

Abstract
Background
Heart failure is a complex syndrome characterized by cardiac contractile impairment with high mortality. Defective intracellular Ca2+ homeostasis is the central cause under this scenario and tightly links to ultrastructural rearrangements of sarcolemmal transverse tubules and the sarcoplasmic reticulum (SR); however, the modulators of the SR architecture remain unknown. The SR has been thought to be a specialized endoplasmic reticulum membrane system. Receptor accessory proteins (REEPs)/DP1/Yop1p are responsible for shaping high‐curvature endoplasmic reticulum tubules. This study aimed to determine the role of REEPs in SR membrane shaping and thus cardiac function.
Methods and Results
We identified REEP5 (receptor accessory protein 5) as more highly expressed than other REEP members in adult rat ventricular myocardium, and it was downregulated in the failing hearts. Targeted inactivation of REEP5 in rats specially deformed the cardiac SR membrane without affecting transverse tubules, and this was visualized by focused ion beam scanning electron microscopy–based 3‐dimensional reconstruction. Accordingly, simultaneous recordings of depolarization‐induced Ca2+ currents and Ca2+ transients in REEP5‐null cardiomyocytes revealed normal L‐type Ca2+ channel currents but a depressed SR Ca2+ release. Consequently, the excitation–contraction coupling gain of cardiomyocytes and consequent cardiac contractility were compromised. REEP5 deficiency did not alter the expression of major proteins involved in Ca2+ handling in the heart.
Conclusions
REEP5 modulates cardiac function by shaping the SR. REEP5 defect deforms the SR architecture to depress cardiac contractility. REEP5‐dependent SR shaping might have potential as a therapeutic target for heart failure.
Keywords: cardiac myocyte, excitation–contraction coupling, electron microscopy, heart failure, sarcoplasmic reticulum
Subject Categories: Heart Failure
Clinical Perspective
What Is New?
REEP5 (receptor accessory protein 5) is an important molecule for normal cardiac sarcoplasmic reticulum shaping and function.
REEP5 ablation results in abnormal Ca2+ metabolism in the heart by disrupting the spatial contiguity between the sarcoplasmic reticulum and transverse tubules, leading to depressed cardiac contractility.
What Are the Clinical Implications?
This discovery helps us further understand the molecular mechanisms for intracellular Ca2+ activity and heart failure and implicates a new therapeutic target for cardiac dysfunction.
The prominent hallmark of heart failure is contractile dysfunction. Intracellular Ca2+ mishandling critically underlies the mechanical defects in failing myocardium.1 A tight junction between specialized domains of the sarcoplasmic reticulum (SR) and sarcolemmal transverse tubules (t‐tubules), called calcium release units, determines intracellular Ca2+ homeostasis and consequent cardiomyocyte contractility.2, 3, 4 Ultrastructural rearrangements to SR and t‐tubule networks represent a major feature in cases of heart failure.5, 6, 7 Several regulators for t‐tubule integrity have been identified, for example, BIN‐1 (bridging integrator 1) and JPH2 (junctophilin 2)8, 9; however, understanding of molecular entities underlying the structure‐based function of SR is still very limited.
The SR has been considered merely a specialized endoplasmic reticulum (ER) network.10 High‐curvature ER tubules are generated and maintained by an integral membrane protein superfamily consisting of DP1 in mammals.11, 12 Receptor accessory proteins (REEPs) are members of the DP1 family.12 Six REEPs (REEP1 to REEP6) have been identified in humans and other mammals.13 They are classified into 2 subfamilies: (1) REEP1 to REEP4 and (2) REEP5 and REEP6. A direct role for REEP5/DP1 in shaping ER tubules has been established.11 Whether REEPs contribute to SR membrane shaping and the associated SR Ca2+ handling and cardiac function remains to be characterized.
In this study, using focused ion beam scanning electron microscopy–based 3‐dimensional (3D) reconstruction, we provided compelling visual evidence that knockout of REEP5, which was highly expressed in intact cardiomyocytes and downregulated in the failing hearts, deformed the SR membrane structure and disrupted the physical coupling of the SR to t‐tubules. Consequently, the uncoupling of the SR from t‐tubules induced the depression of SR Ca2+ release and cardiac contraction.
Methods
The data, analytic methods, and study materials have been made available to other researchers for purposes of reproducing the results or replicating the procedure. The authors declare that all supporting data are available within the article and its online supplementary files.
Generation of REEP5 Knockout Rats and Cardiomyocyte Isolation
All animal protocols were approved by the animal care and use committee at the Tongji University School of Medicine and were conducted according to the Guidelines for the Care and Use of Animals in Research (US National Institutes of Health Publication No. 85‐23, revised 1996).
A CRISPR/Cas9‐based genome‐editing technique was adopted to produce knockout rats by targeting exon 2 of REEP5. We designed single guide RNAs for SpCas9 target selection using the CRISPR design tool (http://crispr.mit.edu/), which directed Cas9 DNA endonuclease to cleave the Reep5 gene and to create a double‐strand break, disrupting the expression of REEP5 by frameshift mutation. To confirm the efficiency of REEP5 deletion, quantitative real‐time polymerase chain reaction (PCR) using TaqMan primer sets spanning the constitutively expressed exon 2 were used to measure total transcription of the REEP5 gene using cDNA from wild‐type (WT) and REEP5‐knockout (REEP5‐KO) rat cardiomyocytes. Quantitative real‐time PCR reactions were performed in a 384‐well format using an ABI 7900HT instrument (Applied Biosystems). Sequencing analysis was performed to detect base pair deletions and frameshift mutations.
Ventricular myocytes were isolated from 8‐week‐old WT and REEP5‐KO littermates using a previously described method.14 Each cellular experiment used cardiomyocytes isolated from 5 sex‐matched rats per genotype.
Immunostaining
Dissociated cardiomyocytes were allowed to attach to glass coverslips precoated with laminin before fixation. For all immunocytochemistry experiments, the cells were fixed in methanol at room temperature for 5 minutes. After fixation, the cardiomyocytes were permeabilized with 0.1% Triton X‐100 for 15 minutes and successively blocked with 10% normal goat serum in PBS for 1 hour at room temperature. Next, the cells were incubated in antibodies (1:50 for REEP5 and RyR2 [ryanodine receptor 2]) overnight at 4°C. After several washes with PBS after primary antibody incubation, the cells were then incubated in goat antimouse and antirabbit IgG conjugated to AlexaFluor 488 and 555, respectively. All cardiomyocytes were imaged by confocal microscopy (Leica TCS SP8) and analyzed using LAS AF and IPP 6.0 software.
Transmission Electron Microscopy
Transmission electron microscopy analysis of the rat ventricular tissue ultrastructure was performed as described previously.15 Tissue sections were imaged at ×20 000 and ×80 000 magnification using a transmission electron microscope (JEOL TEM1230 from the Electron Microscopy Core of the Shanghai Institute of Biological Science at the Chinese Academy of Sciences). For immunogold localization, the sections were incubated with anti‐REEP5 for 3 days and then with antirabbit IgG conjugated to 4‐nm gold particles for 3 hours at 21°C. Finally, the sections were post‐fixed in 1% osmium tetroxide and 2% uranyl acetate, dehydrated, and flat‐embedded in Durcupan ACM (Fluka) for light and electron microscopic examinations.
Focused Ion Beam Scanning Electron Microscopy Imaging and 3D Reconstruction
The hearts of WT and REEP5 KO rats were harvested rapidly after anesthesia and fixed in a 0.1 mol/L phosphate buffer solution containing 4% paraformaldehyde. Focused ion beam scanning electron microscopy (from the Institute of Biophysics at the Chinese Academy of Sciences) was used to obtain continuous sectional slices at a spacing of 20 nm, according to a previous study.16 The 3D reconstruction of SR and t‐tubules was performed using ImageJ 1.51 (US National Institutes of Health) and the Imaris 7.2 software package (Bitplane).
Simultaneous Recording of Depolarization‐Induced Ca2+ Currents and Ca2+ Transients
L‐type Ca2+ channel (LTCC) currents and SR Ca2+ transients were simultaneously measured in acute isolated adult rat ventricular cardiomyocytes loaded with Fluo‐4 pentapotassium salt (10 μmol/L; Thermo Fisher) via a patch‐clamp recording amplifier (EPC‐10 USB; HEKA Elektronik) coupled to a confocal laser scanning microscope (Leica TCS SP8).
Measurement of Cardiac Function
In vivo pressure–volume analysis was performed in 8‐week‐old WT and REEP5‐KO rats, as described previously.17 Briefly, steady‐state data were recorded after 10 to 15 minutes of stabilization. Load‐independent measurements were recorded during transient intrathoracic inferior vena cava occlusion. Data were recorded using LABCHART 7 software (AD Instruments) and analyzed offline using PVAN 3.6 software (Millar Instruments).
Real‐Time PCR
Messenger RNA was extracted from rat heart tissue using TRIzol reagent. The reverse transcription reaction was performed using PrimeScript RT reagent (Takara), and real‐time PCR was performed in triplicate using SYBR Green PCR Master Mix (Applied Biosystems).
Preparation of Subcellular Membrane Fractions From Cardiomyocytes
Membrane fractions of rat cardiomyocytes were prepared according to a sucrose gradient centrifugation–based approach, as described previously.18, 19
Western Blot Analyses and Antibodies
Western blot was performed according to standard protocols. The following antibodies were used: anti‐REEP5, anti‐Cav1.2, anti‐RyR2, anti‐SERCA2α, anti‐IP3R, anti‐PLB, anti‐CaMKII, anti‐CASQ, and anti‐β‐tubulin.
Statistical Analyses
The data are presented as the mean±SEM. Prism 6 software (GraphPad) was used for statistical analyses. For comparison between 2 groups, a paired or unpaired 2‐tailed Student t test was performed. P<0.05 was considered statistically significant.
Results
REEP5 Is Predominantly Concentrated at the Cardiac SR
To ascertain whether REEPs contribute to membrane shaping of the SR in the heart, we first examined the transcriptional profiles of REEPs in cardiomyocytes. REEP5 was more highly expressed than the other members screened in adult rat ventricular myocytes (Figure 1A). Next, we analyzed the subcellular localization of REEP5 in intact cardiomyocytes. Immunocytochemical imaging showed that REEP5 extensively overlapped with RyR2, a representative SR membrane protein (Figure 1B). A sucrose gradient centrifugation–based approach also detected abundant REEP5 in the subcellular fractionations in which RyR2 was enriched (Figure 1C), suggesting the predominant localization of REEP5 at the SR membrane. This finding was further validated by immunogold labeling and transmission electron microscopy (Figure 1D).
Figure 1.
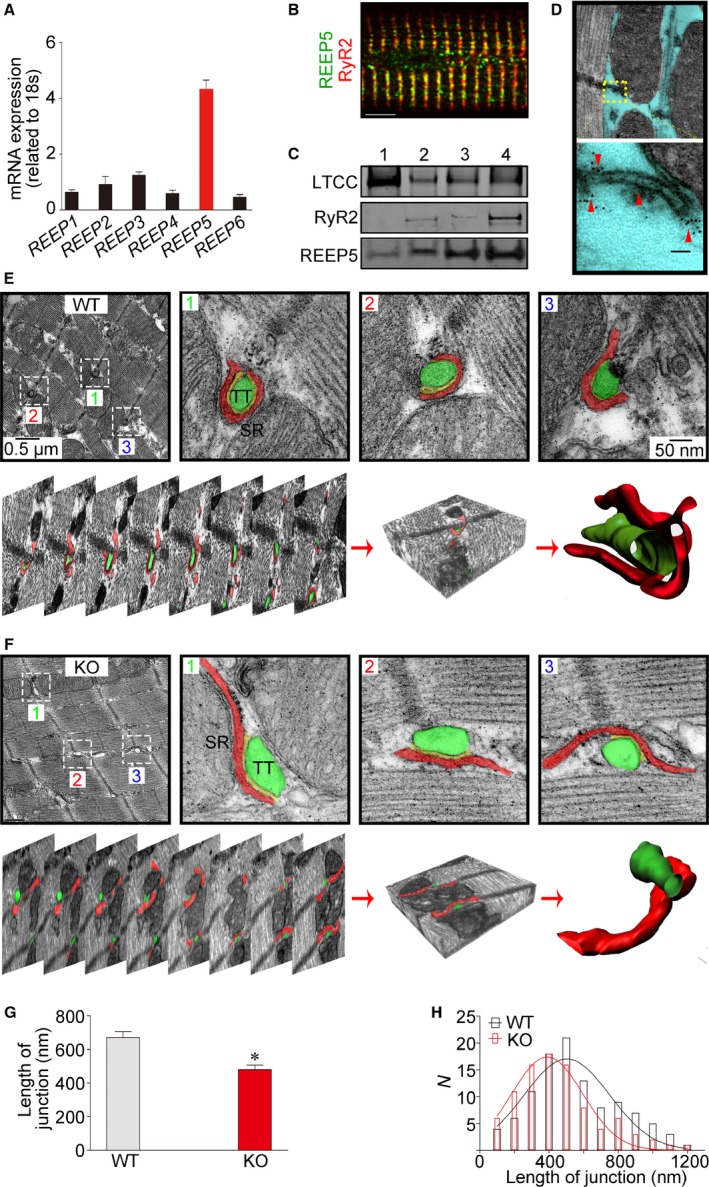
REEP5 mediates membrane shaping of the cardiac SR. A, Quantitative polymerase chain reaction measurements of REEP1 through REEP6 levels in adult rat ventricular myocardium. B, Immunofluorescence imaging of REEP5 in isolated intact adult rat ventricular myocytes. Scale bar: 5 μm. C, Identification of REEP5 proteins in sucrose gradient centrifugation–based membrane fractions. Numbers 1 to 4 indicate <23%, 23% to 29%, 29% to 32%, and 32% to 35% sucrose contents, respectively. D, Two‐dimensional transmission EM images of normal heart sections immunolabeled for REEP5. The yellow box identifies the SR structure, and the red arrowheads denote the REEP5 proteins situated on the SR. E and F, Three‐dimensional reconstruction of SR from serial EM images of WT and REEP5‐KO heart sections. The top panels in (E) and (F) show EM images of axial cross‐sections of heart tissue, and the bottom panels present serial aligned images from the same section. Bottom right shows digitized 3D reconstruction of the SR with an associated t‐tubule from the serial images. The green regions indicate t‐tubules, and the red regions indicate the SR. G and H, The apparent length of SR/t‐tubule junction (G) and its distribution (H). Data are from 90 (WT) and 108 (REEP5‐KO) SR/t‐tubule junction images in 3 independent experiments per group. *P<0.05 compared with WT controls. The blots and images shown are representative of 3 to 5 independent experiments. EM indicates electron microscopy; KO, knockout; LTCC, L‐type Ca2+ channel; REEP, receptor accessory protein; RyR2, ryanodine receptor 2; SR, sarcoplasmic reticulum; t‐tubule, transverse tubule; WT, wild type.
REEP5 Knockout Deforms SR Membranes
We next generated genetically engineered rats using CRISPR/Cas9‐based genome editing to target exon 2 of REEP5 to ablate its expression (Figure S1A). Heterozygous rats were interbred to produce homozygous animals. Offspring exhibited the expected mendelian ratios. Sequencing analyses of the targeted REEP5 locus revealed that 1 founder possessed a mutation comprising an 8‐bp deletion. Western blot analysis confirmed the gene‐targeting efficiency of REEP5, revealing a significant reduction in REEP5 protein expression (Figure S1B and S1C). Homozygous REEP5‐KO rats were viable and fertile and appeared healthy.
To directly visualize the impact of REEP5 deletion on the SR network in situ, we utilized focused ion beam scanning electron microscopy, which allows automatic connectome tracing for serial ultrathick sections of heart tissues to produce data sets optimized for final 3D reconstruction of the SR membrane structures. We found that the gross morphology of SR network is preserved in REEP5‐KO cardiomyocytes. In high‐magnification axial cross‐sectional images of the SR, we observed that, relative to WT SR, REEP5‐KO SR consistently exhibited fewer membrane contortions surrounding t‐tubules. As revealed by 3D tomography (Figure 1E and 1F), the junctional SR membrane folds, which envelop t‐tubules, were significantly outstretched and dissociated from t‐tubules in REEP5‐KO cardiomyocytes.
To quantitatively analyze the connection between SR and t‐tubule structures, we measured the length of the SR/t‐tubules junction from multiple samples (Figure 1G and 1H). We found that the SR/t‐tubule junction length in REEP5‐KO cardiomyocytes was dramatically reduced (Figure 1G), suggesting that REEP5 controls SR membrane shaping and the spatial connection of the SR with t‐tubules in cardiomyocytes.
REEP5‐Defective SR Membrane Remodeling Depresses Depolarization‐Induced SR Ca2+ Release
The physical contiguity between the SR and t‐tubules determines the excitation–contraction (EC) coupling efficiency in ventricular cardiomyocytes.4 This permits the influx of Ca2+ through LTCCs, which rapidly diffuses across the dyadic cleft to activate RyR2, and causes SR Ca2+ release and synchronous myocyte contraction. Simultaneous recording of LTCC current and Ca2+ transients showed that LTCC current in REEP5‐KO myocytes was comparable to that in WT cells (Figure 2A and 2B), but the peak amplitude of the Ca2+ transient was significantly decreased in REEP5‐KO myocytes (Figure 2C), leading to a reduced EC coupling gain (Figure 2D). Notably, the REEP5‐defective SR Ca2+ release seemed not to be correlated with the levels of SR Ca2+‐handling proteins such as RyR2, SERCA2a (sarco/endoplasmic reticulum Ca2+ ATPase cardiac isoform 2a), and CASQ2 (calsequestrin 2), in that REEP5 deficiency did not significantly alter their expression (Figure S2). These results indicate that structural deformation of the SR membrane induced by REEP5 deficiency depresses depolarization‐induced SR Ca2+ release by decreasing the efficiency of the EC coupling in cardiomyocytes.
Figure 2.
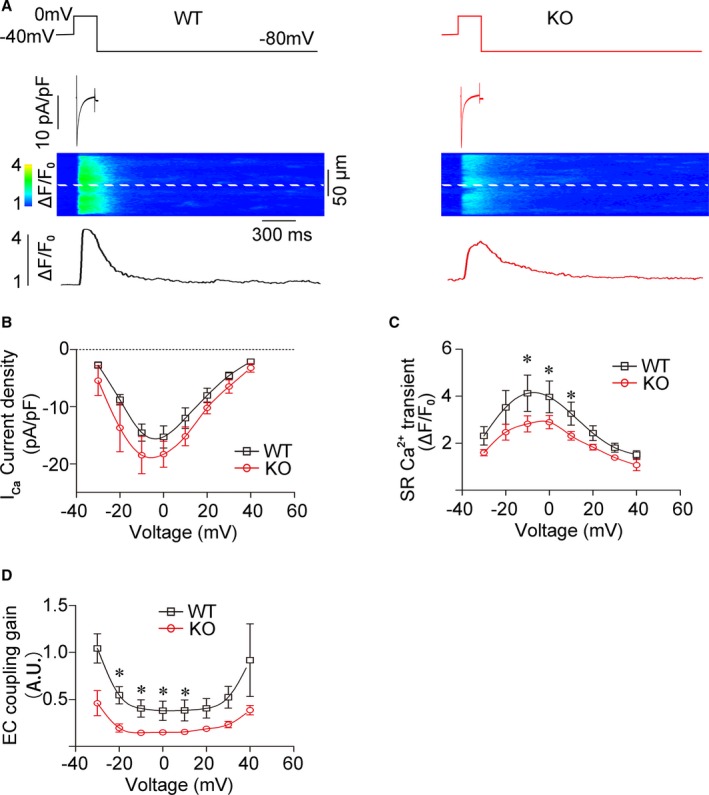
REEP5 ablation depresses depolarization‐induced SR Ca2+ release. Simultaneous recordings of LTCC currents and Ca2+ transients in freshly isolated rat ventricular myocytes. A, A typical LTCC current and a synchronously evoked Ca2+ transient. B through D, Pooled data. n=9 for WT, n=13 for REEP5‐KO. *P<0.05 compared with WT controls. EC indicates excitation–contraction; KO, knockout; LTCC, L‐type Ca2+ channel; REEP, receptor accessory protein; SR, sarcoplasmic reticulum; WT, wild type.
Depressed SR Ca2+ Release Compromises Cardiac Contractility in REEP5‐KO Rats
In the heart, cardiomyocyte SR Ca2+ release is coupled to cardiac contraction.4 In view of the effects of REEP5 reduction on SR Ca2+ release, we further studied in vivo cardiac contractile function in REEP5‐KO rats. We found that the hearts of REEP5‐KO rats were structurally normal (Figure 3A through 3F). However, dynamic measures of intact heart function using a rigorous ventricular pressure–volume catheter technique demonstrated that contractile function (end‐systolic pressure–volume relationship, preload recruitable stroke work, and time‐varying maximal elastance) was significantly compromised in REEP5‐KO animals (Figure 3G through 3J). Collectively, REEP5‐mediated membrane shaping of the SR modulates cardiac contraction.
Figure 3.
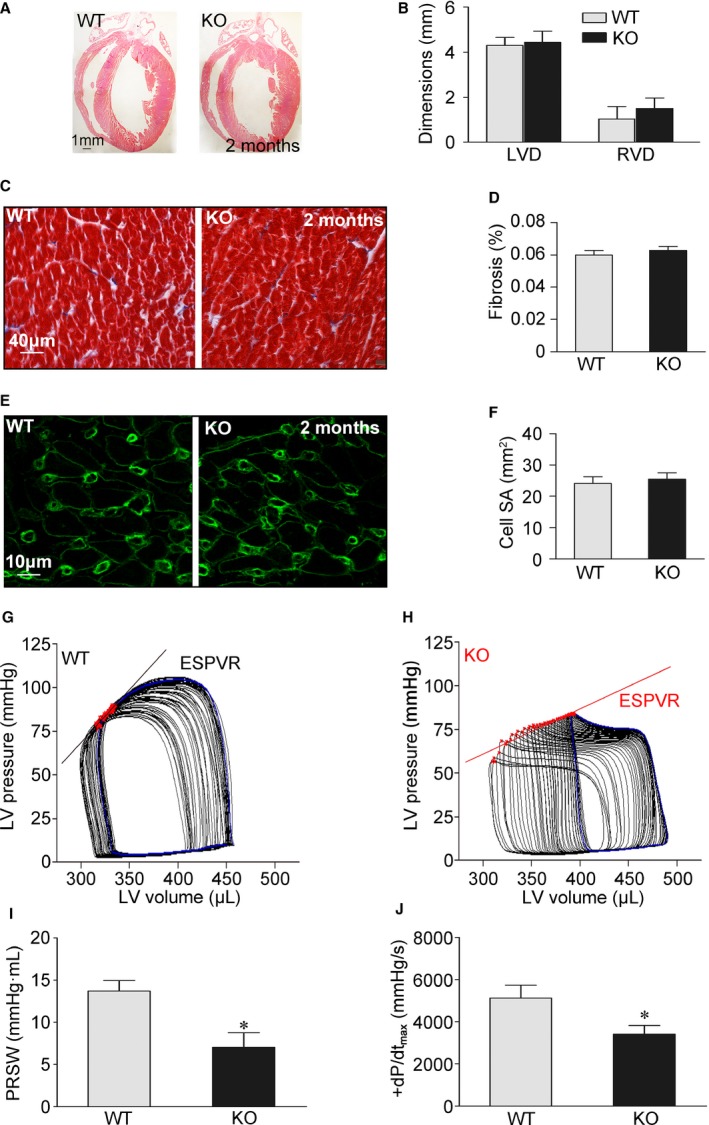
REEP5‐KO compromises cardiac contractility in vivo. A, Representative histological sections of 2‐month‐old rat hearts stained with HE. B, Cardiac wall dimensions were assessed from HE‐stained histological sections. C, Masson's trichrome staining for fibrosis in histological sections of rat hearts. D, Percentage of fibrosis quantified from a Masson trichrome stain. E, Representative WGA fluorescence stains of rat cardiomyocytes. F, Cardiomyocyte surface area calculated from WGA stains. WT, n=5; REEP5‐KO, n=5 (A through F). G and H, Representative in vivo PV loops during transient inferior vena caval occlusion in a REEP5‐KO rat and a WT control. ESPVR relationships are presented. I and J, Steady‐state PV data demonstrating decreased PRSW and increased peak velocities of pressure change (peak+dP/dt) during isovolumic contraction in REEP5‐KO rats. WT, n=8; REEP5‐KO, n=8. *P<0.05 compared with WT controls. ESPVR indicates end‐systolic pressure–volume relationship; HE indicates hematoxylin and eosin; KO, knockout; LV, left ventricular; LVD, left ventricular wall dimension; PRSW, preload recruitable stroke work; PV, pressure–volume; REEP, receptor accessory protein; RVD, right ventricular wall dimension; SR, sarcoplasmic reticulum; WGA, wheat germ agglutinin; WT, wild type.
Discussion
Our studies in both cells and genetically engineered animals have uncovered a novel role of REEP5 in the structure‐based function of the cardiac SR. First, we identified the predominant expression of REEP5 in cardiomyocytes and its spatial localization at the SR. Second, we found that REEP5 ablation induced a reduction of SR membrane folds and disrupted the spatial contiguity between the SR and t‐tubules. Third, the reduction in REEP5 depressed SR Ca2+ release without any effect on the expression of critical SR Ca2+‐handling proteins and sarcolemmal LTCC currents. Finally, knockout of REEP5 compromised cardiac contractility. Therefore, we demonstrated that REEP5 serves as an SR membrane sculptor to control cardiac function.
REEP family members were originally identified by their ability to enhance the membrane expression of G protein–coupled receptors.20 REEP members such as REEP5, a member of the DP1 family, regulate intracellular trafficking by affecting ER membrane structure and cargo capacity and by acting as adapter proteins.12 Contrasting findings regarding the intracellular localization and mechanisms of action of the REEP proteins have been described.20, 21 The present findings expand our understanding of REEP5 as a molecule that shapes the ER. Furthermore, our discovery of the restricted distribution of REEP5 at the SR and of the effects of REEP5 on SR membrane curvature and related SR Ca2+ release reveals an important modulatory mechanism for cardiac EC coupling.
The SR is an extremely motile organelle that is capable of molecular motor protein–driven structural changes.22 Under physiological conditions, SR vesicles move dynamically throughout the cell, approaching or moving away from the myocyte sarcolemma. SR dysfunction is a major feature of cardiac diseases, and its underlying mechanisms have been intensively studied.23, 24, 25 Abnormal SR Ca2+ release occurs in human cardiomyocytes isolated from patients with heart failure, regardless of etiology.26 This mirrors findings concerning SR/t‐tubule coupling disruption in animal models of heart failure that resulted from ischemic, hypertensive, and tachycardic phenotypes and suggests that the disruption of the SR network and its connection to t‐tubules is a common pathway in heart failure. In addition, we found that REEP5 proteins were significantly reduced in the failing hearts (Figure S3). Therefore, our identification of depressed SR Ca2+ release and cardiac contractility in REEP5‐defective rats may yield novel perspectives on the mechanisms underlying heart failure.
In sum, REEP5 serves as an SR membrane sculptor to modulate cardiac function. Its deficiency deforms the SR membranes, with the result that EC coupling of cardiomyocytes and cardiac contractility are compromised. As such, REEP5‐dependent SR shaping could be of pathophysiological significance, and targeting REEP5 appears to be a novel therapeutic approach for heart failure.
Sources of Funding
This work was funded by the National Key Basic Research Program of China (2013CB531100, to Chen), the Key Program of National Natural Science Foundation of China (81530017 to Chen), the National Innovative Research Groups Program of the National Natural Science Foundation of China (81521061, to Chen), the General Program of the National Natural Science Foundation of China (81770397 to Chen; 81670360, to J. Li; 81500252, 81770267 to Liang), the Outstanding Young Talent Training Program of Shanghai Municipal Commission of Health and Family Planning (2017YQ045, to Liang), the Fundamental Research Funds for the Central Universities to Chen.
Disclosures
None.
Supporting information
Figure S1. Validation of REEP5 KO in rats. A, A schematic overview of the CRISPR/Cas9‐based genome‐editing strategy used to generate the REEP5 KO. Bottom, Example chromatogram showing a microdeletion and representative sequences of mutated alleles identified from clonal amplicons. Yellow dashes indicate the deleted bases. B, Western blot analysis of REEP5 expression in the hearts of rats carrying homozygous 8‐bp deletion mutations. Left, representative blots; right, pooled data. WT, n=6; REEP5‐KO, n=6. P<0.05 compared with WT. C, Analysis of the effects of CRISPR/Cas9‐mediated REEP5 gene editing on the mRNA expression of REEPs other than REEP5. n=3 for each group. P<0.05. KO indicates knockout; REEP, receptor expression‐enhancing protein; WT, wild type.
Figure S2. REEP5 deficiency does not affect the expression of SR Ca2+‐handling proteins. Analysis of critical Ca2+‐handling proteins on the SR. Left, typical blots; right, pooled data from 3 separate experiments. REEP indicates receptor expression‐enhancing protein; SR, sarcoplasmic reticulum.
Figure S3. Measurement of REEP5 protein during the development of heart failure. A, Echocardiographic analysis of cardiac function during TAC‐induced heart failure, as demonstrated by ejection fraction. B, Western blotting examination of REEP5 protein in the heart tissues. Top, typical blots; bottom, pooled data. Three to 5 animals were analyzed at indicated time points in WT and REEP5‐KO rats. KO, knockout; REEP, receptor expression‐enhancing protein; TAC, transverse aortic constriction; WT, wild type.
(J Am Heart Assoc. 2018;7:e007205 DOI: 10.1161/JAHA.117.007205.)29431104
Contributor Information
Jun Li, Email: junli@tongji.edu.cn.
Yi‐Han Chen, Email: yihanchen@tongji.edu.cn.
References
- 1. Luo M, Anderson ME. Mechanisms of altered Ca²⁺ handling in heart failure. Circ Res. 2013;113:690–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Fabiato A. Calcium‐induced release of calcium from the cardiac sarcoplasmic reticulum. Am J Physiol. 1983;245:C1–C14. [DOI] [PubMed] [Google Scholar]
- 3. Franzini‐Armstrong C, Protasi F, Ramesh V. Shape, size, and distribution of Ca(2+) release units and couplons in skeletal and cardiac muscles. Biophys J. 1999;77:1528–1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bers DM. Cardiac excitation‐contraction coupling. Nature. 2002;415:198–205. [DOI] [PubMed] [Google Scholar]
- 5. Pinali C, Bennett H, Davenport JB, Trafford AW, Kitmitto A. Three‐dimensional reconstruction of cardiac sarcoplasmic reticulum reveals a continuous network linking transverse‐tubules: this organization is perturbed in heart failure. Circ Res. 2013;113:1219–1230. [DOI] [PubMed] [Google Scholar]
- 6. Guo A, Zhang C, Wei S, Chen B, Song LS. Emerging mechanisms of T‐tubule remodelling in heart failure. Cardiovasc Res. 2013;98:204–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wei S, Guo A, Chen B, Kutschke W, Xie YP, Zimmerman K, Weiss RM, Anderson ME, Cheng H, Song LS. T‐tubule remodeling during transition from hypertrophy to heart failure. Circ Res. 2010;107:520–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Caldwell JL, Smith CE, Taylor RF, Kitmitto A, Eisner DA, Dibb KM, Trafford AW. Dependence of cardiac transverse tubules on the BAR domain protein amphiphysin II (BIN‐1). Circ Res. 2014;115:986–996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Takeshima H, Komazaki S, Nishi M, Iino M, Kangawa K. Junctophilins: a novel family of junctional membrane complex proteins. Mol Cell. 2000;6:11–22. [DOI] [PubMed] [Google Scholar]
- 10. Michalak M, Opas M. Endoplasmic and sarcoplasmic reticulum in the heart. Trends Cell Biol. 2009;19:253–259. [DOI] [PubMed] [Google Scholar]
- 11. Voeltz GK, Prinz WA, Shibata Y, Rist JM, Rapoport TA. A class of membrane proteins shaping the tubular endoplasmic reticulum. Cell. 2006;124:573–586. [DOI] [PubMed] [Google Scholar]
- 12. Hu J, Shibata Y, Voss C, Shemesh T, Li Z, Coughlin M, Kozlov MM, Rapoport TA, Prinz WA. Membrane proteins of the endoplasmic reticulum induce high‐curvature tubules. Science. 2008;319:1247–1250. [DOI] [PubMed] [Google Scholar]
- 13. Park SH, Zhu PP, Parker RL, Blackstone C. Hereditary spastic paraplegia proteins REEP1, spastin, and atlastin‐1 coordinate microtubule interactions with the tubular ER network. J Clin Invest. 2010;120:1097–1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Louch WE, Sheehan KA, Wolska BM. Methods in cardiomyocyte isolation, culture, and gene transfer. J Mol Cell Cardiol. 2011;51:288–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Page E, Upshaw‐Earley J, Goings GE. Localization of atrial natriuretic peptide in caveolae of in situ atrial myocytes. Circ Res. 1994;75:949–954. [DOI] [PubMed] [Google Scholar]
- 16. Hayworth KJ, Xu CS, Lu Z, Knott GW, Fetter RD, Tapia JC, Lichtman JW, Hess HF. Ultrastructurally smooth thick partitioning and volume stitching for large‐scale connectomics. Nat Methods. 2015;12:319–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Pacher P, Nagayama T, Mukhopadhyay P, Bátkai S, Kass DA. Measurement of cardiac function using pressure‐volume conductance catheter technique in mice and rats. Nat Protoc. 2008;3:1422–1434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Charuk JH, Howlett S, Michalak M. Subfractionation of cardiac sarcolemma with wheat‐germ agglutinin. Biochem J. 1989;264:885–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zorzano A, Camps M. Isolation of T‐tubules from skeletal muscle. Curr Protoc Cell Biol. 2006;3:24. [DOI] [PubMed] [Google Scholar]
- 20. Saito H, Kubota M, Roberts RW, Chi Q, Matsunami H. RTP family members induce functional expression of mammalian odorant receptors. Cell. 2004;119:679–691. [DOI] [PubMed] [Google Scholar]
- 21. Züchner S, Wang G, Tran‐Viet KN, Nance MA, Gaskell PC, Vance JM, Ashley‐Koch AE, Pericak‐Vance MA. Mutations in the novel mitochondrial protein REEP1 cause hereditary spastic paraplegia type 31. Am J Hum Genet. 2006;79:365–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Vega AL, Yuan C, Votaw VS, Santana LF. Dynamic changes in sarcoplasmic reticulum structure in ventricular myocytes. J Biomed Biotechnol. 2011;2011:382586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sedej S, Schmidt A, Denegri M, Walther S, Matovina M, Arnstein G, Gutschi EM, Windhager I, Ljubojević S, Negri S, Heinzel FR, Bisping E, Vos MA, Napolitano C, Priori SG, Kockskämper J, Pieske B. Subclinical abnormalities in sarcoplasmic reticulum Ca2+ release promote eccentric myocardial remodeling and pump failure death in response to pressure overload. J Am Coll Cardiol. 2014;63:1569–1579. [DOI] [PubMed] [Google Scholar]
- 24. Wang L, Myles RC, De Jesus NM, Ohlendorf AK, Bers DM, Ripplinger CM. Optical mapping of sarcoplasmic reticulum Ca2+ in the intact heart: ryanodine receptor refractoriness during alternans and fibrillation. Circ Res. 2014;114:1410–1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lenaerts I, Bito V, Heinzel FR, Driesen RB, Holemans P, D'hooge J, Heidbüchel H, Sipido KR, Willems R. Ultrastructural and functional remodeling of the coupling between Ca2+ influx and sarcoplasmic reticulum Ca2+ release in right atrial myocytes from experimental persistent atrial fibrillation. Circ Res. 2009;105:876–885. [DOI] [PubMed] [Google Scholar]
- 26. Fischer TH, Eiringhaus J, Dybkova N, Förster A, Herting J, Kleinwächter A, Ljubojevic S, Schmitto JD, Streckfuß‐Bömeke K, Renner A, Gummert J, Hasenfuss G, Maier LS, Sossalla S. Ca2+/calmodulin‐dependent protein kinase II equally induces sarcoplasmic reticulum Ca2+ leak in human ischaemic and dilated cardiomyopathy. Eur J Heart Fail. 2014;16:1292–1300. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Validation of REEP5 KO in rats. A, A schematic overview of the CRISPR/Cas9‐based genome‐editing strategy used to generate the REEP5 KO. Bottom, Example chromatogram showing a microdeletion and representative sequences of mutated alleles identified from clonal amplicons. Yellow dashes indicate the deleted bases. B, Western blot analysis of REEP5 expression in the hearts of rats carrying homozygous 8‐bp deletion mutations. Left, representative blots; right, pooled data. WT, n=6; REEP5‐KO, n=6. P<0.05 compared with WT. C, Analysis of the effects of CRISPR/Cas9‐mediated REEP5 gene editing on the mRNA expression of REEPs other than REEP5. n=3 for each group. P<0.05. KO indicates knockout; REEP, receptor expression‐enhancing protein; WT, wild type.
Figure S2. REEP5 deficiency does not affect the expression of SR Ca2+‐handling proteins. Analysis of critical Ca2+‐handling proteins on the SR. Left, typical blots; right, pooled data from 3 separate experiments. REEP indicates receptor expression‐enhancing protein; SR, sarcoplasmic reticulum.
Figure S3. Measurement of REEP5 protein during the development of heart failure. A, Echocardiographic analysis of cardiac function during TAC‐induced heart failure, as demonstrated by ejection fraction. B, Western blotting examination of REEP5 protein in the heart tissues. Top, typical blots; bottom, pooled data. Three to 5 animals were analyzed at indicated time points in WT and REEP5‐KO rats. KO, knockout; REEP, receptor expression‐enhancing protein; TAC, transverse aortic constriction; WT, wild type.