

Abstract
Background
Fibronectin in endothelial basement membranes promotes endothelial inflammatory activation and atherosclerosis but also promotes plaque stability and vascular remodeling. The fibronectin receptor α5 subunit is proinflammatory through binding to and activating phosphodiesterase 4D5, which inhibits anti‐inflammatory cyclic adenosine monophosphate and protein kinase A. Replacing the α5 cytoplasmic domain with that of α2 resulted in smaller atherosclerotic plaques. Here, we further assessed plaque phenotype and compensatory vascular remodeling in this model.
Methods and Results
α5/2 mice in the hyperlipidemic apolipoprotein E null background had smaller plaques in the aortic root, with reduced endothelial NF‐κB activation and inflammatory gene expression, reduced leukocyte content, and much lower metalloproteinase expression. However, smooth muscle cell content, fibrous cap thickness, and fibrillar collagen were unchanged, indicating no shift toward vulnerability. In vivo knockdown of phosphodiesterase 4D5 also decreased endothelial inflammatory activation and atherosclerotic plaque size. α5/2 mice showed improved recovery from hindlimb ischemia after femoral artery ligation.
Conclusions
Blocking the fibronectin‐Integrin α5 pathway reduces atherosclerotic plaque size, maintains plaque stability, and improves compensatory remodeling. This pathway is therefore a potential therapeutic target for treatment of atherosclerosis.
Keywords: arteriogenesis, atherosclerosis, fibronectin, inflammation, matrix metalloprotease, phosphodiesterase 4D5, plaque vulnerability
Subject Categories: Atherosclerosis, Coronary Artery Disease, Peripheral Vascular Disease, Vascular Disease
Clinical Perspective
What Is New?
Fibronectin has been implicated in atherosclerosis, but although it promotes inflammatory activation of the endothelium and increases plaque size, it also promotes plaque stability, most likely through its function as a scaffold in collagen fibril formation.
The current study shows that mutating the cytoplasmic domain of its receptor integrin α5, which blocks the proinflammatory effect, reduces plaque size by inhibiting binding to phosphodiesterase 4D5 and improves recovery from hindlimb ischemia without impairing plaque stability.
What Are the Clinical Implications?
These data indicate that a pathway involving integrin α5‐phosphodiesterase 4D5 signaling is a promising therapeutic target for treating coronary and peripheral artery disease.
Introduction
Atherosclerosis is an inflammatory disease of large to midsized arteries that is induced by the convergence of biomechanical, inflammatory, and metabolic risk factors including disturbed fluid shear stress, inflammatory cytokines, oxidative stress, and high low‐density lipoprotein cholesterol.1, 2, 3, 4 Despite medical progress, it remains the major source of illness and death in developed nations.5, 6 Atherosclerotic lesions preferentially develop at regions of arteries where fluid shear stress from blood flow is lower and undergoes complex changes in direction, termed disturbed flow. These flow profiles are sensed by endothelial cells (ECs) and trigger oxidative stress,7 NF‐κB activation, and expression of downstream genes including cytokines and leukocyte recruitment receptors. This inflammatory “priming” sensitizes these regions of arteries to systemic risk factors such as hypercholesterolemia, diabetes mellitus, hypertension, and smoking, to induce atherosclerotic plaques.8 These plaques are characterized by accumulation of lipids, inflammatory cells (monocytes, macrophages, and foam cells), cholesterol crystals, proteoglycans, and extracellular matrix.5 Plaques that become more inflamed, with large necrotic cores and thin fibrous caps, are prone to rupture, which triggers thrombosis and vessel occlusion, the major source of mortality. Recent studies have revealed that plaque vulnerability is also highly correlated with low/disturbed flow as well as collagen degradation by matrix metalloproteinases (MMPs).9, 10 Hence, elucidating the mechanisms that govern plaque formation and stability and vessel remodeling is an important goal.
EC inflammatory activation by disturbed flow, oxidized low‐density lipoprotein, and IL‐1β, which are major factors in atherosclerosis, is strongly modulated by the extracellular matrix to which the cells adhere. Fibronectin (FN) through its receptors, integrin α5β1 and avb3, enhances activation of NF‐κB and other inflammatory pathways in response to disturbed flow, oxidized low‐density lipoproteins and IL‐1β to increase recruitment of inflammatory cells at atheroprone sites, whereas the collagen/laminin receptor integrin α2β1 is anti‐inflammatory.11, 12 These effects were traced to selective activation of the anti‐inflammatory mediators cyclic adenosine monophosphate (cAMP) and protein kinase A in ECs on collagen or basement membrane protein but not FN.7 The role of FN, however, is not entirely deleterious. When plaque phenotype was examined after deletion of plasma FN, plaques were smaller but appeared more vulnerable, with thinner fibrous caps, less collagen and smooth muscle actin, and evidence of plaque rupture and healing.13 Matrix remodeling therefore has complex effects in plaque pathology.
To elucidate the role of integrin‐specific signaling in these processes, we created a chimeric integrin in which the cytoplasmic domain of integrin α5 was replaced with that of integrin α2 (α5/2 chimera). This receptor pairs with the β1 subunit and binds FN but signals as if bound to collagen, activating cAMP and protein kinase A, and inhibiting NF‐κB.14 α5/2 knock‐in mice on the apolipoprotein E (ApoE−/−)background had reduced atherosclerotic plaque size in the aortic arch, supporting an inflammatory role for integrin α5. These effects were linked to an interaction between α5 cytoplasmic domain and cAMP‐specific phosphodiesterase (PDE) 4D5. In the current study, we further investigate the role of FN in atherosclerosis and vascular homeostasis by more closely examining plaque phenotype and blood flow recovery in the integrin α5/2 mouse.
Materials and Methods
All data are available in the article. Reagents will be made available upon reasonable request.
Mice
All mouse protocols were approved by the Yale University Institutional Animal Care & Use Committee. ApoE mice on a pure C57BL6 background were purchased from Jackson Laboratory (Bar Harbor, ME) and maintained in the colony. ApoE mice were crossed with integrin α5/2 knock‐in mice on a pure C57BL6 background. Age‐matched, 8‐week‐old male mice were maintained on a Western diet (RD Western diet #D12079B, Open Source Diet) for 4 weeks (wild‐type [WT];ApoE−/−, n=5 mice; α5/2;ApoE−/− n=5 mice) or 16 weeks (WT;ApoE−/− mice, n=9; α5/2;ApoE−/− mice, n=9) as indicated.
PDE4D small interfering RNA (siRNA) and control luciferase siRNA packaged into endothelial specific nanoparticles were administered intravenously via tail vein into WT (C57BL6) male mice. Mouse aortas were isolated and intimal RNA prepared. Quantitative real‐time polymerase chain reaction analysis was carried out for PDE4D and other inflammation markers as described previously.14 For the atherosclerotic studies, 8‐week‐old male ApoE−/− mice were placed on a high‐fat diet and injected every 2 weeks with PDE4D or control siRNA for 4 injections in total. For all experiments, mice were euthanized according to the Yale University Institutional Animal Care & Use Committee–approved protocol. Mice were perfused with 4% paraformaldehyde via the left ventricle. The heart with ascending aorta was dissected and fixed overnight in 4% paraformaldehyde and used for histological analysis.
Immunofluorescence and Histochemistry
For cryosections, tissue was embedded in optimal cutting temperature, frozen on dry ice, and stored at −80°C. Aortic roots were sectioned on a cryostat to generate 10‐ to 15‐μm sections. Cryosections were fixed in acetone for 10 minutes at −20°C, blocked in IHC Tek antibody diluent (IHC World, Ellicott City, MD) for 1 hour at room temperature, and incubated with the indicated antibodies in IHC Tek antibody diluent buffer. Antibodies were phospho‐NFκB‐P65 (1:100; Cell Signaling Technology, Danvers, MA); FN (1:400; Sigma, St. Louis, MO); VCAM‐1 (1:200; BD Biosciences, San Jose, CA); ICAM‐1 (1:200; Biolegend, San Diego, CA); MMP9 (1:200; Abcam, Cambridge, UK); MMP2 (1:300; Millipore, Burlington, MA); CD45 (1:150; BD Pharmingen); CD68 (1:200; Abcam); SMA (1:200; Sigma); F480 (1:200; Serotec, Hercules, CA). Sections were washed 3 times in PBS and incubated with Alexa fluor 598‐conjugated donkey anti‐rabbit or ‐rat secondary antibody (Invitrogen, Carlsbad, CA) for 1 hour at room temperature. Slides were washed with PBS and mounted in Vectashield with DAPI (Vector Laboratories, Burlingame, CA). Images were acquired using a Nikon 4 laser confocal microscope. For histology, sections were stained with hematoxylin and eosin, Oil Red, or picrosirius red.
Quantification and Statistical Analysis
ImageJ software (National Institutes of Health, Bethesda, MD) was used for morphometric analysis. Mean lesion area and necrotic core area in the aortic root (in mm2) were calculated from the hematoxylin and eosin–stained aortic root sections. All graphs were created using GraphPad Prism software (San Diego, CA) and statistical analysis were performed using a Student's t test or a 2‐way‐ANOVA (multiple comparisons) as described in GraphPad Prism 6. P values below 0.05 are considered significant.
Hindlimb Ischemia
Age‐matched (12‐ to 14‐week‐old) WT (n=9) and α5/2 knock‐in mice (n=8) were subjected to femoral artery ligation, and blood‐flow imaging was carried out by laser Doppler. Vascular anatomy was analyzed by high‐resolution micro‐CT (GE eXplore Locus SP, GE Healthcare, Chicago, IL) as described previously.15, 16
Results
Early Inflammatory Signaling in ApoE−/− Mice
FN is deposited at atherosclerosis‐prone regions in wild type C57/Bl6 mice and increases as plaques develop in hyperlipidemic models.12 To examine the consequences of altered α5β1 signaling in early plaque development, we maintained WT;ApoE−/− and α5/2;ApoE−/− mice on a high‐fat diet for 4 weeks and stained for FN and NF‐κB target genes in the aortic root. α5/2;ApoE−/− mice showed reduced staining for FN, the endothelial leukocyte recruitment receptor VCAM‐1, and inflammatory cells compared with WT;ApoE−/− mice (Figure 1). Thus, the α5 tail promotes high‐fat diet–induced inflammatory signaling at early stages of plaque formation.
Figure 1.
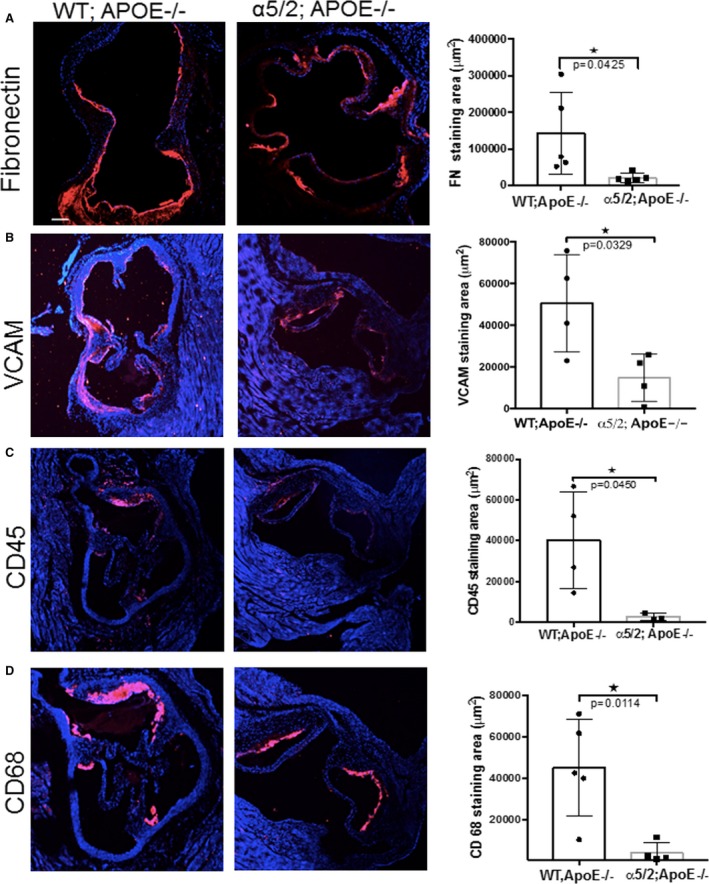
Inflammatory signaling in early atherosclerosis. Sections through the aortic root from ApoE−/− and α5/2;ApoE−/− mice on a high‐fat Western diet for 4 weeks were stained for (A) fibronectin (ApoE−/− mice, n=5; α5/2;ApoE−/− mice, n=5), (B) VCAM‐1 (ApoE−/− mice, n=4; α5/2;ApoE−/− mice, n=4), (C) CD45 (ApoE−/− mice, n=4; α5/2;ApoE−/− mice, n=3), or (D) CD68 (ApoE−/− mice, n=5; α5/2;ApoE−/− mice, n=4). Nuclei were counterstained with DAPI (blue). Right, Quantification of staining. Values are means±SD (*P<0.05 compared with ApoE−/−; unpaired 2‐tailed Student's t test). APOE indicates ApoE; FN, fibronectin; WT, wild type.
Advanced Atherosclerotic Lesions in ApoE−/− Mice
We next examined aortic root lesions in mice on a high‐fat diet for 16 weeks. Morphometric analysis of the hematoxylin and eosin–stained aortic root showed a nearly 50% reduction in aortic root area occupied by plaque in α5/2;ApoE−/− mice compared with WT;ApoE−/− mice (Figure 2A), similar to what was observed in the aortic arch.14 Further, plaques in α5/2;ApoE−/− had significantly smaller necrotic cores compared with WT;ApoE−/− mice, and lipid staining with Oil Red O showed similar reductions (Figure 2B). No differences in plasma lipids or body weight were observed between WT and α5/2 mice.14
Figure 2.
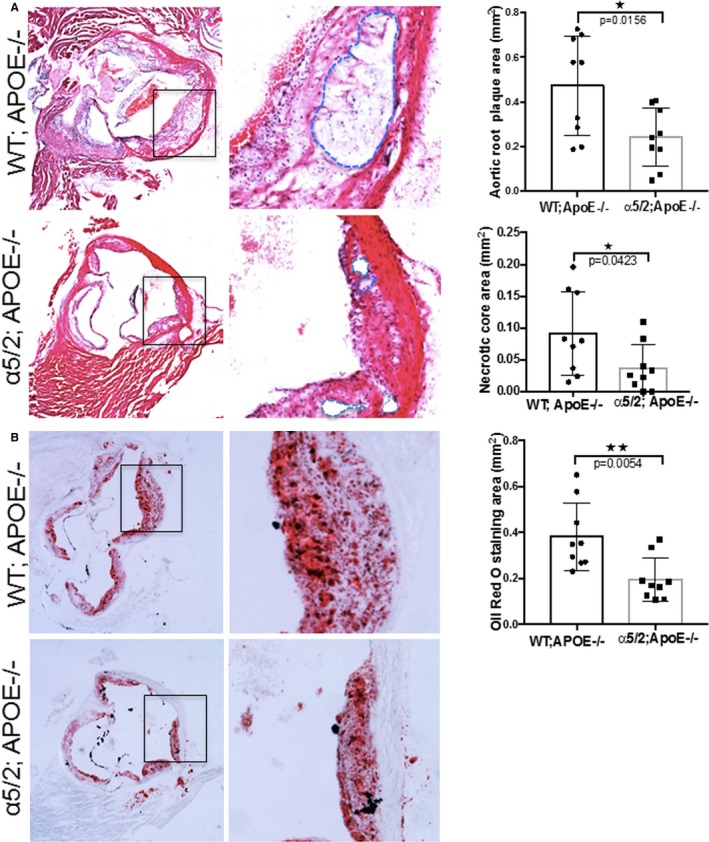
Advanced atherosclerosis. Aortic roots from ApoE−/− and α5/2;ApoE−/− mice on a high‐fat diet for 16 weeks. A, hematoxylin and eosin–stained aortic root sections with quantification of total plaque area (ApoE−/− mice, n=9; α5/2;ApoE−/− mice, n=9); and necrotic core area (ApoE−/− mice, n=9; α5/2;ApoE−/− mice, n=9). Necrotic areas are highlighted with dotted blue lines. B, Oil Red staining of aortic roots and quantification of the stained area (ApoE−/− mice, n=9; α5/2;ApoE−/− mice, n=9). Values are means±SD (*P<0.05, **P<0.01 compared with ApoE−/−; unpaired 2‐tailed Student's t test). APOE indicates ApoE; WT, wild type.
NF‐κB Activation and Leukocyte Accumulation
To gain further insight into the mechanisms involved, we examined activation of NF‐κB and induction of downstream inflammatory genes in mice after 16 weeks of high‐fat feeding. Aortic root sections were stained for phosphorylated NF‐κB subunit p65, a step in NF‐κB activation that accompanies nuclear translocation and promotes expression of target genes. Phospho‐p65 staining in the endothelial layer of the aortic lesion was significantly reduced in the α5/2;ApoE−/− compared with WT;ApoE−/− mice (Figure 3A). The leukocyte recruitment receptors ICAM1 and VCAM1, which are induced in part through NF‐κB, were significantly reduced in atherosclerotic lesions in the α5/2;ApoE−/− compared with WT;ApoE−/− mice (Figure 3B and 3C). To test functional consequences, we stained sections for markers of inflammatory cells. CD45, a general marker for hematopoietic cells, was significantly reduced in α5/2;ApoE−/− aortic root sections (Figure 3D), as were the macrophage markers CD68 (Figure 3E) and F4/80, a marker for mature macrophages (Figure 3F). Together, these results demonstrate that the α5 tail promotes plaque expansion and inflammation.
Figure 3.
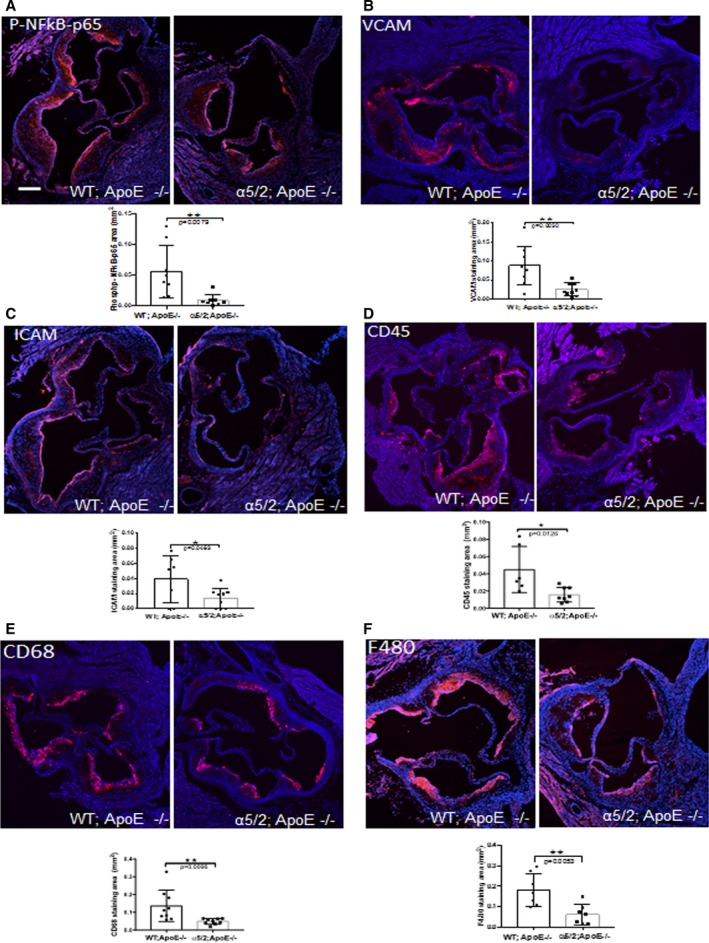
Plaque inflammatory markers. Aortic roots from ApoE−/− and α5/2;ApoE−/− mice on a high‐fat diet for 16 weeks were stained for (A) phospho‐S536 NF‐κB p65 (ApoE−/− mice, n=9; α5/2;ApoE−/− mice, n=9), (B) VCAM‐1; (ApoE−/− mice, n=9; α5/2;ApoE−/− mice, n=9), (C) ICAM‐1 (ApoE−/− mice, n=7; α5/2;ApoE−/− mice, n=9), (D) CD45 (ApoE−/− mice, n=8; α5/2;ApoE−/− mice, n=9), (E) CD68 (ApoE−/− mice, n=9; α5/2;ApoE−/− mice, n=9), and (F) F480 (ApoE−/− mice, n=7; α5/2;ApoE−/− mice, n=7). For quantification, values are means±SD (*P<0.05, **P<0.01 compared with ApoE−/−; unpaired 2‐tailed Student's t test). Nuclei are counterstained with DAPI (blue).
Matrix Composition and Fibrous Cap Thickness
Inflammation and proteolysis of the extracellular matrix are major contributors to plaque vulnerability.17, 18 Plaque is stabilized by a fibrous cap, rich in smooth muscle cells and collagen, and destabilized by inflammatory cells that produce matrix‐degrading metalloproteinases, principally MMP2 and 9.19, 20, 21, 22 We therefore examined these components. When fibrillar collagen, a major determinant of tensile strength of the plaque, was assessed by staining with picrosirius red, there was no detectable difference between plaque in α5/2;ApoE−/− and ApoE−/− mice (Figure 4A). FN staining was also similar (Figure 4B), which may reflect its role in tissue repair and remodeling through the TGFβ23, 24, 25 and β‐catenin pathways.26, 27 By contrast, the major matrix‐degrading enzymes, MMP9 and MMP2, were significantly reduced in the α5/2;ApoE−/− mice (Figure 4C and 4D). Vascular smooth muscle cells synthesize a large fraction of the collagen in the fibrous cap and promote plaque stability.17, 28, 29 When smooth muscle cell content was evaluated by staining for smooth muscle actin, the staining in the plaque core, media, and fibrous cap showed no significant difference between α5/2;ApoE−/− and WT;ApoE−/− mice (Figure 5). Fibrous cap thickness was also similar between WT and α5/2;ApoE−/− mice. Mutation of the integrin α5 tail thus results in less plaque with decreased inflammation and no evidence of vulnerability.
Figure 4.
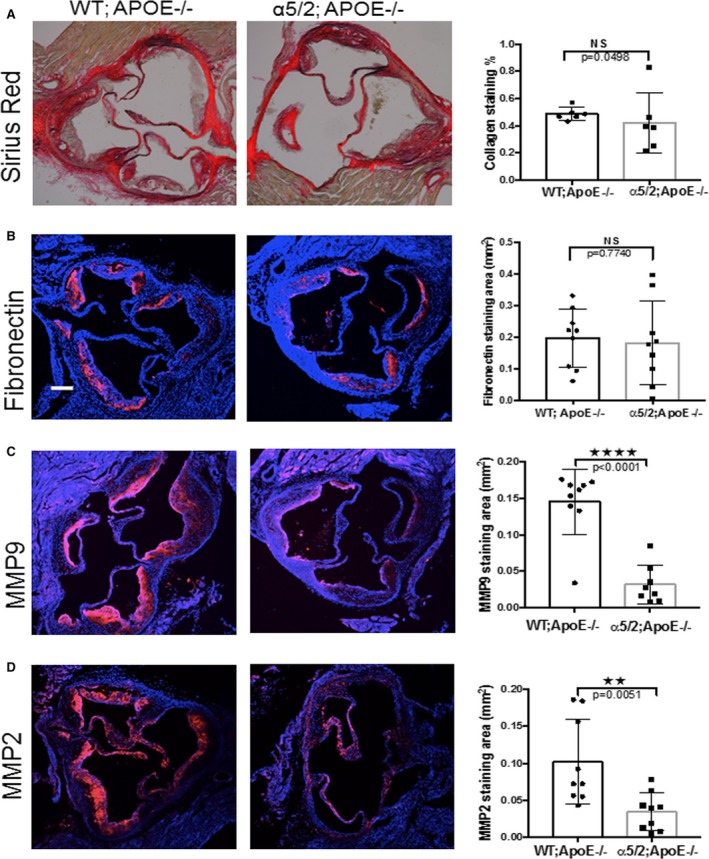
Matrix and matrix remodeling. Aortic root sections from ApoE−/− and α5/2;ApoE−/− mice on a high‐fat diet for 16 weeks were stained as indicated, with nuclei counterstained with DAPI (blue). Adjacent graphs show quantification as means±SD. A, Picrosirius red (ApoE−/− mice, n=6; α5/2;ApoE−/− mice, n=6). B, Anti‐fibronectin (ApoE−/− mice, n=9; α5/2;ApoE−/− mice, n=9). C, Anti‐MMP9 (ApoE−/− mice, n=9; α5/2;ApoE−/− mice, n=8). D, Anti‐MMP2 (ApoE−/− mice, n=9; α5/2;ApoE−/− mice, n=9). NS, not significant; ****P<0.0001; **P<0.005 compared with ApoE−/−; unpaired 2‐tailed Student's t test). MMP indicates matrix metalloproteinase.
Figure 5.
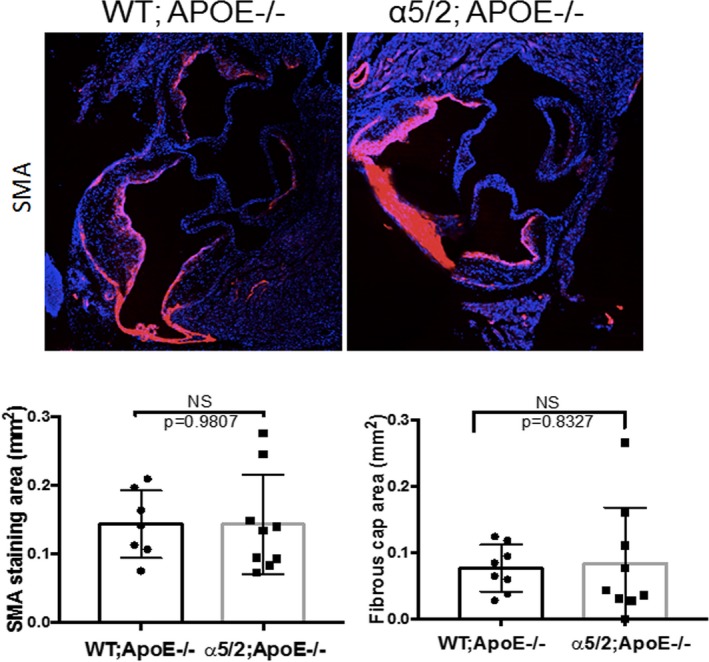
Fibrous caps. Aortic root sections from WT;ApoE−/− and α5/2;ApoE−/− mice on a high‐fat diet for 16 weeks were stained for smooth muscle actin (SMA). Quantification shows the percentage of the plaque area stained positive for SMA (ApoE−/− mice, n=7; α5/2;ApoE−/− mice, n=9) and fibrous cap thickness (ApoE−/− mice, n=8; α5/2;ApoE−/− mice, n=9). Values are means±SD (NS, Not significant compared with ApoE−/−; unpaired 2‐tailed Student's t test). APOE indicates ApoE; WT, wild type.
In Vivo PDE4D Knockdown and Atherosclerosis
Our previous data implicated PDE4D5 as a binding partner for the integrin α5 cytoplasmic domain that mediated the proinflammatory effects of FN.14 We therefore investigated whether its depletion in ECs in vivo reduced atherosclerosis. siRNA packaged into nanoparticles specifically target the endothelium without affecting hepatocytes, hematopoietic cells, or other tissues.30 First, we evaluated the endothelial in vivo knockdown of PDE4D and other proinflammatory markers in intimal RNA, isolated from athero‐prone regions of WT mice injected with PDE4D or control luciferase siRNA, as previously described.14 PDE4D siRNA greatly reduced the mRNA levels of PDE4D, FN, VCAM, and MMP2 compared with luciferase siRNA (Figure 6A). Next, ApoE−/− mice on a high‐fat diet were treated with serial injections of PDE4D or luciferase siRNA as outlined (Figure 6B). Intravenous injection of these particles had no effect on plasma low‐density lipoprotein cholesterol or triglycerides or on body weight (Figure 6C). Examination of aortic roots in these mice after 8 weeks revealed significantly reduced Oil Red O staining in PDE4D‐ compared with control‐injected mice (Figure 6D). The NF‐κB target genes VCAM1 and ICAM1, which in luciferase siRNA‐injected mice were elevated in atherosclerotic plaques compared with uninvolved regions, were significantly reduced after PDE4D knockdown (Figure 7A and 7B). PDE4D siRNA also reduced plaque staining for the leukocyte markers CD45 and CD68 (Figure 7C and 7D). These results are consistent with PDE4D5 as a downstream effector for integrin α5 proinflammatory signaling in atherosclerosis.
Figure 6.
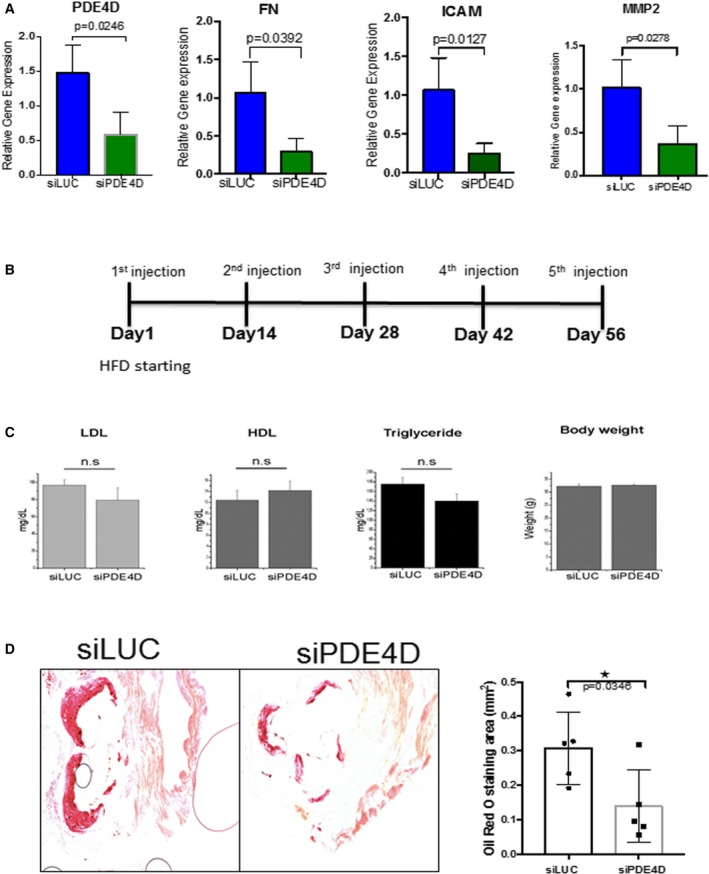
Endothelial in vivo PDE4D5 knockdown. PDE4D siRNA or luciferase siRNA were packaged into endothelial‐specific nanoparticles and administered intravenously into WT (C57BL6) mice. A, Intimal RNA was isolated and assayed for PDE4D and proinflammatory gene expression by quantitative real‐time polymerase chain reaction. B, Outline of PDE4D siRNA protocol for atherosclerosis. ApoE−/− mice on a high‐fat diet were serially injected with nanoparticle‐packaged PDE4D or luciferase siRNA and examined at 8 weeks. C, Plasma lipid levels. D, Aortic root sections were stained for Oil Red O, and the positively stained area was quantified (siLUC; n=5 mice; siPDE4D, n=5 mice). *P<0.05 compared with ApoE−/− by unpaired 2‐tailed Student's t test. FN indicates fibronectin; HDL, high‐density lipoprotein; HFD, high‐fat diet; LDL, low‐density lipoprotein; LUC, luciferase; MMP, matrix metalloproteinase; PDE, phosphodiesterase; si, small interfering [RNA]; WT, wild type.
Figure 7.
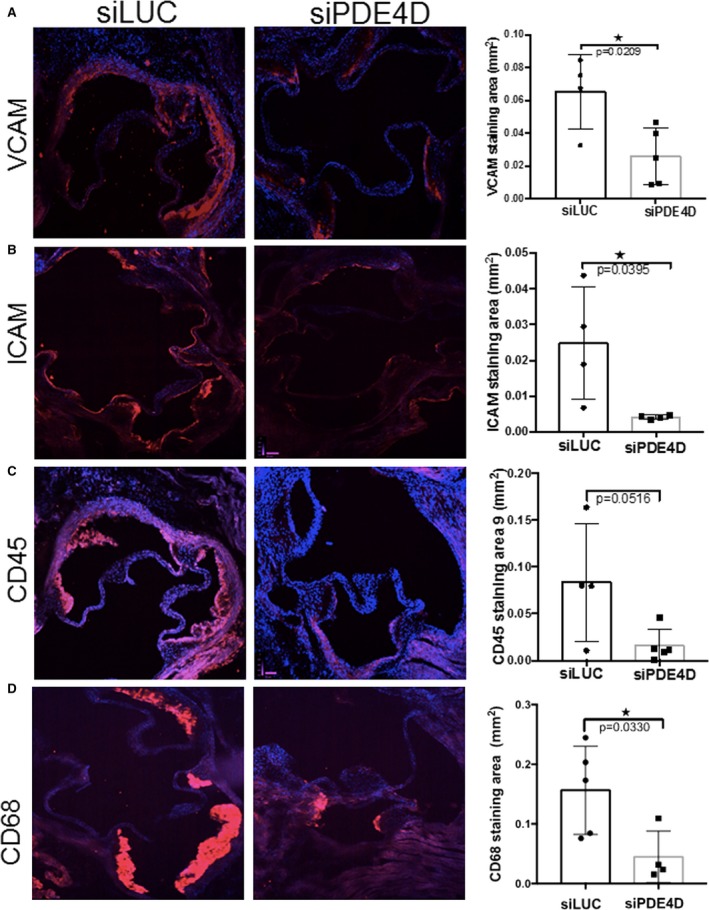
In vivo PDE4D (4D5) knockdown and plaque inflammatory markers. ApoE−/− mice on a high‐fat diet were serially injected with nanoparticle‐packaged PDE4D or luciferase siRNA, and examined at 8 weeks. Aortic root sections were stained for (A) VCAM‐1 (siLUC, n=4 mice; siPDE4D, n=5 mice); (B) ICAM‐1 (siLUC, n=4 mice; siPDE4D, n=4 mice); (C) CD45 (siLUC, n=4 mice; siPDE4D, n=5 mice); and (D) CD68 (siLUC, n=5 mice; siPDE4, n=4 mice), as indicated. For quantification, values are means±SD *P<0.05 compared with ApoE−/− by unpaired 2‐tailed Student's t test. LUC indicates luciferase; PDE, phosphodiesterase; si, small interfering [RNA].
Hindlimb Ischemia in Integrin α5/2 Mice
The major mechanism for adaptation or recovery following an arterial blockage by atherosclerotic plaque is compensatory remodeling. This process is triggered by elevated shear stress in the small vessels parallel to the blocked artery, resulting in their enlargement and arterialization, to provide blood flow to affected tissues downstream. To compare responses of α5/2 and WT mice to arterial blockage, femoral arteries were ligated. Blood flow in the lower limbs was then measured by laser Doppler imaging, with blood flow in the unaffected limb serving as a control. These measurements showed that recovery of flow was accelerated in the α5/2 mice compared with WT (Figure 8A, quantified in 8B). Next, we analyzed the remodeled vasculature. Staining with platelet/endothelial cell adhesion molecule antibody to mark capillaries in the lower leg at 21 days after surgery showed evidence of angiogenesis but no significant difference between WT and α5/2 mice (Figure 9A). Next, larger vessels were visualized by micro CT. In these experiments the vasculature is maximally dilated prior to fixation, followed by injection of contrast agent and imaging. α5/2 mice had decreased vascular density in both the thigh and calf regions compared with WT mice (Figure 9B). To resolve the discrepancy, we also stained for smooth muscle actin in sections from the adductor muscle in the thigh to mark the arteries, fixed without prior dilation. Under these conditions, we observed that the increase in artery diameter triggered by femoral artery ligation was greater in the α5/2 mice (Figure 9C). Together, these data suggest that α5/2 mice have improved recovery from hindlimb ischemia (HLI) mainly due to improved vessel function.
Figure 8.
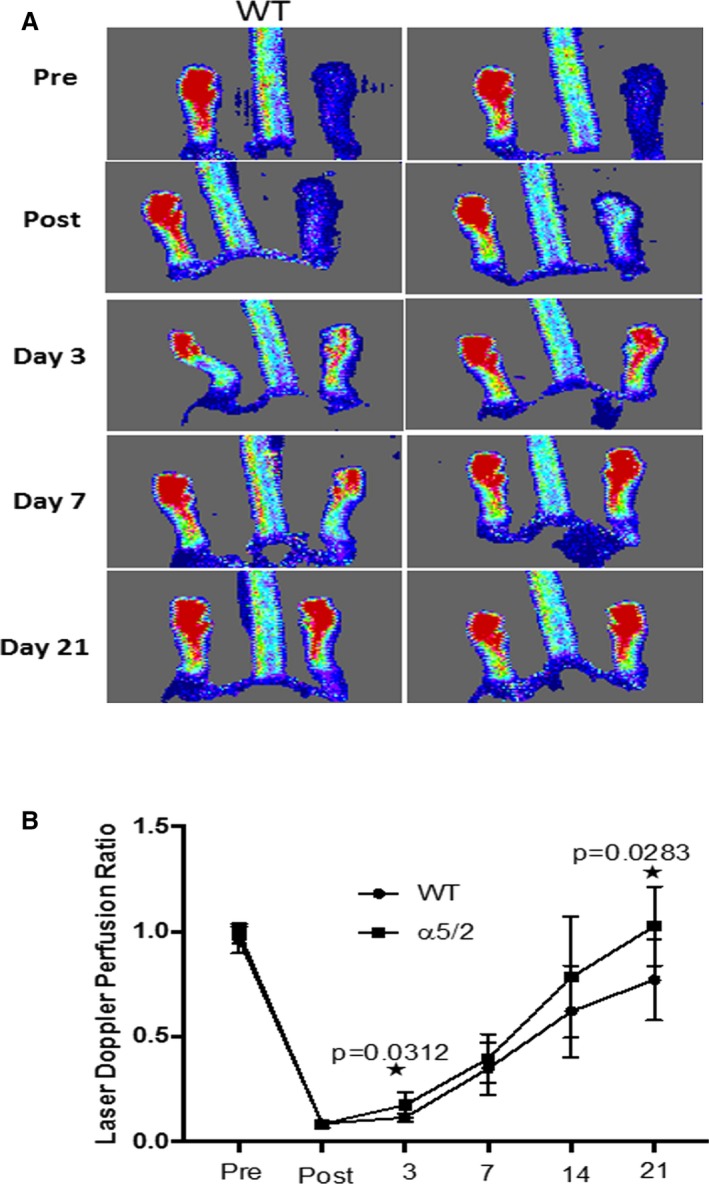
Blood flow recovery after hindlimb ischemia. A, Representative laser Doppler images of flow in the hind limbs before surgery and at the indicated times after femoral artery ligation. B, Quantification from multiple mice (WT mice, n=9; α5/2 mice, n=9). Values are means±SD. *P<0.05 by unpaired 2‐tailed Student's t test. WT indicates wild type.
Figure 9.
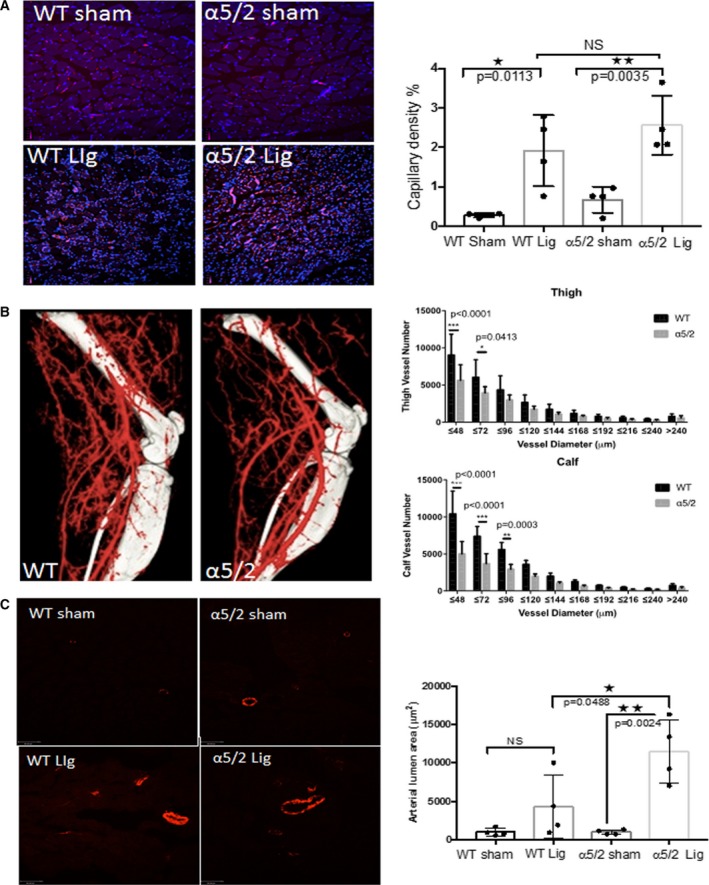
Arterial vasculature after hindlimb ischemia. Analysis of vasculature in WT and mutant mice 21 days after surgery. A, Sections through the calf region were stained with PECAM antibody (CD31) to mark blood vessels in the lower leg. Capillary density was then quantified (WT mice, n=4; α5/2 mice, n=4). B, Representative microCT images of vasculature. The number of vessels within each size range was quantified as described in Methods (WT mice, n=5; α5/2 mice, n=5; *P<0.001 by 2‐way ANOVA, multiple comparisons). C, Sections through the upper leg were stained for smooth muscle actin to label arteries and vessel diameter measured (WT mice, n=4; α5/2 mice, n=4). For all panels, values are means±SD. *P<0.05, **P<0.005 compared with WT mice by unpaired 2‐tailed Student's t test. CT indicates computed tomography; Lig, ligated; PECAM, platelet/endothelial cell adhesion molecule; WT, wild type.
Discussion
Previous studies showed that genetic manipulations that reduced FN in the vessel wall reduced atherosclerosis. However, in the study that examined plaque phenotype,13 deletion of plasma FN resulted in thinning of the fibrous cap and plaque rupture, demonstrating that FN contributes to plaque stabilization. Mechanistic work in our lab showed that the major FN receptor, integrin α5, promotes inflammatory activation of ECs by binding PDE4D5. This interaction promotes PDE enzymatic activity and inhibits the anti‐inflammatory cAMP–protein kinase A pathway, thus sensitizing ECs to inflammatory stimuli.14 This study also showed that replacing the cytoplasmic domain of α5 with that of α2 in mice reduced atherosclerotic plaque size in the aortic arch, supporting a role in vivo. In the current study, we analyzed plaque phenotype in greater detail in α5/2;ApoE−/− mice. We found that these mice had reduced early and late inflammatory activation in the vessel wall and smaller plaques in the aortic root. Importantly, plaques in these mice had lower MMP expression and maintained fibrous cap thickness. Endothelial‐specific in vivo knockdown of PDE4D5 also reduced plaque burden, supporting the notion that this enzyme is an important mediator of the inflammatory, proatherosclerotic effects of FN. However, maintaining knockdown of the enzyme for longer periods is technically difficult, and the shorter time for these experiments precluded analysis of plaque stability.
FN is well known to serve as a scaffold that promotes collagen fibrillogenesis.31, 32 These results lead us to hypothesize that FN has at least 2 distinct functional roles in atherosclerosis. By binding integrin α5β1, it promotes inflammatory signaling and favors progression of the atherosclerotic plaque. However, through its role in collagen fibrillogenesis, FN also promotes plaque stability. Because plaque rupture is thought to be the major cause of clinical events, FN synthesis and matrix assembly are not attractive therapeutic targets.
Results in the HLI model, however, support the α5‐PDE‐cAMP pathway as a potential therapeutic target. These experiments showed that blood flow in the affected limbs of α5/2 mice recovered more rapidly than in WT controls after femoral artery ligation. This outcome is surprising in that C57BL6 recover well from HLI and, indeed, have been used in genetic studies as a prototypical high‐responding strain.33, 34 The improved recovery in a system where remodeling is already rapid strongly encourages examining these effects in genetic backgrounds and disease settings where recovery is poor.
The second surprising result is that the improved flow recovery was associated with less rather than more vascularization. Thus, blocking the FN‐ α5/2‐PDE pathway does not promote vessel remodeling per se. A more likely explanation is that the α5/2 mutation improves vessel function to allow higher blood flow through the existing vasculature. Indeed, when analyzed without prior vasodilation, arteries in the upper leg in α5/2 mice had larger diameters. A similar improved recovery from HLI while decreasing vascular density as measured by microCT was seen following EC‐specific inhibition of NF‐κB.16 Because microCT is done in mice injected with a strong vasodilator before fixation, differences in vessel function would not be detected. This idea is consistent with a report that plating ECs on FN reduced endothelial nitric oxide synthase activation and NO production, and that injection of an antagonist of integrin α5 increased plasma nitrate levels in mice.35 Whatever the mechanism, the improved blood flow recovery after HLI further supports the idea that blocking FN signaling through α5 may be beneficial in vascular disease.
The α5/2 chimera was a whole‐animal knock‐in; thus, multiple cell types could contribute to the observed effects. In vitro experiments with ECs showed reduced NF‐κB activation and inflammatory gene expression,14 which fits well with reduced inflammatory gene expression in the endothelium in early atherosclerosis, supporting these cells as important mediators. However, smooth muscle cells also show modulation of inflammatory pathways by matrix proteins,36, 37 as do monocyte/macrophage lineages.38, 39 Moreover, both of these lineages play roles in both inflammatory processes and fibrous cap phenotype. Further work with tissue‐specific knock‐in will be required to sort out these questions.
In conclusion, these results resolve the dual role of FN in atherosclerosis by demonstrating that signaling through integrin α5β1 mediates the proinflammatory effects but not the effects on plaque stability. Proinflammatory signaling is mediated at least in part through PDE4D5. The data further demonstrate that inhibiting FN‐α5 signaling promotes recovery from arterial restriction. Further study to determine whether this pathway can serve as a therapeutic target in atherosclerosis and possibly other chronic inflammatory diseases therefore seems warranted.
Author Contributions
Budatha designed the study, performed all experiments, and analyzed data except where otherwise noted, and wrote the article. Zhang performed femoral artery ligation surgery. Zhuang performed and analyzed microCT. Yun developed the in vivo siRNA sequences. Dahlman carried out nanoparticle packaging for in vivo knockdowns. Anderson supervised and supported in vivo nanoparticle packaging. Schwartz initiated and supervised the project, co‐wrote the article, and provided financial support.
Sources of Funding
This work was supported by National Institute of Health (RO1 HL75092 and HL107205) to Schwartz.
Disclosures
None.
Acknowledgments
Lipid analysis was done by the Yale Mouse Phenotypic Center supported by U24 DKO59635 grant. We are thankful to Rita Webber and Nicole Copeland for maintaining the mouse colonies used in the study. We thank Tristan Driscoll (Yale University) for advice about statistical analysis.
(J Am Heart Assoc. 2018;7:e007501 DOI: 10.1161/JAHA.117.007501.)29382667
Contributor Information
Madhusudhan Budatha, Email: madhusudhanhcu294@gmail.com.
Martin A. Schwartz, Email: martin.schwartz@yale.edu.
References
- 1. Herrington W, Lacey B, Sherliker P, Armitage J, Lewington S. Epidemiology of atherosclerosis and the potential to reduce the global burden of atherothrombotic disease. Circ Res. 2016;118:535–546. [DOI] [PubMed] [Google Scholar]
- 2. Libby P, Ridker PM, Hansson GK. Progress and challenges in translating the biology of atherosclerosis. Nature. 2011;473:317–325. [DOI] [PubMed] [Google Scholar]
- 3. Virmani R, Kolodgie FD, Burke AP, Farb A, Schwartz SM. Lessons from sudden coronary death: a comprehensive morphological classification scheme for atherosclerotic lesions. Arterioscler Thromb Vasc Biol. 2000;20:1262–1275. [DOI] [PubMed] [Google Scholar]
- 4. Tabas I, Garcia‐Cardena G, Owens GK. Recent insights into the cellular biology of atherosclerosis. J Cell Biol. 2015;209:13–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Naghavi M, Libby P, Falk E, Casscells SW, Litovsky S, Rumberger J, Badimon JJ, Stefanadis C, Moreno P, Pasterkamp G, Fayad Z, Stone PH, Waxman S, Raggi P, Madjid M, Zarrabi A, Burke A, Yuan C, Fitzgerald PJ, Siscovick DS, de Korte CL, Aikawa M, Airaksinen KE, Assmann G, Becker CR, Chesebro JH, Farb A, Galis ZS, Jackson C, Jang IK, Koenig W, Lodder RA, March K, Demirovic J, Navab M, Priori SG, Rekhter MD, Bahr R, Grundy SM, Mehran R, Colombo A, Boerwinkle E, Ballantyne C, Insull W Jr, Schwartz RS, Vogel R, Serruys PW, Hansson GK, Faxon DP, Kaul S, Drexler H, Greenland P, Muller JE, Virmani R, Ridker PM, Zipes DP, Shah PK, Willerson JT. From vulnerable plaque to vulnerable patient: a call for new definitions and risk assessment strategies: Part II. Circulation. 2003;108:1772–1778. [DOI] [PubMed] [Google Scholar]
- 6. Dave T, Ezhilan J, Vasnawala H, Somani V. Plaque regression and plaque stabilisation in cardiovascular diseases. Indian J Endocrinol Metab. 2013;17:983–989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Funk SD, Yurdagul A Jr, Green JM, Jhaveri KA, Schwartz MA, Orr AW. Matrix‐specific protein kinase A signaling regulates p21‐activated kinase activation by flow in endothelial cells. Circ Res. 2010;106:1394–1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. DeBakey ME, Glaeser DH. Patterns of atherosclerosis: effect of risk factors on recurrence and survival—analysis of 11,890 cases with more than 25‐year follow‐up. Am J Cardiol. 2000;85:1045–1053. [DOI] [PubMed] [Google Scholar]
- 9. Papafaklis MI, Takahashi S, Antoniadis AP, Coskun AU, Tsuda M, Mizuno S, Andreou I, Nakamura S, Makita Y, Hirohata A, Saito S, Feldman CL, Stone PH. Effect of the local hemodynamic environment on the de novo development and progression of eccentric coronary atherosclerosis in humans: insights from PREDICTION. Atherosclerosis. 2015;240:205–211. [DOI] [PubMed] [Google Scholar]
- 10. Andreou I, Antoniadis AP, Shishido K, Papafaklis MI, Koskinas KC, Chatzizisis YS, Coskun AU, Edelman ER, Feldman CL, Stone PH. How do we prevent the vulnerable atherosclerotic plaque from rupturing? Insights from in vivo assessments of plaque, vascular remodeling, and local endothelial shear stress. J Cardiovasc Pharmacol Ther. 2015;20:261–275. [DOI] [PubMed] [Google Scholar]
- 11. Yurdagul A Jr, Green J, Albert P, McInnis MC, Mazar AP, Orr AW. α5β1 integrin signaling mediates oxidized low‐density lipoprotein‐induced inflammation and early atherosclerosis. Arterioscler Thromb Vasc Biol. 2014;34:1362–1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Orr AW, Sanders JM, Bevard M, Coleman E, Sarembock IJ, Schwartz MA. The subendothelial extracellular matrix modulates NF‐κB activation by flow: a potential role in atherosclerosis. J Cell Biol. 2005;169:191–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Rohwedder I, Montanez E, Beckmann K, Bengtsson E, Duner P, Nilsson J, Soehnlein O, Fassler R. Plasma fibronectin deficiency impedes atherosclerosis progression and fibrous cap formation. EMBO Mol Med. 2012;4:564–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Yun S, Budatha M, Dahlman JE, Coon BG, Cameron RT, Langer R, Anderson DG, Baillie G, Schwartz MA. Interaction between integrin α5 and PDE4D regulates endothelial inflammatory signalling. Nat Cell Biol. 2016;18:1043–1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Conway DE, Coon BG, Budatha M, Arsenovic PT, Orsenigo F, Wessel F, Zhang J, Zhuang Z, Dejana E, Vestweber D, Schwartz MA. VE‐cadherin phosphorylation regulates endothelial fluid shear stress responses through the polarity protein LGN. Curr Biol. 2017;27:2727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Tirziu D, Jaba IM, Yu P, Larrivee B, Coon BG, Cristofaro B, Zhuang ZW, Lanahan AA, Schwartz MA, Eichmann A, Simons M. Endothelial nuclear factor‐κB‐dependent regulation of arteriogenesis and branching. Circulation. 2012;126:2589–2600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Falk E, Shah PK, Fuster V. Coronary plaque disruption. Circulation. 1995;92:657–671. [DOI] [PubMed] [Google Scholar]
- 18. Libby P. The molecular mechanisms of the thrombotic complications of atherosclerosis. J Intern Med. 2008;263:517–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Galis ZS, Sukhova GK, Kranzhofer R, Clark S, Libby P. Macrophage foam cells from experimental atheroma constitutively produce matrix‐degrading proteinases. Proc Natl Acad Sci USA. 1995;92:402–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Galis ZS, Sukhova GK, Lark MW, Libby P. Increased expression of matrix metalloproteinases and matrix degrading activity in vulnerable regions of human atherosclerotic plaques. J Clin Invest. 1994;94:2493–2503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Galis ZS, Muszynski M, Sukhova GK, Simon‐Morrissey E, Unemori EN, Lark MW, Amento E, Libby P. Cytokine‐stimulated human vascular smooth muscle cells synthesize a complement of enzymes required for extracellular matrix digestion. Circ Res. 1994;75:181–189. [DOI] [PubMed] [Google Scholar]
- 22. Schneider F, Sukhova GK, Aikawa M, Canner J, Gerdes N, Tang SM, Shi GP, Apte SS, Libby P. Matrix‐metalloproteinase‐14 deficiency in bone‐marrow‐derived cells promotes collagen accumulation in mouse atherosclerotic plaques. Circulation. 2008;117:931–939. [DOI] [PubMed] [Google Scholar]
- 23. Ignotz RA, Endo T, Massague J. Regulation of fibronectin and type I collagen mRNA levels by transforming growth factor‐β. J Biol Chem. 1987;262:6443–6446. [PubMed] [Google Scholar]
- 24. Raghow R, Postlethwaite AE, Keski‐Oja J, Moses HL, Kang AH. Transforming growth factor‐beta increases steady state levels of type I procollagen and fibronectin messenger RNAs posttranscriptionally in cultured human dermal fibroblasts. J Clin Invest. 1987;79:1285–1288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ignotz RA, Massague J. Transforming growth factor‐beta stimulates the expression of fibronectin and collagen and their incorporation into the extracellular matrix. J Biol Chem. 1986;261:4337–4345. [PubMed] [Google Scholar]
- 26. Gradl D, Kuhl M, Wedlich D. The Wnt/Wg signal transducer β‐catenin controls fibronectin expression. Mol Cell Biol. 1999;19:5576–5587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gelfand BD, Meller J, Pryor AW, Kahn M, Bortz PD, Wamhoff BR, Blackman BR. Hemodynamic activation of β‐catenin and T‐cell‐specific transcription factor signaling in vascular endothelium regulates fibronectin expression. Arterioscler Thromb Vasc Biol. 2011;31:1625–1633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bennett MR, Sinha S, Owens GK. Vascular smooth muscle cells in atherosclerosis. Circ Res. 2016;118:692–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Loree HM, Kamm RD, Stringfellow RG, Lee RT. Effects of fibrous cap thickness on peak circumferential stress in model atherosclerotic vessels. Circ Res. 1992;71:850–858. [DOI] [PubMed] [Google Scholar]
- 30. Dahlman JE, Barnes C, Khan OF, Thiriot A, Jhunjunwala S, Shaw TE, Xing Y, Sager HB, Sahay G, Speciner L, Bader A, Bogorad RL, Yin H, Racie T, Dong Y, Jiang S, Seedorf D, Dave A, Singh Sandhu K, Webber MJ, Novobrantseva T, Ruda VM, Lytton‐Jean AK, Levins CG, Kalish B, Mudge DK, Perez M, Abezgauz L, Dutta P, Smith L, Charisse K, Kieran MW, Fitzgerald K, Nahrendorf M, Danino D, Tuder RM, von Andrian UH, Akinc A, Panigrahy D, Schroeder A, Koteliansky V, Langer R, Anderson DG. In vivo endothelial siRNA delivery using polymeric nanoparticles with low molecular weight. Nat Nanotechnol. 2014;9:648–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kadler KE, Hill A, Canty‐Laird EG. Collagen fibrillogenesis: fibronectin, integrins, and minor collagens as organizers and nucleators. Curr Opin Cell Biol. 2008;20:495–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kubow KE, Vukmirovic R, Zhe L, Klotzsch E, Smith ML, Gourdon D, Luna S, Vogel V. Mechanical forces regulate the interactions of fibronectin and collagen I in extracellular matrix. Nat Commun. 2015;6:8026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Chalothorn D, Clayton JA, Zhang H, Pomp D, Faber JE. Collateral density, remodeling, and VEGF‐A expression differ widely between mouse strains. Physiol Genom. 2007;30:179–191. [DOI] [PubMed] [Google Scholar]
- 34. Sealock R, Zhang H, Lucitti JL, Moore SM, Faber JE. Congenic fine‐mapping identifies a major causal locus for variation in the native collateral circulation and ischemic injury in brain and lower extremity. Circ Res. 2014;114:660–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Yurdagul A Jr, Chen J, Funk SD, Albert P, Kevil CG, Orr AW. Altered nitric oxide production mediates matrix‐specific PAK2 and NF‐κB activation by flow. Mol Biol Cell. 2013;24:398–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Orr AW, Lee MY, Lemmon JA, Yurdagul A Jr, Gomez MF, Bortz PD, Wamhoff BR. Molecular mechanisms of collagen isotype‐specific modulation of smooth muscle cell phenotype. Arterioscler Thromb Vasc Biol. 2009;29:225–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Orr AW, Hastings NE, Blackman BR, Wamhoff BR. Complex regulation and function of the inflammatory smooth muscle cell phenotype in atherosclerosis. J Vasc Res. 2010;47:168–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Roman J, Ritzenthaler JD, Fenton MJ, Roser S, Schuyler W. Transcriptional regulation of the human interleukin 1β gene by fibronectin: role of protein kinase C and activator protein 1 (AP‐1). Cytokine. 2000;12:1581–1596. [DOI] [PubMed] [Google Scholar]
- 39. Reyes‐Reyes M, Mora N, Gonzalez G, Rosales C. β1 and β2 integrins activate different signalling pathways in monocytes. Biochem J. 2002;363:273–280. [DOI] [PMC free article] [PubMed] [Google Scholar]