sham–operated mice (Figure 3B). Because platelets represent a large storage pool for the cytokine transforming growth factor‐β1 (TGF‐β1),22 we compared plasma levels by ELISA in mice that underwent sham or AAC surgery at postsurgical day 1. Although we found no difference in total TGF‐β1 levels, we made the striking observation that active TGF‐β1 presence was increased by AAC, but not detectable in mice that were treated with rhADAMTS13 (Figure 3C).
Figure 3.
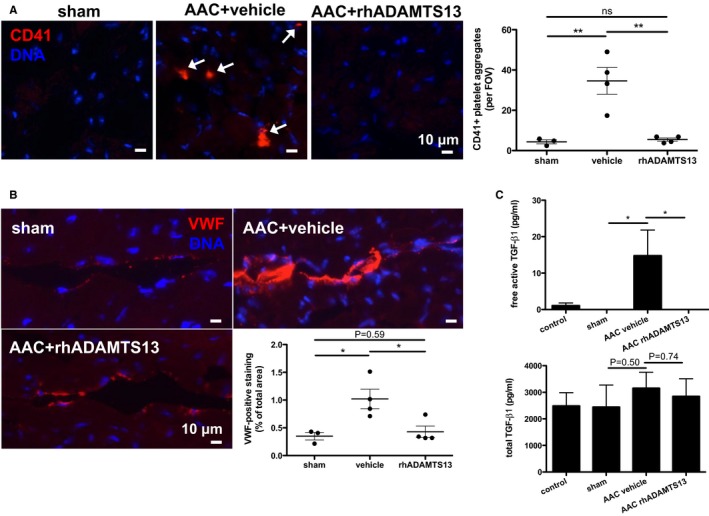
Recombinant human a disintegrin‐like metalloproteinase with thrombospondin motif type 1 member 13 (rhADAMTS13) treatment reduces platelet recruitment to the myocardium and plasma levels of active transforming growth factor (TGF)‐β1. Myocardial platelet aggregate accumulation (white arrows; A) and endothelial von Willebrand factor (VWF) adherence (VWF‐positive staining; B) 24 hours after surgery is significantly attenuated by high‐dose rhADAMTS13 treatment. Quantification was performed in 5 fields of view (FOVs) per mouse (sham, n=3; vehicle, n=4; rhADAMTS13, n=4) by an investigator (K.M.) blinded to treatment conditions. The number of platelet aggregates per field of view was averaged and, therefore, significance was assessed using a 1‐way ANOVA with Holm‐Sidak multiple‐comparisons test with multiplicity‐adjusted P values. VWF‐positive staining was summed and normalized to measured area and, therefore, expressed as a proportion of area. C, Plasma levels of total TGF‐β1 and the active form of TGF‐β1 were measured at day 1 after surgery by ELISA. Although total levels of TGF‐β1 were not different among the groups, the active form was significantly elevated in the ascending aortic constriction (AAC) vehicle‐treated group (n=4) compared with sham–operated (n=3) and unoperated control (n=5) groups. Active TGF‐β1 levels were lower than the detection limit in mice that underwent AAC and were given rhADAMTS13 (n=4). Mann‐Whitney U tests were performed to determine statistical significance. NS indicates not significant. *P<0.05, **P<0.01.
rhADAMTS13 Treatment Preserves Systolic Function and Reduces Cardiac Fibrosis in Mice Exposed to Chronic LV Pressure Overload
Mice were treated with rhADAMTS13 or vehicle for the first 7 days after AAC surgery. Aortic constriction resulted in LV wall motion disturbances, especially of the posterior wall (Figure 4A), and severe heart failure at day 3 after AAC, without a significant improvement with rhADAMTS13 injections (Figure 4B). However, by day 7, the rhADAMTS13‐treated group showed better recovery of contractility, resulting in a significantly better LV ejection fraction compared with the vehicle group (Figure 4B). After 4 weeks, the average LV ejection fraction of vehicle‐treated animals remained significantly worse than that of rhADAMTS13‐treated mice. No difference was found in left ventricle diameter between the groups (Figure 4C). Hearts that underwent banding showed hypertrophic walls compared with sham mice, confirming successful application of LV pressure overload (Figure 4D and 4E). The septal diameter of the left ventricle was bigger at 28 days in the vehicle group compared with the ADAMTS13 group (Figure 4D). No differences in posterior LV wall thickness were found between the 2 groups (Figure 4E). Heart weights were significantly elevated in both vehicle‐ and ADAMTS13‐treated mice that underwent AAC compared with sham–operated mice (sham median, 126.1 g [IQR, 111.8–141.2 g]; vehicle AAC median, 234.5 g [IQR, 204.8–288.4 g]; ADAMTS13 AAC median, 187.0 g [IQR, 175.3–228.1 g]; P=0.02 for sham versus vehicle AAC and P=0.006 for sham versus ADAMTS13 AAC by Mann‐Whitney U test). Although there was a trend toward decreased heart weight with ADAMTS13 treatment compared with vehicle treatment, this did not reach statistical significance (P=0.10 by Mann‐Whitney U test). Survival during the first 4 weeks after surgery was 45.5% for the vehicle group and 70% for rhADAMTS13 group. However, the difference did not reach significance (log‐rank test, P=0.08; data not shown).
Figure 4.
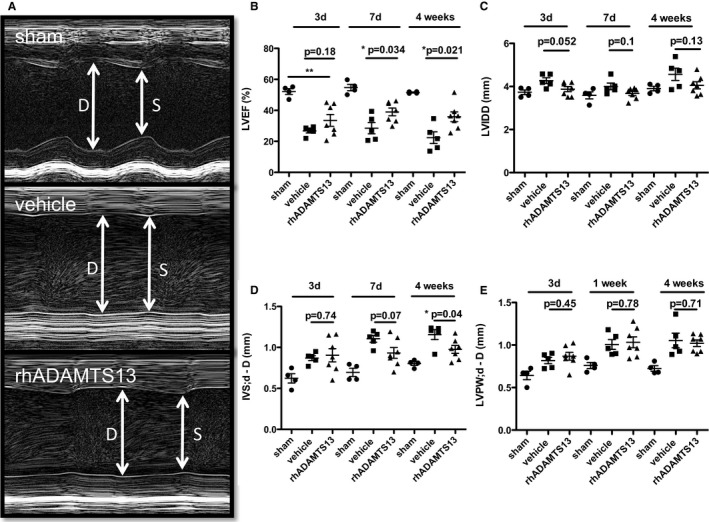
Recombinant human a disintegrin‐like metalloproteinase with thrombospondin motif type 1 member 13 (rhADAMTS13) treatment attenuates the left ventricular functional decline and hypertrophy resulting from chronic left ventricular (LV) pressure overload induced by ascending aortic constriction (AAC). A, Representative LV ejection fraction (LVEF) measurements in M‐mode echocardiography (sham, n=4; vehicle, n=5; rhADAMTS13, n=7). B, Mice that received rhADAMTS13 treatment for the first 7 days had significantly preserved left ventricular systolic function at 7 and 28 days. C, There was no significant difference in left ventricular diastolic diameters (LVDDs). D, Interventricular septum (IVS) hypertrophy was reduced in the rhADAMTS13 group compared with vehicle at 28 days. E, LV posterior wall (LVPW) hypertrophy was not different in the 2 groups that underwent AAC. d denotes diameter; D, denotes Diastole
Cardiac Endothelial Expression of ICAM‐1 Is Increased in Mice That Underwent AAC But Ameliorated by rhADAMTS13 Treatment
We performed histological analysis of the hearts from mice 28 days after surgery to assess endothelial activation and remodeling. ICAM‐1 was recently reported to be essential in this model for the inflammatory response leading to collagen deposition and fibrosis.23, 24 ICAM‐1 is upregulated in activated and dysfunctional endothelium,25 and we did confirm an increase in vehicle‐treated mice that underwent AAC compared with shams. Interestingly, substantial ICAM‐1 upregulation was not seen in rhADAMTS13‐treated animals in either coronary microvessels or the endocardium, indicating that endothelial cells may be less activated with rhADAMTS13 treatment in this model (Figure 5A). Differences in staining intensity in vehicle‐ versus rhADAMTS13‐treated mice were significant, as assessed by a blinded investigator's (T.W.) scoring system (median vehicle score, 4.0 [IQR, 3.0–4.0] [n=5]; median rhADAMTS13 score, 1.75 [IQR, 1.5–2.625] [n=6]; P=0.0164 by Wilcoxon rank‐sum test). In addition, Masson trichrome staining was used for quantification of collagen content within the heart. We observed significantly less collagen deposition (blue) in the rhADAMTS13 group compared with vehicle 28 days after AAC in midventricular and apical cross sections (Figure 5B). This difference in fibrosis likely contributes to the functional protection seen by rhADAMTS13 treatment.
Figure 5.
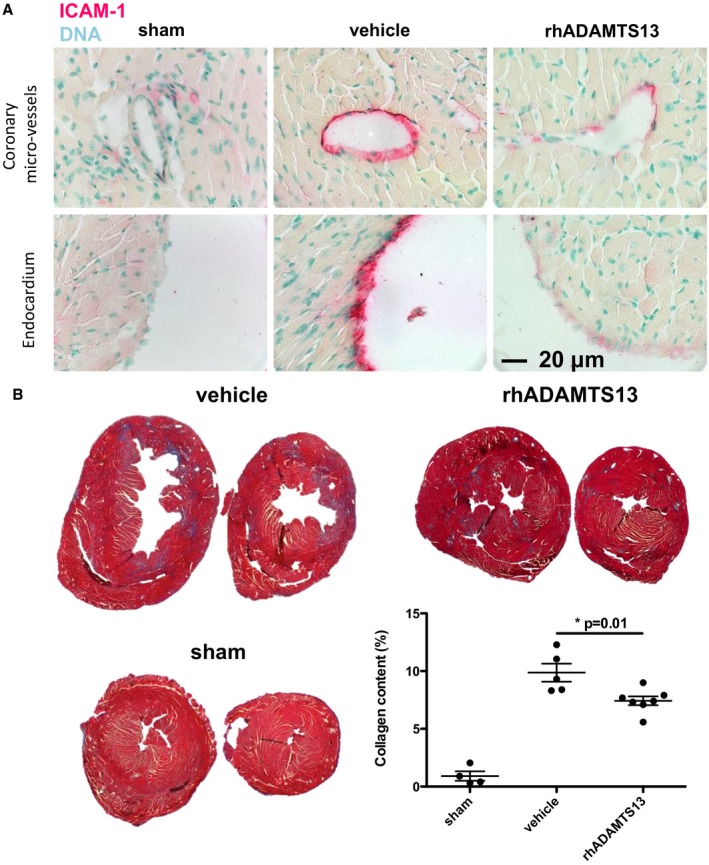
Fibrotic and inflammatory changes of the myocardium resulting from left ventricular pressure overload are decreased in mice treated with recombinant human a disintegrin‐like metalloproteinase with thrombospondin motif type 1 member 13 (rhADAMTS13). A, Immunohistochemical staining for intercellular adhesion molecule‐1 (ICAM‐1), a marker for endothelial activation, demonstrated stronger staining in coronary vessel endothelium and endocardium of vehicle‐treated mice compared with rhADAMTS13‐treated mice 28 days after ascending aortic constriction (AAC), suggesting an anti‐inflammatory effect of rhADAMTS13. B, Collagen deposition in the heart 28 days after AAC was significantly lower in rhADAMTS13‐ than vehicle‐treated mice (sham, n=4; vehicle, n=5; rhADAMTS13, n=7).
rhADAMTS13 Treatment or VWF Deficiency Protect From Coronary Perivascular Collagen Deposition Caused by AngII Infusion
Next, we wanted to investigate whether VWF, the substrate of ADAMTS13, promotes cardiac fibrosis during chronic pressure overload by using VWF−/− animals. Because the open‐chest surgical pressure overload model is not feasible in VWF−/− animals because of bleeding, we used a nonsurgical model of continuous infusion of AngII for 28 days. VWF−/− mice had no bleeding as a result of the subcutaneous osmotic pump implantation. This model resulted in an increase in heart weight in vehicle‐ and rhADAMTS13‐treated AngII‐infused mice compared with mice receiving a saline‐filled osmotic pump (saline control median, 127.7 g [IQR, 122.7–137.6 g]; P=0.0028 versus vehicle and P=0.019 versus rhADAMTS13). However, the rhADAMTS13‐treated group had smaller heart weights compared with vehicle‐treated mice with AngII infusion (vehicle median, 161.4 g [IQR, 152.6–182.0 g]; rhADAMTS13 median, 148.9 g [IQR, 138.3–155.2 g]; P=0.018 by Mann‐Whitney U test). On histological examination for fibrotic remodeling, we saw a protective effect of rhADAMTS13 treatment on perivascular (Figure 6A), but not total, collagen deposition compared with vehicle treatment (Figure 6B) in mice receiving AngII. Similarly to rhADAMTS13 treatment, VWF−/− mice had significantly less collagen surrounding vessels compared with WT mice (Figure 6C). Total collagen deposition in the heart was less in VWF‐deficient animals compared with WT (Figure 6D). The fibrotic response in this model was weaker compared with the AAC model, which can be explained by the less abrupt onset of injury and overall less severe damage to the myocardium. Furthermore, the fibrotic response was more pronounced around the vessels than in the interstitium in this model. Because VWF−/− mice lack Weibel‐Palade body in endothelial cells, it was expected that the protective phenotype observed was stronger compared with rhADAMTS13 treatment in WT mice. In summary, we show that VWF contributes to pressure overload‐mediated cardiac damage, and rhADAMTS13 reduced heart failure caused by fibrotic LV remodeling.
Figure 6.
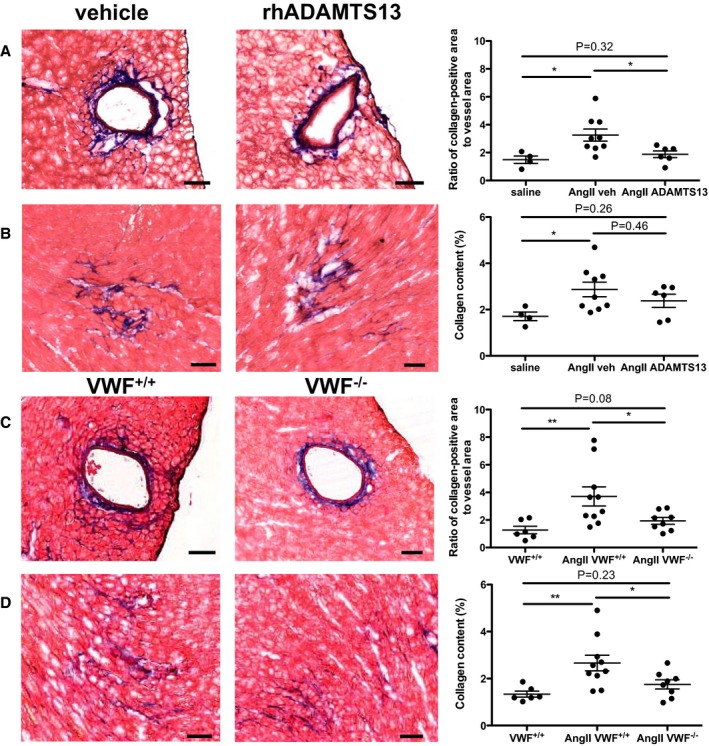
Fibrosis induced by angiotensin II (AngII) infusion is attenuated with recombinant human a disintegrin‐like metalloproteinase with thrombospondin motif type 1 member 13 (rhADAMTS13) treatment or in von Willebrand factor–deficient (VWF−/−) mice. A and B, Wild‐type mice were injected for 7 days after osmotic pump implantation with vehicle or rhADAMTS13. Control mice were implanted with osmotic pumps containing saline. A, Masson trichrome staining shows areas of interstitial collagen in hearts after 28 days of AngII infusion. The percentage of collagen‐positive area/total area was determined using color thresholding. B, Perivascular fibrosis was imaged and determined as a ratio of the collagen‐positive area/vessel interior area (saline, n=4; AngII vehicle, n=9; AngII rhADAMTS13, n=6). Perivascular (C) and interstitial (D) collagen were similarly analyzed in VWF+/+ or VWF−/− mice (VWF+/+, n=6; AngII VWF+/+, n=10; AngII VWF−/−, n=8). Bar=100 μm. Mann‐Whitney U tests were used to determine statistical significance. NS indicates not significant. *P<0.05, **P<0.01.
Discussion
Heart failure is a major cause for hospitalization, and it decreases life expectancy substantially. One of the main reasons for heart failure development is the loss of cardiomyocytes, which are replaced by fibrotic scar tissue, leading to a decrease in pump function and/or a decrease in elasticity. Decreased elasticity, in turn, impairs the filling of the left ventricle. Cardiomyocyte death and scar tissue formation may result from acute diseases, like myocardial infarction, leading to ischemic cardiomyopathy. During a life span, fibrotic tissue accumulates within the myocardium, increasing the incidence of congestive heart failure.1 Although aging itself can be viewed as an inflammatory process,21 chronic or repeated injuries to the heart, as with viral myocarditis, may contribute to this phenomenon. In the elderly population especially, arterial hypertension and aortic valve stenosis lead to LV hypertrophy, fibrotic remodeling, and decreased elasticity from chronically increased LV afterload stress.
In the cardiovascular field, it is becoming widely accepted that the balance between VWF and ADAMTS13 is of major importance. Numerous clinical studies established a link between low ADAMTS13 and adverse cardiovascular events,26, 27, 28 including heart failure,29 or high VWF levels.30, 31, 32
Several experimental studies suggested beneficial short‐term effects of rhADAMTS13 after acute injury caused by myocardial ischemia reperfusion injury.12, 13 Therefore, we were curious whether this effect could also be found long‐term in a more chronic injury to the myocardium. Increased afterload burden to the left ventricle by aortic stenosis leads to myocardial hypertrophy, extracellular matrix deposition, and heart failure in humans and mice.21, 33 Past animal studies have shown that aortic constriction and the subsequent afterload stress lead to upregulation of inflammatory cytokines34 and inflammatory cell recruitment,5 indicating that endothelial activation might play a role in the detrimental cardiac remodeling that leads to heart failure.
As a first approach, we tested whether endothelial activation with VWF secretion and inflammatory cell recruitment were present in the applied AAC model. Indeed, we found a substantial increase of VWF in the affected cardiac tissue and a significant accumulation of platelets and neutrophils in the myocardium. The rhADAMTS13 we used worked within minutes after intravenous injection and had a highly significant effect on platelet string persistence in ADAMTS13−/− mice, but only when used in the highest dose that we tested. Therefore, we hypothesized that this pharmacological dose would have a protective effect in a model of cardiac hypertrophy in which inflammation is a key driver. Furthermore, AAC did not lead to a rapid decrease in ADAMTS13 activity, indicating there was no effect to be expected by administering a physiological dose of rhADAMTS13, which would be effective in rescuing a deficiency, such as in TTP, but not necessarily effective in an anti‐inflammatory capacity. Therefore, we chose to use the highest dose to study a potential therapeutic effect.
This study has some possible limitations. Mice studied in the intravital microscopy experiments were younger than those in the fibrosis experiments; therefore, the responses to rhADAMTS13 dosing may not be identical. Furthermore, because many hypothesis tests were performed in this study, the authors cannot rule out that some statistically significant results may have occurred by chance alone.
ADAMTS13 deficiency is associated with thrombotic microangiopathies,10 and rhADAMTS13 recently underwent phase 1 clinical trials for the treatment of severe hereditary TTP, where plasma ADAMTS13 levels are <6%.35 Interestingly, there is evidence that TTP may be associated with cardiac damage and heart failure4, 36 and that troponin T at hospital admission is associated with increased ADAMTS13‐inhibitory antibodies and mortality.37 These clinical observations make the use of rhADAMTS13 in clinical trials for cardiac diseases highly interesting.
In this study, we observed an effect of rhADAMTS13 treatment on coronary platelet recruitment. Platelets activate leukocytes promoting coagulation38 and possess immune functions and the ability to release proinflammatory cytokines. TGF‐β1 is stored in platelet granules in high concentrations and is involved in fibrotic cardiac remodeling after aortic constriction. Platelet TGF‐β1–null mice were partially protected from developing cardiac hypertrophy, fibrosis, and systolic dysfunction in response to aortic constriction.39 Hence, the protective effect seen in rhADAMTS13 treatment may potentially be the result of decreased platelet recruitment by ultralarge VWF and, consequently, reduced presence of cardiac TGF‐β1. Future studies using ADAMTS13‐deficient animals would be of interest to investigate if platelet TGF‐β1 levels and cardiac fibrosis are increased in the absence of endogenous ADAMTS13.
ADAMTS13 has high substrate specificity, known to cleave only one substrate (VWF) and only at a specific sequence in the A2 domain.40 The positive pharmacological effects of ADAMTS13 on the outcome of ischemia reperfusion injury were shown to be VWF dependent in myocardial infarction11 and stroke.17 In a nonsurgical model of cardiac pressure overload, used herein to avoid bleeding, we show that VWF−/− mice develop significantly less fibrosis than control WT mice. So, what does VWF do to promote fibrotic response? VWF released to the endothelial surface directly recruits platelets to form platelet strings. This is the obvious, and likely insufficient, impact of VWF, because these are short‐lived in WT mice. In a recent independent study, we observed that neutrophil extracellular traps (NETs) are key for the fibrotic response in the AAC model.21 VWF may affect NETs. VWF directly recruits leukocytes by binding to neutrophil receptors, P‐selectin glycoprotein ligand‐1, and β2 integrins.41 Signaling through P‐selectin glycoprotein ligand‐1 by P‐selectin can induce NET formation,42 and VWF could have a similar direct effect. In addition, we have shown that VWF binds to the NET scaffold and appears to anchor it to the vessel wall in a thrombus.43 Therefore, VWF may help to adhere NETs at the sites of stimulated endothelium. NETs, in turn, can immensely amplify platelet recruitment44 and activation, promoting the vicious circle of inflammation.38
In conclusion, the dual effect of rhADAMTS13 on inflammation and microvessel obstruction after AAC likely preserved the long‐term ejection fraction and reduced myocardial fibrosis. We showed that rhADAMTS13 should be given in therapeutic doses that result in far higher plasma activity than what can be considered physiological to have an anti‐inflammatory effect. The protective effect is striking, and we propose a possible interrelation between VWF/ADAMTS13 and the proinflammatory active TGF‐β1 produced by platelets in this model. Our data further support the therapeutic potential of rhADAMTS13, which has undergone clinical trials for treatment of TTP,35 in cardiovascular diseases. Given its minor impact on bleeding risk15 and the highly beneficial protective effect seen in models of cardiac damage, ADAMTS13 should be considered a treatment option for conditions characterized by coronary microvessel dysfunction.
Sources of Funding
This work was supported by the National Heart, Lung, and Blood Institute of the National Institutes of Health grants R01HL102101 and R35HL135765 (Wagner) and 5T32HL066987‐13 (Martinod); a basic research grant from Baxalta Innovations GmbH (Wagner); and a fellowship of the German Cardiac Society (Deutsche Gesellschaft für Kardiologie; Witsch). Martinod is the recipient of a Fonds Wetenschappelijk Onderzoek Vlaanderen (PEGASUS)2 Marie Skłodowska‐Curie Fellowship (12S9917N), with funding from the European Union's Horizon 2020 research and innovation program under the Marie Skłodowska‐Curie grant agreement 665501. Portier is a PhD fellow of Flanders Innovation and Entrepreneurship (IWT.141178).
Disclosures
Wagner received a research grant from Baxalta Innovations GmbH. The remaining authors have no disclosures to report.
Acknowledgments
We thank Hanspeter Rottensteiner and Friedrich Scheiflinger (Baxalta Innovations GmbH, Vienna, Austria) for kindly providing recombinant human a disintegrin‐like metalloproteinase with thrombospondin motif type 1 member 13; Haley Larson for English editing; and Belén Fernández Castilla for assistance with statistical analyses.
(J Am Heart Assoc. 2018;7:e007004 DOI: 10.1161/JAHA.117.007004.)29367415
References
- 1. Bui AL, Horwich TB, Fonarow GC. Epidemiology and risk profile of heart failure. Nat Rev Cardiol. 2011;8:30–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hausenloy DJ, Yellon DM. Myocardial ischemia‐reperfusion injury: a neglected therapeutic target. J Clin Invest. 2013;123:92–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lorell BH, Carabello BA. Left ventricular hypertrophy: pathogenesis, detection, and prognosis. Circulation. 2000;102:470–479. [DOI] [PubMed] [Google Scholar]
- 4. Hawkins HK, Entman ML, Zhu JY, Youker KA, Berens K, Dore M, Smith CW. Acute inflammatory reaction after myocardial ischemic injury and reperfusion: development and use of a neutrophil‐specific antibody. Am J Pathol. 1996;148:1957–1969. [PMC free article] [PubMed] [Google Scholar]
- 5. Weisheit C, Zhang Y, Faron A, Kopke O, Weisheit G, Steinstrasser A, Frede S, Meyer R, Boehm O, Hoeft A, Kurts C, Baumgarten G. Ly6C(low) and not Ly6C(high) macrophages accumulate first in the heart in a model of murine pressure‐overload. PLoS One. 2014;9:e112710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Yang F, Dong A, Mueller P, Caicedo J, Sutton AM, Odetunde J, Barrick CJ, Klyachkin YM, Abdel‐Latif A, Smyth SS. Coronary artery remodeling in a model of left ventricular pressure overload is influenced by platelets and inflammatory cells. PLoS One. 2012;7:e40196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Takawale A, Fan D, Basu R, Shen M, Parajuli N, Wang W, Wang X, Oudit GY, Kassiri Z. Myocardial recovery from ischemia‐reperfusion is compromised in the absence of tissue inhibitor of metalloproteinase 4. Circ Heart Fail. 2014;7:652–662. [DOI] [PubMed] [Google Scholar]
- 8. Heymans S, Schroen B, Vermeersch P, Milting H, Gao F, Kassner A, Gillijns H, Herijgers P, Flameng W, Carmeliet P, Van de Werf F, Pinto YM, Janssens S. Increased cardiac expression of tissue inhibitor of metalloproteinase‐1 and tissue inhibitor of metalloproteinase‐2 is related to cardiac fibrosis and dysfunction in the chronic pressure‐overloaded human heart. Circulation. 2005;112:1136–1144. [DOI] [PubMed] [Google Scholar]
- 9. Wagner DD. Cell biology of von Willebrand factor. Annu Rev Cell Biol. 1990;6:217–246. [DOI] [PubMed] [Google Scholar]
- 10. Moake JL. Thrombotic microangiopathies. N Engl J Med. 2002;347:589–600. [DOI] [PubMed] [Google Scholar]
- 11. Gandhi C, Motto DG, Jensen M, Lentz SR, Chauhan AK. ADAMTS13 deficiency exacerbates VWF‐dependent acute myocardial ischemia/reperfusion injury in mice. Blood. 2012;120:5224–5230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. De Meyer SF, Savchenko AS, Haas MS, Schatzberg D, Carroll MC, Schiviz A, Dietrich B, Rottensteiner H, Scheiflinger F, Wagner DD. Protective anti‐inflammatory effect of ADAMTS13 on myocardial ischemia/reperfusion injury in mice. Blood. 2012;120:5217–5223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Savchenko AS, Borissoff JI, Martinod K, De Meyer SF, Gallant M, Erpenbeck L, Brill A, Wang Y, Wagner DD. VWF‐mediated leukocyte recruitment with chromatin decondensation by PAD4 increases myocardial ischemia/reperfusion injury in mice. Blood. 2014;123:141–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chauhan AK, Kisucka J, Brill A, Walsh MT, Scheiflinger F, Wagner DD. ADAMTS13: a new link between thrombosis and inflammation. J Exp Med. 2008;205:2065–2074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zhao BQ, Chauhan AK, Canault M, Patten IS, Yang JJ, Dockal M, Scheiflinger F, Wagner DD. von Willebrand factor‐cleaving protease ADAMTS13 reduces ischemic brain injury in experimental stroke. Blood. 2009;114:3329–3334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Fujioka M, Hayakawa K, Mishima K, Kunizawa A, Irie K, Higuchi S, Nakano T, Muroi C, Fukushima H, Sugimoto M, Banno F, Kokame K, Miyata T, Fujiwara M, Okuchi K, Nishio K. ADAMTS13 gene deletion aggravates ischemic brain damage: a possible neuroprotective role of ADAMTS13 by ameliorating postischemic hypoperfusion. Blood. 2010;115:1650–1653. [DOI] [PubMed] [Google Scholar]
- 17. Khan MM, Motto DG, Lentz SR, Chauhan AK. ADAMTS13 reduces VWF‐mediated acute inflammation following focal cerebral ischemia in mice. J Thromb Haemost. 2012;10:1665–1671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Martinod K, Demers M, Fuchs TA, Wong SL, Brill A, Gallant M, Hu J, Wang Y, Wagner DD. Neutrophil histone modification by peptidylarginine deiminase 4 is critical for deep vein thrombosis in mice. Proc Natl Acad Sci U S A. 2013;110:8674–8679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Tarnavski O, McMullen JR, Schinke M, Nie Q, Kong S, Izumo S. Mouse cardiac surgery: comprehensive techniques for the generation of mouse models of human diseases and their application for genomic studies. Physiol Genomics. 2004;16:349–360. [DOI] [PubMed] [Google Scholar]
- 20. Reininger AJ, Heijnen HF, Schumann H, Specht HM, Schramm W, Ruggeri ZM. Mechanism of platelet adhesion to von Willebrand factor and microparticle formation under high shear stress. Blood. 2006;107:3537–3545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Martinod K, Witsch T, Erpenbeck L, Savchenko A, Hayashi H, Cherpokova D, Gallant M, Mauler M, Cifuni SM, Wagner DD. Peptidylarginine deiminase 4 promotes age‐related organ fibrosis. J Exp Med. 2017;214:439–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Assoian RK, Komoriya A, Meyers CA, Miller DM, Sporn MB. Transforming growth factor‐beta in human platelets: identification of a major storage site, purification, and characterization. J Biol Chem. 1983;258:7155–7160. [PubMed] [Google Scholar]
- 23. Nevers T, Salvador AM, Grodecki‐Pena A, Knapp A, Velazquez F, Aronovitz M, Kapur NK, Karas RH, Blanton RM, Alcaide P. Left ventricular T‐cell recruitment contributes to the pathogenesis of heart failure. Circ Heart Fail. 2015;8:776–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Salvador AM, Nevers T, Velazquez F, Aronovitz M, Wang B, Abadia Molina A, Jaffe IZ, Karas RH, Blanton RM, Alcaide P. Intercellular adhesion molecule 1 regulates left ventricular leukocyte infiltration, cardiac remodeling, and function in pressure overload‐induced heart failure. J Am Heart Assoc. 2016;4:e003126 DOI: 10.1161/JAHA.115.003126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Liao JK. Linking endothelial dysfunction with endothelial cell activation. J Clin Invest. 2013;123:540–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Chion CK, Doggen CJ, Crawley JT, Lane DA, Rosendaal FR. ADAMTS13 and von Willebrand factor and the risk of myocardial infarction in men. Blood. 2007;109:1998–2000. [DOI] [PubMed] [Google Scholar]
- 27. Kaikita K, Soejima K, Matsukawa M, Nakagaki T, Ogawa H. Reduced von Willebrand factor‐cleaving protease (ADAMTS13) activity in acute myocardial infarction. J Thromb Haemost. 2006;4:2490–2493. [DOI] [PubMed] [Google Scholar]
- 28. Sonneveld MA, de Maat MP, Portegies ML, Kavousi M, Hofman A, Turecek PL, Rottensteiner H, Scheiflinger F, Koudstaal PJ, Ikram MA, Leebeek FW. Low ADAMTS13 activity is associated with an increased risk of ischemic stroke. Blood. 2015;126:2739–2746. [DOI] [PubMed] [Google Scholar]
- 29. Gombos T, Mako V, Cervenak L, Papassotiriou J, Kunde J, Harsfalvi J, Forhecz Z, Pozsonyi Z, Borgulya G, Janoskuti L, Prohaszka Z. Levels of von Willebrand factor antigen and von Willebrand factor cleaving protease (ADAMTS13) activity predict clinical events in chronic heart failure. Thromb Haemost. 2009;102:573–580. [DOI] [PubMed] [Google Scholar]
- 30. Fuchigami S, Kaikita K, Soejima K, Matsukawa M, Honda T, Tsujita K, Nagayoshi Y, Kojima S, Nakagaki T, Sugiyama S, Ogawa H. Changes in plasma von Willebrand factor‐cleaving protease (ADAMTS13) levels in patients with unstable angina. Thromb Res. 2008;122:618–623. [DOI] [PubMed] [Google Scholar]
- 31. Crawley JT, Lane DA, Woodward M, Rumley A, Lowe GD. Evidence that high von Willebrand factor and low ADAMTS‐13 levels independently increase the risk of a non‐fatal heart attack. J Thromb Haemost. 2008;6:583–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Maino A, Siegerink B, Lotta LA, Crawley JT, le Cessie S, Leebeek FW, Lane DA, Lowe GD, Peyvandi F, Rosendaal FR. Plasma ADAMTS‐13 levels and the risk of myocardial infarction: an individual patient data meta‐analysis. J Thromb Haemost. 2015;13:1396–1404. [DOI] [PubMed] [Google Scholar]
- 33. Neubauer S, Bull S. Myocardial fibrosis in aortic stenosis. JACC Cardiovasc Imaging. 2017;10:1334–1336. [DOI] [PubMed] [Google Scholar]
- 34. Sun M, Chen M, Dawood F, Zurawska U, Li JY, Parker T, Kassiri Z, Kirshenbaum LA, Arnold M, Khokha R, Liu PP. Tumor necrosis factor‐alpha mediates cardiac remodeling and ventricular dysfunction after pressure overload state. Circulation. 2007;115:1398–1407. [DOI] [PubMed] [Google Scholar]
- 35. Scully M, Knobl P, Kentouche K, Rice L, Windyga J, Schneppenheim R, Hovinga JAK, Kajiwara M, Fujimura Y, Maggiore C, Doralt J, Hibbard C, Martell L, Ewenstein B. Recombinant ADAMTS‐13: first‐in‐human pharmacokinetics and safety in congenital thrombotic thrombocytopenic purpura. Blood. 2017;130:2055–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Gandhi K, Aronow WS, Desai H, Amin H, Sharma M, Lai HM, Singh P. Cardiovascular manifestations in patients with thrombotic thrombocytopenic purpura: a single‐center experience. Clin Cardiol. 2010;33:213–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hughes C, McEwan JR, Longair I, Hughes S, Cohen H, Machin S, Scully M. Cardiac involvement in acute thrombotic thrombocytopenic purpura: association with troponin T and IgG antibodies to ADAMTS 13. J Thromb Haemost. 2009;7:529–536. [DOI] [PubMed] [Google Scholar]
- 38. Wagner DD, Frenette PS. The vessel wall and its interactions. Blood. 2008;111:5271–5281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Meyer A, Wang W, Qu J, Croft L, Degen JL, Coller BS, Ahamed J. Platelet TGF‐beta1 contributions to plasma TGF‐beta1, cardiac fibrosis, and systolic dysfunction in a mouse model of pressure overload. Blood. 2012;119:1064–1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kremer Hovinga JA, Coppo P, Lammle B, Moake JL, Miyata T, Vanhoorelbeke K. Thrombotic thrombocytopenic purpura. Nat Rev Dis Primers. 2017;3:17020. [DOI] [PubMed] [Google Scholar]
- 41. Pendu R, Terraube V, Christophe OD, Gahmberg CG, de Groot PG, Lenting PJ, Denis CV. P‐selectin glycoprotein ligand 1 and beta2‐integrins cooperate in the adhesion of leukocytes to von Willebrand factor. Blood. 2006;108:3746–3752. [DOI] [PubMed] [Google Scholar]
- 42. Etulain J, Martinod K, Wong SL, Cifuni SM, Schattner M, Wagner DD. P‐selectin promotes neutrophil extracellular trap formation in mice. Blood. 2015;126:242–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Fuchs TA, Brill A, Duerschmied D, Schatzberg D, Monestier M, Myers DD Jr, Wrobleski SK, Wakefield TW, Hartwig JH, Wagner DD. Extracellular DNA traps promote thrombosis. Proc Natl Acad Sci U S A. 2010;107:15880–15885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Martinod K, Wagner DD. Thrombosis: tangled up in NETs. Blood. 2014;123:2768–2776. [DOI] [PMC free article] [PubMed] [Google Scholar]