

Introduction
Heart failure (HF) is a culmination of pathological processes presenting with debilitating symptoms that highlight a complex interplay between immunological, hormonal, and metabolic systems resulting in impaired cardiac function. HF has a major impact on the quality of life and longevity of the affected patients.1 Inflammation has been shown to play a pivotal role in the pathogenesis of HF within animal studies, but there has been limited translation of these findings into human research.2, 3
Monocytes and monocyte‐derived macrophages play a role in the development of HF. In human immunology, monocytes undertake phagocytosis to provide protection from foreign pathogens such as bacteria and viruses. Nevertheless, there are distinct subsets of monocytes with potential for beneficial or detrimental effects on HF pathogenesis, although intimate details of the involved processes are not yet fully determined.4, 5 Of importance is the fact that the role of monocytes in cardiovascular diseases is complex and includes inflammation, which subsequently contributes to processes of regeneration, repair, and modulation of the prothrombotic state.6, 7 All these functions are highly relevant to patients with HF, who show progressive impairment of cardiac function and frequently develop atrial fibrillation (AF), an arrhythmia with a high risk of thrombotic complications.
Therefore, of equal interest is the role of the immune system in AF, a very common arrhythmia in HF, which has been strongly linked to inflammation.8 Atrial and ventricular fibrosis have been documented in patients with AF, with monocytes playing a role in these processes, based on animal studies.9, 10 Indeed, inflammation in the myocardium clearly predisposes to cardiac fibrosis.11
Why is this important? Cardiac fibrosis is an active process that is part of the remodeling of the myocardium in response to mechanical, chemical, and electrical stressors, along with the inflammation.12 Myocardial fibrosis reduces left ventricular (LV) compliance and increases filling pressures and atrial load. This, in turn, promotes LV and atrial fibrosis, predisposing to AF and thus completing the vicious circle.13 HF and AF should therefore not necessarily be considered as pathophysiologically unrelated entities, and indeed the pathological/inflammatory processes underpinning both conditions seem to overlap and this may, in turn, guide therapeutic options.
The aim of this review article is to identify the role of monocytes and their associated inflammatory cells in the pathogenesis of cardiac fibrosis. Particular focus is given to monocyte subsets and their associated immune response cells in the inflammation process of HF and AF. Considerations on possible therapeutic targets related to these cells in the treatment of HF are also discussed.
Etiology and Pathogenesis of HF
A wide range of cardiac conditions, inherited defects, and systemic disorders contribute to the pathogenesis of HF. Ischemic heart disease is a major cause of HF attributed to chronic ischemia (atherosclerosis/coronary calcification) and acute myocardial necrosis (atherothrombosis).14 Hypertensive cardiac disease leads to the mechanical myocardial stress and neurohormonal changes that are detrimental to cardiac myocytes. Definitive studies have not only found significant lifetime risk of HF in people with blood pressure of over 160/90, but also provided evidence of improved blood pressure control contributing to reduced incidence of HF.15, 16 Valvular heart disease plays a lesser role as a cause of HF in the developed world attributed to improved living conditions and medical and surgical therapy. Globally, however, rheumatic heart disease is still a major cause for HF.17
LV dysfunction secondary to myocardial infarction (MI) is the most studied HF etiology. Cardiac remodeling post‐MI includes stretching of cardiac myocytes attributed to the raised intraventricular pressures found in acute cardiac ischemia.18 The noninfarcted myocardium attempts to compensate for the area of myocardial loss. Remodeling of the unaffected myocardium is a consequence of the expanding myocardial collagen scar, as well as the response to neuroendocrine stimuli and increased wall stress.18
MI is accompanied by an inflammatory process, involving the migration of macrophages, monocytes, and neutrophils into the necrotic and ischemic areas. The subsequent signaling cascade and neurohormonal activation is responsible for the recruitment of inflammatory cells to site of tissue injury.19
The early phase of postinfarct remodeling (within 72 hours) predominantly involves the infarct zone itself and can be associated with the zone expansion. Late remodeling involves the left ventricle globally and is associated with dilatation, changes in ventricular morphology, and hypertrophy. Adverse remodeling leads to cardiac failure attributed to inability to prevent or reverse progressive ventricular dilatation, expansion of the myocardial scar, and deterioration in contractile function.20
The role of monocytes/macrophages in postinfarct remodeling has been demonstrated in numerous studies.19, 21, 22 There exists a fine balance between excessive and prolonged infiltration of inflammatory macrophages into the infarct myocardium, causing a detrimental inflammatory response with subsequent cardiac fibrosis, dysfunction, and adverse ventricular remodeling.23, 24 In contrast, monocytes/macrophages are also essential to wound healing and tissue repair through phagocytosis, angiogenesis, and favorable remodeling of the extracellular matrix in the infarcted area.4 It remains unclear how the balance of contrasting roles of monocyte/macrophages is achieved. However, the role of monocyte subset populations may be a logical explanation for the diversity in function.
The hypothesis of monocyte subset heterogeneity and function in MI is formed on the basis of their role in modulating chemokine expression in mice, which, in turn, recruit Ly‐6Chigh and Ly‐6Clow monocyte subset through C‐C chemokine receptor type 2 (CCR2) and C‐X3‐C motif chemokine receptor (CX3CR) 1, respectively. Ly‐6Chigh monocytes dominate early and exhibit a proinflammatory function. Ly‐6Clow monocytes dominate later. Ly‐6Clow monocytes promote myocardial healing through myofibroblast accumulation, angiogenesis, and collagen deposition.4
Therefore, targeted therapy toward monocyte subsets is an attractive therapeutic option to facilitate favorable cardiac remodeling. The existence of monocyte/macrophage subset heterogeneity and their step‐wise contribution to cardiac remodeling provides an opportunity for specific target intervention in the future.
What Do Monocytes Do?
Monocytes are a type of white blood cell present in the peripheral circulation. The primary roles of monocytes are in the participation of innate immunity and to maintain or replenish different types of macrophages and dendritic cells, which aid in phagocytosis of pathogens.25 Monocytes make up to 8% of the peripheral blood white cells and play a central role in the host response to exogenous and endogenous pathogen species, such as bacteria and viruses. Additionally, they modulate the inflammatory processes, producing both pro‐ and anti‐inflammatory cytokines and developing macrophages with pro‐ and anti‐inflammatory phenotype.26
Monocytes are derived from macrophage dendritic cell precursors that originate from the bone marrow under normal homeostatic conditions. Common myeloid progenitor cells, derived from the bone marrow, are responsible for differentiation of precursor progenitor cells into monocytes.26 Macrophage dendritic cell precursors mature to form either dendritic cells or macrophages. This process is dependent upon stimulation by cytokines and/or microbial molecules.27 Evidence to date suggests that both monocytes and dendritic cells diverge at a very early multipotent progenitor stage.26 Common myeloid progenitor cells give rise to the granulocyte‐macrophage lineage, which, in turn, give rise to macrophage dendritic cell precursors and subsequently, the committed monocyte precursor.28 Control of monocyte/macrophage differentiation is guided by a multitude of transcription factors, the complexity of which is beyond the scope of this review article.29
In the 1970s, studies highlighted the increase in monocyte proliferation within the bone marrow in response to inflammatory stimuli, allowing for monocytosis.30 During steady state, circulating monocytes have a half‐life of ≈3 days.31 Monocytes are mobilized from the bone marrow at times of tissue injury and differentiate into macrophages or dendritic cells while mounting an immune response. However, they are also implicated in diseases with proinflammatory shift such as heart failure and atherosclerosis.32, 33 Multiple animal studies have shown a diverse and complex function of monocytes depending upon the inflammatory environment, central to which is the ability of monocytes to be mobilized to site of injury.34
With regard to atherosclerosis, monocyte‐derived “foam cell” macrophages act as a substrate and thus facilitate the progress to MI. Monocyte counts are further highly increased in other forms of acute cardiovascular pathology.2, 33, 35
Overall, monocytes have been used as indicators of prognosis in humans, with their high numbers being associated with increased risk of recurrent MI, hospitalization, and cardiac death. Available data indicate that monocyte mobilization in acute cardiac disease does not simply reflect response to cardiac damage, but its active involvement in the pathological process.5, 36
Human Monocyte Heterogeneity
Monocyte subsets were first isolated in 1988 using flow cytometry.37 Expression of CD14 (lipopolysaccharide receptor) and CD16 (Fc receptor) is used to define human monocyte subsets. It should be noted that changes in expression of monocyte subsets are limited to cell‐surface protein expression assessed by flow cytometry, with changes in gene expression being up‐ or downregulated dependent on functional properties.38
Human monocyte subsets do not follow their mouse counterparts, in which initial studies in this field were undertaken. As such, nomenclature for human and mouse monocytes is not directly interchangeable and thus should not be directly compared. Human monocytes do not express Ly6C and description of their subsets is primarily based on expression of CD14 (lipopolysaccharide receptor) and CD16 (FC gamma III receptor; Table 1).39, 40, 41, 42, 43, 44, 45, 46, 47, 48, 49, 50, 51
Table 1.
Phenotypic and Functional Differences Between Monocyte Subsets
| Human (Mon1) | Mouse (Mon1) | Human (Mon2) | Mouse (Mon2) | Human (Mon3) | Mouse (Mon3) | |
|---|---|---|---|---|---|---|
| Proportion of total monocytes, %39, 40 | 85 | 40 to 45 | 5 | 5 to 32 | 10 | 26 to 50 |
| Functional properties39, 40, 41 | High phagocytic activity | High phagocytic activity, proinflammatory | High phagocytic activity. T‐ cell proliferation and stimulation, angiogenesis, superior ROS production | High phagocytic activity, proinflammatory | Low phagocytic activity, high “patrolling” activity (in vivo), T‐cell proliferation and stimulation | Low phagocytic activity, patrolling function, tissue repair |
| Surface markers present39, 42, 43, 44 | CD62L, CCR2, CLEC4D, CLEC5A, IL13Rα1, CXCR1, CXCR2 | CCR2, CD11b, CD115, CCR5 | CCR2, CD74, HLA‐DR, Tie‐2, ENG | CCR2, CD11b, CD115 | Siglec10, CD43, SLAN | CX3CR1, CD11b, CD115, CCR5 |
| Surface markers absent39, 41, 45 | CX3CR1, CD123, p2rx1, Siglec10 | CX3CR1 (low) | CD62L, CXCR1, CXCR2, CLEC4D IL13Rα1 | CX3CR1 (low) | CCR5, CD62L, CXCR1, CXCR2, CD163, CLEC4D, IL13Rα1 | CCR2 (low) |
| Response to LPS39, 42, 46, 47, 48 | IL‐10, G‐CSF, CCL2, RANTES, IL‐6, IL‐8 | ROS, TNFα, nitric oxide, IL‐1β, IL10 (low levels), IFN‐1, VLA‐4, IL‐6, CD62L | IL‐6, IL‐8 | ROS, TNFα, nitric oxide, IL‐1β, IL10 (low levels), IFN‐1, VLA‐4, IL‐6, CCR7, CCR8 | TNFα, IL‐1β, IL‐6, IL‐8 | IL‐10 (high levels) |
| Increased gene expression28, 39, 42, 49, 50, 51 | Wound healing and anticoagulation, S‐100 proteins, scavenger receptors, C‐type lectin receptors, antiapoptosis, response to stimuli (CCR2, THBS1, CD163, RNASE4, EDG3, S100A12, CLEC4D, VEGFA, F5, RNASE2, RNASE6, F13A1, CRISPLD2, PLA2G7CES1, EREG, QPCT) | CD177, FN1, Sell, Mmp8, F13a1, Atrnl1, Ly‐6c, Chi313 | MHC Class II, presentation and processing (CD14, CSPG2, SLC2A3, CD9, CD163, PLA2G7, MCEMP1, CLEC10A, EVA1, RNASE2, GFRA2, ALDH1A1, GALS2, MARCO, ALOX5AP, S100A12, QPCT, FOLR3, OSM, EGR1, CYP27A1, OLFM1, PAD14, HLADOA, ANG, H19, SCD, calgranulin B, S100A9DDIT4 | Inconclusive data | Cytoskeletal arrangement, complement components, proapoptosis, downregulation of transcription (FMNL2, CDKN1C, FCGR3A/B) | Vegfc, G0s2, Ikzf3, Tgfbr3, Cd83, Eno3, Tgm2, Itgax, CD36, Dusp16, Slc12a2, Fabp4 |
Human monocytes are dominated by “classical” CD14++CD16− (Mon1) monocytes (ie, 85%). Humans have at least 2 types of nonclassical monocytes. The CD14++CD16+ (Mon2) subset makes up around 6% of monocytes in humans. CD14+CD16++ (Mon3) human monocytes make up 9% of all monocytes.4, 52 There are many significant differences between Mon2 and Mon3, and, overall Mon2 is phenotypically and functionally closer to Mon1 than to Mon3 (discussed in more detail below). Earlier studies analyzed these 2 subsets together, and such data need to be interpreted with care.
A consensus opinion on the nomenclature of human monocytes in 2010 classed monocyte subsets as classical (CD14++CD16−), intermediate (CD14++CD16+), and nonclassical (CD14+CD16++).53 However, to avoid ambiguity the phenotypic definition and numerical designation (ie, Mon1, Mon2, and Mon3) have been incorporated into the most recent consensus document on monocytes subsets.54
Although direct correlation between human monocyte subsets is difficult, their differentiation and role in innate immunity are comparable. In fact, both Mon2 and Mon3 have reduced phagocytic activity, reduced production of reactive oxygen species along with lower levels of CCR2 expression.55 Several studies have highlighted the presence of raised levels of Mon2 in human inflammatory diseases.56
Mon1 is characterized by high expression of CD14, interleukin (IL)‐6 receptor, CD64, CCR2, and CD163, with less‐dense expression of vascular cell adhesion molecule and CD204. Intracellular adhesion molecule receptor, C‐X‐C chemokine receptor type 4, CD163, and vascular endothelial growth factor receptor 1 have the highest expression on Mon2. Mon3 has maximal expression of CD16, vascular cell adhesion molecule 1 receptor, and CD204, with much lower expression of CD14, IL‐6 receptor, CD64, CCR2, and CD163 that Mon257 (Table 2).42, 55, 58, 59, 60, 61, 62, 63, 64, 65, 66, 67, 68, 69, 70, 71, 72, 73, 74, 75, 76, 77, 78, 79, 80, 81, 82, 83, 84, 85, 86, 87
Table 2.
Differential Expression of Surface Markers Between Monocyte Subset
| Human | Mouse | |
|---|---|---|
| Mon1 | ||
| Proportion of total monocytes, %39 | 8539 | 40 to 4539 |
| Functional properties39 | High phagocytic activity39 | High phagocytic activity, proinflammatory39 |
| Surface markers/receptors present | ||
| CD1449, 58 | High | High |
| CD1649, 58 | Low | Low |
| CCR259, 60, 61 | High (increased 26‐fold) | High |
| CX3CR149, 55, 59, 60 | Low | Low |
| CXCR149, 55, 62 | High (increased 5‐fold) | Low |
| CXCR249, 62 | High (increased 7‐fold) | – |
| CD11b49, 61, 63, 64, 65 | Low | High |
| CD11549, 66 | – | High |
| CD62L49, 58, 65 | High (increased 3‐fold) | High |
| CLEC4D49, 67 | High (increased 4‐fold) | – |
| CLEC5A42, 49, 67 | High (increased 3‐fold) | – |
| IL13Rα149 | High (increased 9‐fold) | – |
| CD5449, 68 | Low (decreased 2‐fold) | – |
| CD4049, 65 | High (increased 6‐fold) | – |
| CD3642, 49, 69 | High (increased 2‐fold) | – |
| CD9949, 70 | High (increased 2‐fold) | – |
| CCR149, 71 | High (increased 2‐fold) | – |
| P2XR149 | Low (4‐fold) | – |
| HLA‐ABC49, 72 | Low (decreased 1‐fold) | – |
| CLEC10A49 | Low (decreased 6‐fold) | – |
| GFRA249 | Low (decreased 6‐fold) | – |
| HLA‐DR42, 49, 72, 73 | Low (decreased 8‐fold) | – |
| CD16349, 74, 75 | Low (decreased 1‐fold) | – |
| CD11545, 49, 76 | Low (decreased 1‐fold) | High |
| SLAN49, 77, 78 | High (increased 2‐fold) | – |
| CD1d49 | High (increased 1‐fold) | – |
| CCR549, 79, 80 | Low (decreased 1‐fold) | – |
| CD29449 | Low (decreased 1‐fold) | – |
| Siglec1049, 81, 82 | Low (decreased 7‐fold) | – |
| Mon2 | ||
| Proportion of total monocytes, % | 539 | 5 to 3239 |
| Functional properties | High phagocytic activity, T‐cell proliferation and stimulation, angiogenesis, superior ROS production39 | High phagocytic activity, proinflammatory39 |
| Surface markers/receptors present | ||
| CD1442, 49, 58 | High | High |
| CD1642, 49, 58 | Low | Low |
| CCR249, 60, 83 | High (increased 8‐fold) | High |
| CX3CR149, 55, 60 | Low | Low |
| CXCR149, 55, 62 | High (increased 4‐fold) | Low |
| CXCR246, 49, 62 | High (increased 3‐fold) | – |
| CD11b49, 61, 65 | High | High |
| CD11542, 45, 49 | Low | High |
| CD62L49, 65, 72 | High (increased 1.3‐fold) | – |
| CLEC4D49 | High (increased 18‐fold) | – |
| CLEC5A42, 49 | High (increased 5‐fold) | – |
| IL13Rα149 | High (increased 2‐fold) | – |
| CD5449, 68 | High (increased 1‐fold) | – |
| CD4049, 65 | High (increased 1‐fold) | – |
| CD3649, 69 | High (increased 5‐fold) | – |
| CD9949, 70 | High (increased 5‐fold) | – |
| P2XR149 | Low (decreased 5‐fold) | – |
| HLA‐ABC49, 72 | High (increased 1‐fold) | – |
| CLEC10A49 | High (increased 4‐fold) | – |
| GFRA249 | High (increased 3‐fold) | – |
| HLA‐DR49, 72, 73 | High (increased 2‐fold) | – |
| CD16349, 75 | High (increased 6‐fold) | – |
| SLAN49, 77, 78 | Low (decreased 3‐fold) | – |
| CD1d49 | Low (decreased 5‐fold) | – |
| CCR549, 79, 80 | High (increased 7‐fold) | – |
| CD29449 | Low (decreased 3‐fold) | – |
| Siglec1049, 81, 82 | Low (decreased 21‐fold) | – |
| Mon3 | ||
| Proportion of total monocytes, % | 1039 | 26 to 5039 |
| Functional properties | Low phagocytic activity, high “patrolling” activity (in vivo), T‐cell proliferation and stimulation39 | Low phagocytic activity, patrolling function, tissue repair39 |
| Surface markers/receptors present | ||
| CD1449, 58 | Low | Low |
| CD1649, 58 | High | High |
| CCR249, 60, 61 | Low | Low/– |
| CX3CR145, 49, 60, 65 | High | High |
| CD11b49, 61, 84 | High | High |
| CD62L58, 65, 85 | Low | Low |
| P2XR149 | High (increased 1.2‐fold) | – |
| HLA‐ABC49, 72 | Low (–) | – |
| CLEC10A49 | Low (–) | – |
| GFRA249 | Low (–) | – |
| HLA‐DR49, 72, 73 | Low (–) | – |
| CD16349, 74, 86 | High (increased 7‐fold) | – |
| CD11545, 49, 76 | Low (decreased 2‐fold) | High |
| SLAN49, 77, 78, 87 | Low (decreased 7‐fold) | – |
| CD1d49 | High (increased 4‐fold) | – |
| CCR549, 79, 80 | High (increased 8‐fold) | – |
| CD29449 | Low (decreased 2‐fold) | – |
| Siglec1049, 81, 82 | Low (decreased 3‐fold) | – |
(–) indicates evidence lacking or under‐reported.
Monocyte subpopulations differ in the range of cytokines they can produce in response to stimulation. Mon1 has been shown to preferentially express cytokines IL‐1β, IL‐6, monocyte chemoattractant protein 1 (MCP‐1), an inhibitor of nuclear factor kappa β kinase, whereas Mon2 produces anti‐inflammatory IL‐10. Interestingly, Mon3 stimulates cytokine production in response to viral rather than bacterial load, further emphasizing the functional differences between monocyte subsets.40 However, recent experiments showed specific release of IL‐6 and IL‐8 cytokines by Mon2 and Mon3 in response to bacterial endotoxemia88 (Table 3).79, 89, 90, 91, 92, 93, 94
Table 3.
Differential Cytokines Production by Monocyte Subsets in Response to LPS in Human and Mouse Models Relative to Mon2
| Plasma Cytokine | Mon1 | Mon2 | Mon3 |
|---|---|---|---|
| G‐CSF49, 89, 90 | High | Low | Low |
| IL‐1040, 49, 91, 92 | High | Low | Low |
| CCL227, 49, 93 | High | Low | Low |
| RANTES49, 79 | High | Low | Low |
| IL‐627, 49, 65, 88 | High | Intermediate | Intermediate |
| IL‐840, 49, 88, 90 | High | High | High |
| IL 1‐β27, 49, 65 | Intermediate | Intermediate | High |
| TNF‐α49, 65, 94 | Intermediate | Low | High |
CCR2 indicates C‐C chemokine receptor type 2; G‐CSF, granulocyte colony‐stimulating factor; IL, interleukin; LPS, lipopolysaccharide; RANTES, regulated on activation, normal T cell expressed and secreted; TNF‐α, tumor necrosis factor alpha.
Mouse Monocyte Heterogeneity
Distinct mouse monocyte subsets were initially distinguished based on the differential expression of a chemokine receptor, CCR2 (receptor to MCP‐1). CCR2+ monocytes showed a higher migratory and infiltratory capacity compared with CCR2− cells, most recently being studied in postinfarct cardiac remodeling.42, 95, 96 Differential expression of an inflammatory monocyte marker, Ly6C, allowed for better mouse subset characterization.97 Analysis of the 2 principle mouse monocyte subsets (Ly6C+ and Ly6C−) is commonly used in experimental research; there is accumulating evidence for the existence of a subset with intermediate phenotype, which resemble human “intermediate” Mon2 subset.66, 76 The subsets differ in expression of surface markers, for example: CD11b and CD115 have a high density of CCR2 and only small numbers of CX3CR1 are present on Ly6C+ monocytes. In contrast, Ly6C− monocytes virtually lack CCR2, but express high levels of CX3CR1.97
Ly‐6C+ monocytes have phagocytic and proinflammatory characteristics. In acute MI, they accumulate promptly in areas of myocardial injury, along with macrophages providing a proinflammatory environment.98, 99 Ly‐6C− monocytes, on the other hand, have been found to have anti‐inflammatory properties and this subset promotes post‐MI myocardial healing through the processes of myoblast activation, angiogenesis, and collagen formation.66, 100, 101 Overall, the Ly‐6C+ subset is associated with detrimental effects to myocardium and their high levels in the acute phase of MI delay myocardial healing.102
Functional studies have demonstrated that Ly6C+ cells release reactive oxygen species, nitric oxide, and inflammatory cytokines (eg, tumor necrosis factor α [TNFα], IL‐1β) in response to bacterial infection.27 The subset migration is potentiated through the CCR2 receptor that initiates a change in the ligand for vascular cell adhesion molecule 1 (VLA 4). Studies have found that Ly6C+ monocytes preferentially migrate into the sites of vascular inflammation and CCR2 is central to this process, also promoting the subset maturation toward M1 macrophage phenotype.96, 103
In the absence of inflammation, Ly6C+ transforms into Ly6C−, which predominates in the circulation, binding to vascular endothelium using CX3CR1 receptors.51 In response to bacterial infection, Ly6C− cells release anti‐inflammatory cytokines (namely, IL‐10). The response to inflammation triggers the differentiation of monocytes into M2 macrophages, which, in turn, release anti‐inflammatory cytokines central to tissue repair.97, 104, 105
Release of Monocyte Subsets From the Bone Marrow and Spleen
All 3 subsets are present in the bone marrow.25, 66 After maturation, Mon1 leaves the bone marrow, entering the peripheral circulation through CCR2 chemokine receptors.59 Previous studies had suggested the ability of Mon1 to differentiate further into Mon2 upon migration from the bone marrow, which enter the circulation.59 Most recent studies have shown the initial release of Mon1 in response to endotoxemia with the subsequent differentiation into Mon2 and Mon3 subsets.106 However, analysis of bone marrow samples indicates that cells with Mon2 phenotype are already present in human bone marrow.107 In fact, cells with the Mon2 phenotype were the dominant monocytic cells within the bone marrow.57
Monocyte numbers have been found to follow a circadian rhythm controlled by Arnt‐1, which is a key gene in regulating the molecular circadian clock.108 In contrast to the presence of CCR2 ligands present on monocytes during homeostasis, the release of patrolling monocytes is dependent upon the G‐protein‐coupled receptor for sphingosine‐1‐phosphate, the deficiency of which leads to an inability of Mon3 to redistribute from the bone marrow.109
Under certain inflammatory conditions (eg, cancer, HF, MI, and stroke), mouse models have shown the spleen to be capable of extramedullary monopoiesis.110 Release of monocytes from the spleen is dependent upon angiotensin II as opposed to CCR2 in the bone marrow.63 Furthermore, in cardiovascular disease, there is a dependency upon a separate chemokine, chemokine (C‐X‐C motif) ligand 1, to direct the release of monocytes from both the splenic and bone marrow reservoirs.111
Roles of Circulating Monocyte Subsets
Monocyte studies involving gene analysis highlight the preferential expression of genes involved in angiogenesis, wound healing, and coagulation (namely, Mon3).49 Alternatively, Mon1 have a higher capability to produce IL‐1β and TNFα in response to bacterial lipopolysaccharides.40 During the inflammatory process, both Mon1 and Mon2 bind to MCP‐1, thus allowing monocytes to invade into human tissue and perpetuate the inflammatory cascade.112
In contrast, Mon3 bind to CX3CR/chemokine (C‐C motif) ligand 3 receptors through the leucocyte functional antigen 1, subsequently stimulating the release of IL‐1β and TNFα. Such pathophysiology has been implicated in autoimmune conditions, such as rheumatoid arthritis.40
Role of Monocytes in Cardiovascular Disease and Heart Failure
Inflammation plays a pivotal role in the pathogenesis of HF. Cytokines, such as IL‐6 and TNFα, are important markers of active disease and prognosis.113, 114 Compromise of the myocardium has multiple etiologies, ranging from ischemic heart disease, hypertension, cardiac arrhythmias, and metabolic diseases. Neopterin, which is a metabolite of guanosine triphosphate, has been found to be elevated in patients with HF and this is said to be a marker of monocyte activation.115
The transmigration of cells to the site of tissue injury relies upon specific cell‐surface molecules, namely monocytes and cell adhesion molecules, that respond to signaling by cytokines released from the injured vessel wall.4 Once inside the vessel wall/myocardium, monocytes will differentiate into macrophages, which promote tissue repair. The complex interactions within injured myocardial cells lead to formation of both pro‐ and anti‐inflammatory cells. In pathological conditions, there is an override of tissue homeostasis and uncontrolled inflammation leads to the exaggerated release of macrophages, which, instead of healing tissue, cause tissue damage with adverse remodeling. Therefore, trying to regulate the monocyte/macrophage balance is a logical therapeutic strategy.
As discussed, under specific stimuli monocytes will differentiate into macrophages. Macrophages play a vital role in the phagocytosis and removal of pathogens.116 Although inflammation aims to protect against infection, it can cause damage to the vascular endothelium, activation of tissue macrophage, and activation of cytokine pathway migration of smooth muscle cells to the intima of the arterial wall, thus accelerating the process of atherosclerosis.117
Hypoxia and myocardial necrosis drive an inflammatory process in the injured myocardium, which involves activation of monocytes/macrophages. These cells, in turn, are capable of producing cytokines, chemokines, and growth factors.118 In patients with diabetes mellitus who suffer an ST‐elevation MI, Mon2 subsets were elevated and their high counts predict recurrent cardiovascular events and death.119
In ischemic HF, Mon1 have similar counts to controls with coronary artery disease without HF, but their numbers are increased during HF decompensation. In contrast, Mon2 is the only subset increased in patients with stable HF and it shows a further sharp increase in acute HF.119, 120 Of interest, high Mon2 counts were associated with better survival in that study, using a combined outcome of death and rehospitalization. This suggests a presence of potentially protective properties of this subset in patients with failing hearts. Analysis of their functional status was not analyzed in the study, and it is difficult to be certain what drives possible benefits or dangers associated with the subset.
There is some controversy on the role of Mon3 in HF given that both their depletion or no change were observed.119, 120 This may be attributed to differences in etiology of the studied patients, for example, an accelerated homing of Mon3 in patients with nonischemic HF, as observed in the study with mixed HF etiology. A notable limitation in some studies is the lack of control for comorbidities that may be responsible for the abnormal release of monocytes. Despite this potential limitation, one should still recognize the importance of monocytes in the inflammatory process that happens in HF with further research in human subjects, particularly focusing on Mon2 being justified.5
A compromised myocardium provides multiple stimuli for monocyte recruitment in patients with HF. The presence of excessive LV and atrial stretch in experimental studies of mice with HF with preserved ejection fraction on the background of hypertension has shown to stimulate myocardial resident macrophages to signal the release monocyte chemoattractants (including MCP‐1, interleukins).121 Monocyte recruitment is further amplified by the presence of tissue hypoxia and ischemia122, 123 (Table 4).124, 125, 126, 127, 128, 129, 130, 131, 132, 133, 134, 135, 136
Table 4.
Changes in Mon2 and Mon3 in Cardiovascular Disease States
| Condition | Mon2 | Mon3 |
|---|---|---|
| Stable coronary artery disease33, 124, 125 | No change vs healthy control | No change vs health control |
| Acute myocardial infarction126, 127, 128 | 2.5‐fold increase, positively correlates with troponin T level | No change, no correlation with troponin T level |
| Unstable angina129, 130, 131 | Increased (in intermediate‐high‐risk patients' vs low‐risk cohort) | No change (no difference with risk severity) |
| Acute heart failure5, 132, 133 | Increased, raised CD41 count relative to mon3 | No change |
| Chronic heart failure120, 134 | Increased expression, correlates with NYHA class/LVEF/NT‐proBNP | Increased but no correlation to NYHA class/LVEF/NT‐proBNP |
| Chronic heart failure135 |
No change vs healthy control No association with end‐diastolic dimension |
Increased percentage vs health controls Inverse relationship with end‐diastolic dimension |
| Abdominal aortic aneurysm136 | Increased vs healthy controls | Increase count vs healthy controls |
LVEF indicates left ventricular ejection fraction; NT‐proBNP, N‐terminal pro‐B‐type natriuretic peptide; NYHA, New York Heart Association.
Excessive monocyte/macrophage cardiac recruitment leads to a vicious circle of myocardial damage and remodeling. This process involves apoptosis of cardiomyocytes.122, 123 It has been shown that monocyte TNFα triggers production of the inducible type of nitric oxide synthase, uncontrolled oxidative stress, and, consequently, apoptosis and tissue necrosis.122 In the inflammatory process, cytokine release by stimulated monocytes attracts even more monocytes to the compromised myocardium, thus contributing to the vicious circle (Figure 1).
Figure 1.
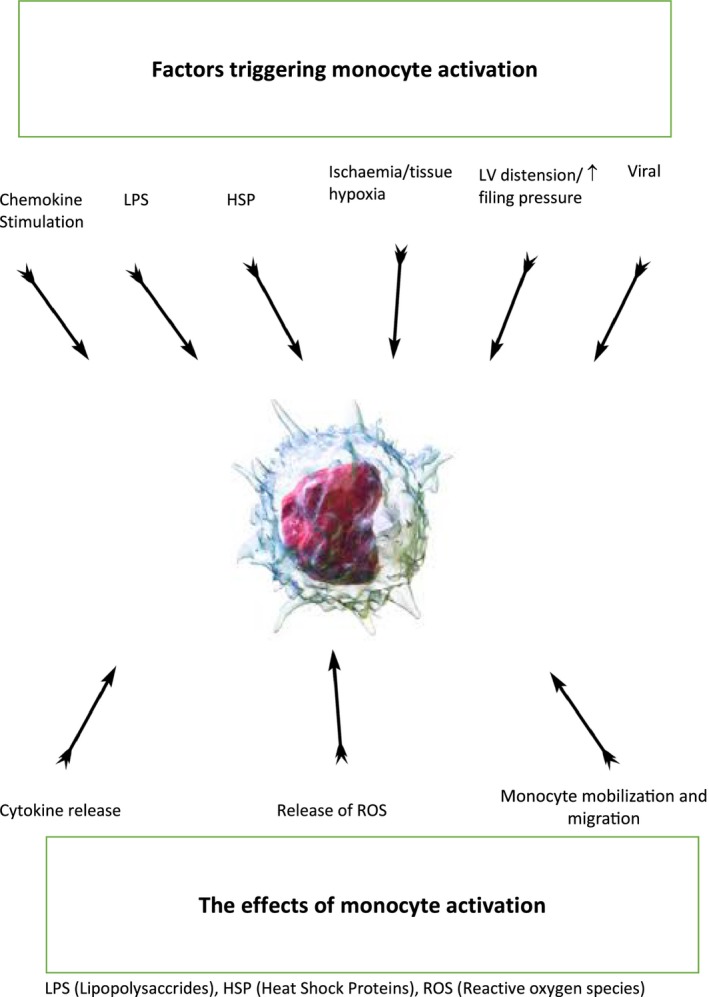
Triggers for monocyte activation and subsequent function. HSP indicates xxx; LPS, lipopolysaccharide; LV, left ventricular; ROS, reactive oxygen species.
Monocyte Activation in Cardiac Fibrosis
Cardiac fibrosis is the consequence of an activated monocyte/macrophage cascade in HF. Both cellular and extracellular processes are involved in cardiac fibrosis. Within the extracellular matrix, cardiac fibroblasts make up ≈60% of all cells, in fact, outnumbering cardiomyocytes. They are relatively scarce in a healthy adult heart, and a rise in the cell population occurs during a pathological process. This is suggested, for example, by evidence from mice models where tissue injury led to a rise in IL‐β production and, consequently, to fibroblast expansion.137 This, in turn, propagates inflammatory cell infiltration and further cytokine production in the site of tissue injury.137 In such cases, there is an increase in rate of differentiation of precursor cells (eg, monocytes, endothelial progenitors, pericytes, and bone marrow circulating progenitor cells) into fibroblasts.138, 139
The process by which monocytes can potentiate the cardiac inflammatory response leading to fibrosis is reliant on monocyte cell‐surface receptors. One such group of receptors is termed the Toll‐like receptors (TLRs), which are members of the pattern recognition system, but also able to respond to endogenic stimuli.140 Although TLR4 is present on different types of cells, its highest density has been noted on monocytes, reflective of their vital role in innate immunity. In addition, monocyte density of CD14 has also been found to be higher in patients with moderate‐severe heart failure in comparison with normal or mild LV impairment.141, 142
Expression of TLR4 on monocytes is linked to the degree of cardiac injury and remodeling.143 Evidence thus far points to the enhanced recruitment of TLR4‐expressing monocytes into a compromised myocardium in both human and mouse studies.144, 145 The remodeled myocardium has a higher count of TLR4+ monocytes compared with a healthy myocardium,146 thus creating a proinflammatory environment. Indeed, TLR4‐deficient mice have a lower inflammatory burden post–acute ischemia and reduced apoptosis of cardiomyocytes.147
Further evidence has pointed to several mechanisms by which monocyte activation takes place in HF. With respect to the immune response found in cardiovascular disease, lipopolysaccharides that are found on Gram‐negative bacteria act as the ligand component for the activation of monocytes (eg, binding to CD14 and TLR4 as previously mentioned) and triggering cytokine release. This endotoxin‐cytokine hypothesis centers on bacterial transition into the circulation through entrance through a permeable bowel membrane.148 This is promoted by venous congestion developed through HF increasing membrane permeability. The overexpression of inflammatory cytokines amplifies the process leading to a pathological loop often culminating in symptomatic HF.142
An alternative mechanism to the introduction of bacteria into the circulation in HF involves activation of the sympathetic system, a common feature of HF.149 The sympathetic activity is thought to redistribute blood flow away from the splanchnic circulation, which, in turn, leads to transient ischemia in the bowel. This causes an increase in endothelial permeability and entry of the proinflammatory bowel contaminants into the circulation. This mobilizes inflammatory cells from bone marrow (and the spleen depot), and numerous studies have found an increase in blood leucocytes in patients with advanced HF, thus supporting this hypothesis.150, 151
It is likely that no 1 single hypothesis fully explains the process by which monocytes and their surface receptors are stimulated to propagate cardiac fibrosis. Both systemic and local cascades of inflammatory pathways exist to enhance the stimuli for monocyte‐triggered cardiac fibrosis.
Is There a Different Role of Monocytes in HF With Preserved Ejection Fraction?
HF with preserved ejection fraction (HFpEF) is a common condition that constitutes half of all cases of HF. Its diagnosis is based on the presence of a “normal” ejection fraction together with signs and symptoms suggestive of HF and evidence of diastolic dysfunction in the form of prolonged LV relaxation and filling, increased diastolic stiffness, and elevated LV end‐diastolic pressures.152 An aging population, along with multiple comorbidities, predisposes to the increasing rates of HFpEF. Limited advancement has been made in the treatment of these patients. The complexity in the pathogenesis of LV remodeling in HFpEF with insufficient understanding of its mechanisms likely contribute to the limited progress in this field.
HF symptoms in patients with diastolic dysfunction are attributed to reduced stroke volume secondary to increased cardiac stiffness. This reflects changes in the extracellular matrix with cardiac fibrosis and cardiomyocyte hypertrophy.153 Majority of such patients have a prolonged history of hypertension with an accelerated accumulation of monocytes in the presence of pressure overload.154, 155 Monocytes have been found to mediate the inflammatory process by releasing chemokines, such MCP‐1, TNFα, and transforming growth factor‐β (TGF‐β), which, in turn, play proinflammatory and ‐fibrotic roles.156, 157 MCP‐1 knockout mice studies have shown significant suppression of fibrosis and macrophage activation.158 In relation to cytokines, TNFα, IL‐1, IL‐6, IL‐8, and MCP‐1 in the peripheral circulation of patients with chronic HF are shown to be elevated.159, 160, 161
Monocytes are 1 of the major sources of these inflammatory cells. Such cells have been shown to be of prognostic importance in patients developing HFpEF.162 This is unsurprising given that the comorbidities of patients with HFpEF have long established an inflammatory origin in their etiology, for example, chronic vascular inflammation found in patients with coronary artery disease.
The processes described lead to structural and mechanical remodeling of the heart muscle through the interstitial deposition of extracellular matrix proteins such as collagen.163 The resulting hypertrophic changes of the myocardium cause the findings of diastolic dysfunction. It is this inflammatory and fibrotic process that underpins the changes observed in patients with diastolic dysfunction and HFpEF.
However, until very recently, all pathophysiological evidence for monocytes in HF were based on HF with reduced ejection fraction. More recently, patients with HFpEF have been found to have raised monocyte counts, specifically the Mon2 subset.164, 165 Data on macrophage/monocyte polarization in human patient cardiac tissue showed increased presence of TGF‐β‐expressing leucocytes, resembling blood Mon2 subset.166 In agreement with this, animal studies have shown cardiac upregulation of M2 macrophages, which contribute to cardiac fibrosis in hypertension.167 Healthy monocytes exposed to serum from HFpEF patients preferentially develop M2 macrophage profibrotic features.154 In this study, patients with HFpEF had elevated cytokine levels (eg, TNFα, IL‐6, and IL‐12) and MCP‐1, which paralleled high monocyte counts. Furthermore, there was a positive correlation between monocyte numbers and worsening parameters of diastolic dysfunction.154 An acute ischemic injury is accompanied by conversion from M1 to M2 macrophages, a type of macrophage known to provide a profibrotic inflammatory environment.168
Despite the relative scarcity of the data, it is likely that this monocyte/macrophage‐driven inflammatory environment is largely responsible for the fibrotic changes in HFpEF. Their increase number causes an increase in collagen deposition and conversion of cardiac fibroblasts to myofibroblasts (Figure 2).
Figure 2.
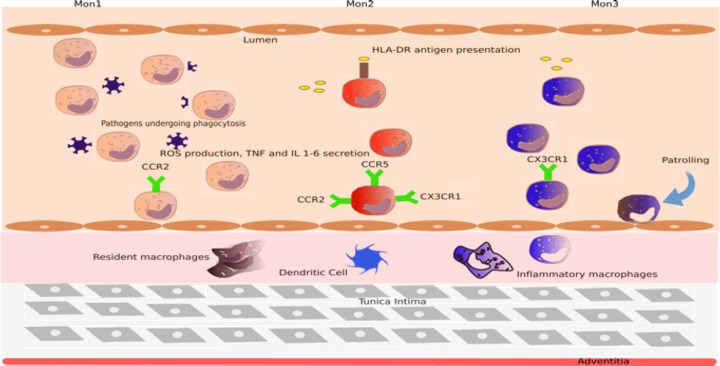
Role of monocyte subsets in heart failure. Human monocytes are classified as Mon1, Mon2, and Mon3, respectively, based on their levels expression of CD14 and CD16. Mon2 are increased in patients with heart failure and are recruited to the myocardium in times of tissue injury, whereas Mon3 serve more of a patrolling function and are not so rapidly recruited. Monocyte subsets then differentiate into dendritic cells and inflammatory macrophages to further potentiate the fibrosis. CCR2/5 indicates C‐C chemokine receptor type 2/5; CX3CR1, C‐X3‐C motif chemokine receptor 1; HLA‐DR, human leukocyte antigen – antigen D related; IL, interleukin; ROS, reactive oxygen species; TNF, tumor necrosis factor.
What About Atrial Fibrillation?
Atrial fibrosis is a hallmark of the structural cardiac remodeling that takes place in AF, causing an increase in the frequency of AF paroxysms, which, in turn, increase likelihood of progression to permanent AF.169 Atrial fibrosis has been observed in biopsies from patients with AF148 as well as in patients with specific risk factors predisposing to AF, such as valvular heart disease,170 dilated and hypertrophic cardiomyopathy,171 and advanced age.172
Structural heart remodeling in aging and heart disease is associated with fibrosis. With aging, there is a progressive enlargement of the extracellular compartment in the atrial septum attributed to accumulation of connective tissue fibers.173 This process is even more prominent in an HF model,174 where larger areas of fibrosis were observed, similar to the “replacement fibrosis” observed after tissue damage and cell death. Atrial fibrosis may, in itself, be sufficient to increase susceptibility to AF, as shown in mice with atrial fibrosis attributed to overexpression of TGF‐β1.175
Although there are strong indications from animal models that atrial fibrosis can be proarrhythmic,8 some questions regarding the role of atrial fibrosis as a substrate of AF are still unresolved. Experimental data linking inflammation and atrial fibrosis have been conflicting. Most recent data point to the upregulation of profibrotic factors, such as TGF‐β, and accumulation of collagen in the atrial interstitium.176 However, previous studies showed preserved interstitium despite changes in atrial architecture and myocyte characteristics.177 The discrepancy between the data can be partly explained by the findings that profibrotic factors may not accumulate over shorter time periods found in some studies, and that increased gene expression of markers of fibrosis may be the first sign of later fibrosis.178 Another proinflammatory peptide, TNFα, has been shown to be elevated in patients with chronic AF. Released largely by monocytes and macrophages, TNFα has been found at higher levels in patients with nonvalvular AF than those in sinus rhythm.179 This correlates with a more‐significant leuckocyte infiltration and more‐advanced fibrotic changes in the atria.
Some, but not all, human studies have confirmed excessive atrial fibrosis in chronic AF patients compared with those with sinus rhythm.180, 181 The degree of atrial fibrosis and profibrogenic status correlates with the persistence of AF.182 However, from these studies, it is unclear whether the fibrosis is caused by underlying structural disease leading to AF or by AF itself. Given that the degree of the underlying heart disease is not well documented in every study, it is currently difficult to establish the magnitude of effects of particular conditions to the development of atrial fibrosis in AF patients. Some insights into profibrotic effects of background cardiac pathology versus AF come from a comparison of structural heart disease patients with and without AF.170 In this study, AF itself has not been found to be associated with atrial fibrosis, but is instead related to the severity of the structural heart disease. Given the significant differences in AF pathogenesis among patients with or without structural heart disease, studies dedicated to nonvalvular AF would be essential to shed further light to the interactions between AF and connective tissue deposition in the atria.
The question therefore remains of how important atrial fibrosis is as a causative factor for AF in humans. Most animal models show that atrial dilation is accompanied by both atrial fibrosis and conduction disturbances, although conduction disturbances could also be observed in the absence of atrial fibrosis.183, 184 However, frequently used mice models of AF have significant limitations attributed to the fact that this species has a high physiological heart rate and thus AF induced in mice may not accurately reflect pathological processes in humans. In patients undergoing open heart surgery, degree of fibrosis does correlate with the occurrence of postoperative AF185 and with the recurrence of AF.181
Similar to AF, cardiac fibrosis is related to myocardial inflammation and oxidative stress secondary to infiltration of inflammatory cells, thus suggesting further pathophysiological links between the 2.186 The oxidative stress observed in these conditions is further amplified by stimulation of the renin angiotensin‐aldosterone system, which aids NADH oxidase release.187 IL‐1, IL‐6, TNFα, and MCP‐1 are all upregulated in AF predisposing to fibrotic changes and the related electrical and structural remodeling, typical of AF. The role of inflammation in AF development is highlighted by the correlation with C‐reactive protein (CRP) and has been found, in postoperative patients, to be a surrogate marker for predictor of new‐onset AF.188 Also, postablation CRP levels can be used as a marker for risk of recurrence.189
Further evidence on the role of cardiac fibrosis in AF comes from experimental and clinical studies demonstrating that prevention of atrial fibrosis can delay the development of AF. Several treatments (eg, statins, angiotensin‐converting enzyme inhibitors, AT1‐receptor blockers, fish oil, and glucocorticoids) have been proven to effectively delay the structural remodeling process and reduce AF burden in a variety of experimental models.190, 191, 192, 193, 194, 195 Several post‐hoc analyses of clinical trials and small‐scale, proof‐of‐principle studies indicate utility of such approaches in humans, but improvement of the patients' hemodynamics with normalization of atrial pressures might also have contributed to the beneficial effects of these compounds.196
The role of ventricular fibrosis in AF is less established. Patients with AF have more‐marked ventricular fibrosis than those with sinus rhythm.197 Although atrial and ventricular fibrosis are likely to share a common mechanism, there are much more‐limited findings of profibrotic gene expression in ventricular fibrosis in comparison with the atrium.198 TGF‐β seems to play a major role in ventricular fibrosis in AF, but further data are needed to establish the mechanisms that trigger its expression in the myocardium and the role of monocytes and macrophages as a source of TGF‐β in the heart.199
Conclusion
Monocytes represent an essential component of the innate immune system and play a vital role in cardiovascular health. The beneficial effects of monocytes include their contribution to cardiac remodeling in response to physiological and pathological changes in hemodynamics, elimination of pathogens, involvement in apoptosis, and phagocytosis of necrotic tissues. However, excessive inflammatory response to cardiac insult can be harmful to the human body and can lead to cardiac fibrosis and heart failure. There is a fine balance between monocytes that predisposes to beneficial or deleterious effects. Existence of several subsets of monocytes is likely to explain the diversity of the monocyte effects in health and disease. To date, few studies have specifically looked at monocytes subsets and their key characteristics as contributors to cardiac fibrosis and AF.
Monocytes trigger an inflammatory cascade involving the release of cytokines. Such cytokines migrate to the myocardium and adhere to the endothelial wall. Infiltration into the myocardium is a complex process, but 1 that ultimately leads to fibrosis and symptoms of HF. To better identify therapeutic targets, the role of monocytes and their individual subsets, in the pathophysiology of AF and its complications, such as HF, must be determined.
Disclosures
Lip serves as a consultant for Bayer/Janssen, BMS/Pfizer, Biotronik, Medtronic, Boehringer Ingelheim, Novartis, Verseon, and Daiichi‐Sankyo and a speaker for Bayer, BMS/Pfizer, Medtronic, Boehringer Ingelheim, and Daiichi‐Sankyo. Shahid and Shantsila have no conflicts to disclose.
J Am Heart Assoc. 2018;7:e007849 DOI: 10.1161/JAHA.117.007849.29419389
References
- 1. Yancy CW, Jessup M, Bozkurt B, Butler J, Casey DE Jr, Drazner MH, Fonarow GC, Geraci SA, Horwich T, Januzzi JL, Johnson MR, Kasper EK, Levy WC, Masoudi FA, McBride PE, McMurray JJ, Mitchell JE, Peterson PN, Riegel B, Sam F, Stevenson LW, Tang WH, Tsai EJ, Wilkoff BL; American College of Cardiology, American Heart Association Task Force on Practice Guidelines . 2013 ACCF/AHA guideline for the management of heart failure: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol. 2013;62:e147–e239. [DOI] [PubMed] [Google Scholar]
- 2. Swirski FK, Pittet MJ, Kircher MF, Aikawa E, Jaffer FA, Libby P, Weissleder R. Monocyte accumulation in mouse atherogenesis is progressive and proportional to extent of disease. Proc Natl Acad Sci USA. 2006;103:10340–10345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hunter M, Wang Y, Eubank T, Baran C, Nana‐Sinkam P, Marsh C. Survival of monocytes and macrophages and their role in health and disease. Front Biosci (Landmark Ed). 2009;14:4079–4102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Nahrendorf M, Swirski FK, Aikawa E, Stangenberg L, Wurdinger T, Figueiredo JL, Libby P, Weissleder R, Pittet MJ. The healing myocardium sequentially mobilizes two monocyte subsets with divergent and complementary functions. J Exp Med. 2007;204:3037–3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wrigley BJ, Shantsila E, Tapp LD, Lip GY. CD14++CD16+ monocytes in patients with acute ischaemic heart failure. Eur J Clin Invest. 2013;43:121–130. [DOI] [PubMed] [Google Scholar]
- 6. Robbins CS, Chudnovskiy A, Rauch PJ, Figueiredo JL, Iwamoto Y, Gorbatov R, Etzrodt M, Weber GF, Ueno T, Van Rooijen N, Mulligan‐Kehoe MJ, Libby P, Nahrendorf M, Pittet MJ, Weissleder R, Swirski FK. Extramedullary hematopoiesis generates Ly‐6C(high) monocytes that infiltrate atherosclerotic lesions. Circulation. 2012;125:364–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hulsmans M, Sam F, Nahrendorf M. Monocyte and macrophage contributions to cardiac remodeling. J Mol Cell Cardiol. 2016;93:149–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Burstein B, Nattel S. Atrial fibrosis: mechanisms and clinical relevance in atrial fibrillation. J Am Coll Cardiol. 2008;51:802–809. [DOI] [PubMed] [Google Scholar]
- 9. Li D, Fareh S, Leung TK, Nattel S. Promotion of atrial fibrillation by heart failure in dogs: atrial remodeling of a different sort. Circulation. 1999;100:87–95. [DOI] [PubMed] [Google Scholar]
- 10. Li J, Solus J, Chen Q, Rho YH, Milne G, Stein CM, Darbar D. Role of inflammation and oxidative stress in atrial fibrillation. Heart Rhythm. 2010;7:438–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Frangogiannis NG. Regulation of the inflammatory response in cardiac repair. Circ Res. 2012;110:159–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kania G, Blyszczuk P, Eriksson U. Mechanisms of cardiac fibrosis in inflammatory heart disease. Trends Cardiovasc Med. 2009;19:247–252. [DOI] [PubMed] [Google Scholar]
- 13. Shantsila E, Shantsila A, Blann AD, Lip GY. Left ventricular fibrosis in atrial fibrillation. Am J Cardiol. 2013;111:996–1001. [DOI] [PubMed] [Google Scholar]
- 14. Cleland JGF. Screening for left ventricular dysfunction and chronic heart failure. Dis Manag Health Outcomes. 1997;1:169–184. [Google Scholar]
- 15. Lloyd‐Jones DM, Larson MG, Leip EP, Beiser A, D'Agostino RB, Kannel WB, Murabito JM, Vasan RS, Benjamin EJ, Levy D. Lifetime risk for developing congestive heart failure: the Framingham Heart Study. Circulation. 2002;106:3068–3072. [DOI] [PubMed] [Google Scholar]
- 16. Moser M, Hebert PR. Prevention of disease progression, left ventricular hypertrophy and congestive heart failure in hypertension treatment trials. J Am Coll Cardiol. 1996;27:1214–1218. [DOI] [PubMed] [Google Scholar]
- 17. Essop MR, Nkomo VT. Rheumatic and nonrheumatic valvular heart disease: epidemiology, management, and prevention in Africa. Circulation. 2005;112:3584–3591. [DOI] [PubMed] [Google Scholar]
- 18. Cleland JG, Puri S. How do ACE inhibitors reduce mortality in patients with left ventricular dysfunction with and without heart failure: remodelling, resetting, or sudden death? Br Heart J. 1994;72:S81–S86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Dewald O, Zymek P, Winkelmann K, Koerting A, Ren G, Abou‐Khamis T, Michael LH, Rollins BJ, Entman ML, Frangogiannis NG. CCL2/monocyte chemoattractant protein‐1 regulates inflammatory responses critical to healing myocardial infarcts. Circ Res. 2005;96:881–889. [DOI] [PubMed] [Google Scholar]
- 20. Van Loon RB, Veen G, Kamp O, Baur LH, Van Rossum AC. Left ventricular remodeling after acute myocardial infarction: the influence of viability and revascularization—an echocardiographic substudy of the VIAMI‐trial. Trials. 2014;15:329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Frangogiannis NG, Dewald O, Xia Y, Ren G, Haudek S, Leucker T, Kraemer D, Taffet G, Rollins BJ, Entman ML. Critical role of monocyte chemoattractant protein‐1/CC chemokine ligand 2 in the pathogenesis of ischemic cardiomyopathy. Circulation. 2007;115:584–592. [DOI] [PubMed] [Google Scholar]
- 22. Leuschner F, Dutta P, Gorbatov R, Novobrantseva TI, Donahoe JS, Courties G, Lee KM, Kim JI, Markmann JF, Marinelli B, Panizzi P, Lee WW, Iwamoto Y, Milstein S, Epstein‐Barash H, Cantley W, Wong J, Cortez‐Retamozo V, Newton A, Love K, Libby P, Pittet MJ, Swirski FK, Koteliansky V, Langer R, Weissleder R, Anderson DG, Nahrendorf M. Therapeutic siRNA silencing in inflammatory monocytes in mice. Nat Biotechnol. 2011;29:1005–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hayashidani S, Tsutsui H, Shiomi T, Ikeuchi M, Matsusaka H, Suematsu N, Wen J, Egashira K, Takeshita A. Anti‐monocyte chemoattractant protein‐1 gene therapy attenuates left ventricular remodeling and failure after experimental myocardial infarction. Circulation. 2003;108:2134–2140. [DOI] [PubMed] [Google Scholar]
- 24. Kaikita K, Hayasaki T, Okuma T, Kuziel WA, Ogawa H, Takeya M. Targeted deletion of CC chemokine receptor 2 attenuates left ventricular remodeling after experimental myocardial infarction. Am J Pathol. 2004;165:439–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Auffray C, Sieweke MH, Geissmann F. Blood monocytes: development, heterogeneity, and relationship with dendritic cells. Annu Rev Immunol. 2009;27:669–692. [DOI] [PubMed] [Google Scholar]
- 26. Akashi K, Traver D, Miyamoto T, Weissman IL. A clonogenic common myeloid progenitor that gives rise to all myeloid lineages. Nature. 2000;404:193–197. [DOI] [PubMed] [Google Scholar]
- 27. Serbina NV, Jia T, Hohl TM, Pamer EG. Monocyte‐mediated defense against microbial pathogens. Annu Rev Immunol. 2008;26:421–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hettinger J, Richards DM, Hansson J, Barra MM, Joschko AC, Krijgsveld J, Feuerer M. Origin of monocytes and macrophages in a committed progenitor. Nat Immunol. 2013;14:821–830. [DOI] [PubMed] [Google Scholar]
- 29. Sieweke MH, Allen JE. Beyond stem cells: self‐renewal of differentiated macrophages. Science. 2013;342:1242974. [DOI] [PubMed] [Google Scholar]
- 30. Levy GA, Edgington TS. Lymphocyte cooperation is required for amplification of macrophage procoagulant activity. J Exp Med. 1980;151:1232–1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ziegler‐Heitbrock L. Reprint of: monocyte subsets in man and other species. Cell Immunol. 2014;291:11–15. [DOI] [PubMed] [Google Scholar]
- 32. Woollard KJ, Geissmann F. Monocytes in atherosclerosis: subsets and functions. Nat Rev Cardiol. 2010;7:77–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Shantsila E, Tapp LD, Wrigley BJ, Pamukcu B, Apostolakis S, Montoro‐Garcia S, Lip GY. Monocyte subsets in coronary artery disease and their associations with markers of inflammation and fibrinolysis. Atherosclerosis. 2014;234:4–10. [DOI] [PubMed] [Google Scholar]
- 34. Yasaka T, Mantich NM, Boxer LA, Baehner RL. Functions of human monocyte and lymphocyte subsets obtained by countercurrent centrifugal elutriation: differing functional capacities of human monocyte subsets. J Immunol. 1981;127:1515–1518. [PubMed] [Google Scholar]
- 35. Maekawa Y, Anzai T, Yoshikawa T, Asakura Y, Takahashi T, Ishikawa S, Mitamura H, Ogawa S. Prognostic significance of peripheral monocytosis after reperfused acute myocardial infarction: a possible role for left ventricular remodeling. J Am Coll Cardiol. 2002;39:241–246. [DOI] [PubMed] [Google Scholar]
- 36. Kervinen H, Manttari M, Kaartinen M, Makynen H. Prognostic usefulness of plasma monocyte/macrophage and T‐lymphocyte activation markers in patients with acute coronary syndromes. Am J Cardiol. 2004;94:993–996. [DOI] [PubMed] [Google Scholar]
- 37. Ziegler‐Heitbrock HW, Passlick B, Flieger D. The monoclonal antimonocyte antibody My4 stains B lymphocytes and two distinct monocyte subsets in human peripheral blood. Hybridoma. 1988;7:521–527. [DOI] [PubMed] [Google Scholar]
- 38. Zhao C, Zhang H, Wong WC, Sem X, Han H, Ong SM, Tan YC, Yeap WH, Gan CS, Ng KQ, Koh MB, Kourilsky P, Sze SK, Wong SC. Identification of novel functional differences in monocyte subsets using proteomic and transcriptomic methods. J Proteome Res. 2009;8:4028–4038. [DOI] [PubMed] [Google Scholar]
- 39. Wong KL, Yeap WH, Tai JJY, Ong SM, Dang TM, Wong SC. The three human monocyte subsets: implications for health and disease. Immunol Res. 2012;53:41–57. [DOI] [PubMed] [Google Scholar]
- 40. Cros J, Cagnard N, Woollard K, Patey N, Zhang SY, Senechal B, Puel A, Biswas SK, Moshous D, Picard C, Jais JP, D'Cruz D, Casanova JL, Trouillet C, Geissmann F. Human CD14dim monocytes patrol and sense nucleic acids and viruses via TLR7 and TLR8 receptors. Immunity. 2010;33:375–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Zang YC, Skinner SM, Robinson RR, Li S, Rivera VM, Hutton GJ, Zhang JZ. Regulation of differentiation and functional properties of monocytes and monocyte‐derived dendritic cells by interferon beta in multiple sclerosis. Mult Scler. 2004;10:499–506. [DOI] [PubMed] [Google Scholar]
- 42. Ingersoll MA, Spanbroek R, Lottaz C, Gautier EL, Frankenberger M, Hoffmann R, Lang R, Haniffa M, Collin M, Tacke F, Habenicht AJ, Ziegler‐Heitbrock L, Randolph GJ. Comparison of gene expression profiles between human and mouse monocyte subsets. Blood. 2010;115:e10–e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Zawada AM, Rogacev KS, Rotter B, Winter P, Marell RR, Fliser D, Heine GH. Supersage evidence for CD14++CD16+ monocytes as a third monocyte subset. Blood. 2011;118:e50–e61. [DOI] [PubMed] [Google Scholar]
- 44. Schakel K, Von Kietzell M, Hansel A, Ebling A, Schulze L, Haase M, Semmler C, Sarfati M, Barclay AN, Randolph GJ, Meurer M, Rieber EP. Human 6‐sulfo LacNAc‐expressing dendritic cells are principal producers of early interleukin‐12 and are controlled by erythrocytes. Immunity. 2006;24:767–777. [DOI] [PubMed] [Google Scholar]
- 45. Si Y, Tsou CL, Croft K, Charo IF. CCR2 mediates hematopoietic stem and progenitor cell trafficking to sites of inflammation in mice. J Clin Invest. 2010;120:1192–1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Frankenberger M, Sternsdorf T, Pechumer H, Pforte A, Ziegler‐Heitbrock HW. Differential cytokine expression in human blood monocyte subpopulations: a polymerase chain reaction analysis. Blood. 1996;87:373–377. [PubMed] [Google Scholar]
- 47. Rossol M, Kraus S, Pierer M, Baerwald C, Wagner U. The CD14(bright) CD16+ monocyte subset is expanded in rheumatoid arthritis and promotes expansion of the TH17 cell population. Arthritis Rheum. 2012;64:671–677. [DOI] [PubMed] [Google Scholar]
- 48. Skrzeczynska‐Moncznik J, Bzowska M, Loseke S, Grage‐Griebenow E, Zembala M, Pryjma J. Peripheral blood CD14high CD16+ monocytes are main producers of IL‐10. Scand J Immunol. 2008;67:152–159. [DOI] [PubMed] [Google Scholar]
- 49. Wong KL, Tai JJ, Wong WC, Han H, Sem X, Yeap WH, Kourilsky P, Wong SC. Gene expression profiling reveals the defining features of the classical, intermediate, and nonclassical human monocyte subsets. Blood. 2011;118:e16–e31. [DOI] [PubMed] [Google Scholar]
- 50. Coffelt SB, Tal AO, Scholz A, De Palma M, Patel S, Urbich C, Biswas SK, Murdoch C, Plate KH, Reiss Y, Lewis CE. Angiopoietin‐2 regulates gene expression in TIE2‐expressing monocytes and augments their inherent proangiogenic functions. Cancer Res. 2010;70:5270–5280. [DOI] [PubMed] [Google Scholar]
- 51. Yona S, Kim KW, Wolf Y, Mildner A, Varol D, Breker M, Strauss‐Ayali D, Viukov S, Guilliams M, Misharin A, Hume DA, Perlman H, Malissen B, Zelzer E, Jung S. Fate mapping reveals origins and dynamics of monocytes and tissue macrophages under homeostasis. Immunity. 2013;38:79–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Tsujioka H, Imanishi T, Ikejima H, Kuroi A, Takarada S, Tanimoto T, Kitabata H, Okochi K, Arita Y, Ishibashi K, Komukai K, Kataiwa H, Nakamura N, Hirata K, Tanaka A, Akasaka T. Impact of heterogeneity of human peripheral blood monocyte subsets on myocardial salvage in patients with primary acute myocardial infarction. J Am Coll Cardiol. 2009;54:130–138. [DOI] [PubMed] [Google Scholar]
- 53. Zawada AM, Rogacev KS, Schirmer SH, Sester M, Bohm M, Fliser D, Heine GH. Monocyte heterogeneity in human cardiovascular disease. Immunobiology. 2012;217:1273–1284. [DOI] [PubMed] [Google Scholar]
- 54. Weber C, Shantsila E, Hristov M, Caligiuri G, Guzik T, Heine GH, Hoefer IE, Monaco C, Peter K, Rainger E, Siegbahn A, Steffens S, Wojta J, Lip GY. Role and analysis of monocyte subsets in cardiovascular disease. Joint consensus document of the European Society of Cardiology (ESC) working groups “atherosclerosis & vascular biology” and “thrombosis”. Thromb Haemost. 2016;116:626–637. [DOI] [PubMed] [Google Scholar]
- 55. Geissmann F, Jung S, Littman DR. Blood monocytes consist of two principal subsets with distinct migratory properties. Immunity. 2003;19:71–82. [DOI] [PubMed] [Google Scholar]
- 56. Thomas G, Tacke R, Hedrick CC, Hanna RN. Nonclassical patrolling monocyte function in the vasculature. Arterioscler Thromb Vasc Biol. 2015;35:1306–1316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Shantsila E, Wrigley B, Tapp L, Apostolakis S, Montoro‐Garcia S, Drayson MT, Lip GY. Immunophenotypic characterization of human monocyte subsets: possible implications for cardiovascular disease pathophysiology. J Thromb Haemost. 2011;9:1056–1066. [DOI] [PubMed] [Google Scholar]
- 58. Ziegler‐Heitbrock L, Ancuta P, Crowe S, Dalod M, Grau V, Hart DN, Leenen PJ, Liu YJ, MacPherson G, Randolph GJ, Scherberich J, Schmitz J, Shortman K, Sozzani S, Strobl H, Zembala M, Austyn JM, Lutz MB. Nomenclature of monocytes and dendritic cells in blood. Blood. 2010;116:e74–e80. [DOI] [PubMed] [Google Scholar]
- 59. Palframan RT, Jung S, Cheng G, Weninger W, Luo Y, Dorf M, Littman DR, Rollins BJ, Zweerink H, Rot A, Von Andrian UH. Inflammatory chemokine transport and presentation in HEV: a remote control mechanism for monocyte recruitment to lymph nodes in inflamed tissues. J Exp Med. 2001;194:1361–1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Grip O, Bredberg A, Lindgren S, Henriksson G. Increased subpopulations of CD16(+) and CD56(+) blood monocytes in patients with active Crohn's disease. Inflamm Bowel Dis. 2007;13:566–572. [DOI] [PubMed] [Google Scholar]
- 61. Weber C, Belge KU, Von Hundelshausen P, Draude G, Steppich B, Mack M, Frankenberger M, Weber KS, Ziegler‐Heitbrock HW. Differential chemokine receptor expression and function in human monocyte subpopulations. J Leukoc Biol. 2000;67:699–704. [DOI] [PubMed] [Google Scholar]
- 62. Ancuta P, Rao R, Moses A, Mehle A, Shaw SK, Luscinskas FW, Gabuzda D. Fractalkine preferentially mediates arrest and migration of CD16+ monocytes. J Exp Med. 2003;197:1701–1707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Swirski FK, Nahrendorf M, Etzrodt M, Wildgruber M, Cortez‐Retamozo V, Panizzi P, Figueiredo JL, Kohler RH, Chudnovskiy A, Waterman P, Aikawa E, Mempel TR, Libby P, Weissleder R, Pittet MJ. Identification of splenic reservoir monocytes and their deployment to inflammatory sites. Science. 2009;325:612–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Varol C, Landsman L, Fogg DK, Greenshtein L, Gildor B, Margalit R, Kalchenko V, Geissmann F, Jung S. Monocytes give rise to mucosal, but not splenic, conventional dendritic cells. J Exp Med. 2007;204:171–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Boyette LB, Macedo C, Hadi K, Elinoff BD, Walters JT, Ramaswami B, Chalasani G, Taboas JM, Lakkis FG, Metes DM. Phenotype, function, and differentiation potential of human monocyte subsets. PLoS One. 2017;12:e0176460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Sunderkotter C, Nikolic T, Dillon MJ, Van Rooijen N, Stehling M, Drevets DA, Leenen PJ. Subpopulations of mouse blood monocytes differ in maturation stage and inflammatory response. J Immunol. 2004;172:4410–4417. [DOI] [PubMed] [Google Scholar]
- 67. Gren ST, Rasmussen TB, Janciauskiene S, Håkansson K, Gerwien JG, Grip O. A single‐cell gene‐expression profile reveals inter‐cellular heterogeneity within human monocyte subsets. PLoS One. 2015;10:e0144351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Frankenberger M, Passlick B, Hofer T, Siebeck M, Maier KL, Ziegler‐Heitbrock LH. Immunologic characterization of normal human pleural macrophages. Am J Respir Cell Mol Biol. 2000;23:419–426. [DOI] [PubMed] [Google Scholar]
- 69. Moniuszko M, Kowal K, Rusak M, Pietruczuk M, Dabrowska M, Bodzenta‐Lukaszyk A. Monocyte CD163 and CD36 expression in human whole blood and isolated mononuclear cell samples: influence of different anticoagulants. Clin Vaccine Immunol. 2006;13:704–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Schenkel AR, Mamdouh Z, Chen X, Liebman RM, Muller WA. CD99 plays a major role in the migration of monocytes through endothelial junctions. Nat Immunol. 2002;3:143–150. [DOI] [PubMed] [Google Scholar]
- 71. Tsou C‐L, Gladue RP, Carroll LA, Paradis T, Boyd JG, Nelson RT, Neote K, Charo IF. Identification of C‐C chemokine receptor 1 (CCR1) as the monocyte hemofiltrate C‐C chemokine (HCC)‐1 receptor. J Exp Med. 1998;188:603–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Italiani P, Boraschi D. From monocytes to M1/M2 macrophages: phenotypical vs. functional differentiation. Front Immunol. 2014;5:514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Poehlmann H, Schefold JC, Zuckermann‐Becker H, Volk HD, Meisel C. Phenotype changes and impaired function of dendritic cell subsets in patients with sepsis: a prospective observational analysis. Crit Care. 2009;13:R119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Tippett E, Cheng WJ, Westhorpe C, Cameron PU, Brew BJ, Lewin SR, Jaworowski A, Crowe SM. Differential expression of CD163 on monocyte subsets in healthy and HIV‐1 infected individuals. PLoS One. 2011;6:e19968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Buechler C, Ritter M, Orsó E, Langmann T, Klucken J, Schmitz G. Regulation of scavenger receptor CD163 expression in human monocytes and macrophages by pro‐ and anti inflammatory stimuli. J Leukoc Biol. 2000;67:97–103. [PubMed] [Google Scholar]
- 76. Qu C, Edwards EW, Tacke F, Angeli V, Llodra J, Sanchez‐Schmitz G, Garin A, Haque NS, Peters W, Van Rooijen N, Sanchez‐Torres C, Bromberg J, Charo IF, Jung S, Lira SA, Randolph GJ. Role of CCR8 and other chemokine pathways in the migration of monocyte‐derived dendritic cells to lymph nodes. J Exp Med. 2004;200:1231–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. de Baey A, Mende I, Riethmueller G, Baeuerle PA. Phenotype and function of human dendritic cells derived from M‐DC8(+) monocytes. Eur J Immunol. 2001;31:1646–1655. [DOI] [PubMed] [Google Scholar]
- 78. Hofer TP, Zawada AM, Frankenberger M, Skokann K, Satzl AA, Gesierich W, Schuberth M, Levin J, Danek A, Rotter B, Heine GH, Ziegler‐Heitbrock L. Slan‐defined subsets of CD16‐positive monocytes: impact of granulomatous inflammation and M‐CSF receptor mutation. Blood. 2015;126:2601–2610. [DOI] [PubMed] [Google Scholar]
- 79. Raghu H, Lepus CM, Wang Q, Wong HH, Lingampalli N, Oliviero F, Punzi L, Giori NJ, Goodman SB, Chu CR, Sokolove JB, Robinson WH. CCL2/CCR2, but not CCL5/CCR5, mediates monocyte recruitment, inflammation and cartilage destruction in osteoarthritis. Ann Rheum Dis. 2017;76:914–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Costa C, Traves SL, Tudhope SJ, Fenwick PS, Belchamber KBR, Russell REK, Barnes PJ, Donnelly LE. Enhanced monocyte migration to CXCR3 and CCR5 chemokines in COPD. Eur Respir J. 2016;47:1093. [DOI] [PubMed] [Google Scholar]
- 81. Macauley MS, Crocker PR, Paulson JC. Siglec‐mediated regulation of immune cell function in disease. Nat Rev Immunol. 2014;14:653–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Delputte PL, Van Gorp H, Favoreel HW, Hoebeke I, Delrue I, Dewerchin H, Verdonck F, Verhasselt B, Cox E, Nauwynck HJ. Porcine sialoadhesin (CD169/SIGLEC‐1) is an endocytic receptor that allows targeted delivery of toxins and antigens to macrophages. PLoS One. 2011;6:e16827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Lauvau G, Chorro L, Spaulding E, Soudja SM. Inflammatory monocyte effector mechanisms. Cell Immunol. 2014;291:32–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Castano D, Garcia LF, Rojas M. Increased frequency and cell death of CD16+ monocytes with mycobacterium tuberculosis infection. Tuberculosis (Edinb). 2011;91:348–360. [DOI] [PubMed] [Google Scholar]
- 85. Gordon S, Taylor PR. Monocyte and macrophage heterogeneity. Nat Rev Immunol. 2005;5:953–964. [DOI] [PubMed] [Google Scholar]
- 86. Moniuszko M, Bodzenta‐Lukaszyk A, Kowal K, Lenczewska D, Dabrowska M. Enhanced frequencies of CD14++CD16+, but not CD14+CD16+, peripheral blood monocytes in severe asthmatic patients. Clin Immunol. 2009;130:338–346. [DOI] [PubMed] [Google Scholar]
- 87. Ancuta P. A slan‐based nomenclature for monocytes? Blood. 2015;126:2536. [DOI] [PubMed] [Google Scholar]
- 88. Thaler B, Hohensinner PJ, Krychtiuk KA, Matzneller P, Koller L, Brekalo M, Maurer G, Huber K, Zeitlinger M, Jilma B, Wojta J, Speidl WS. Differential in vivo activation of monocyte subsets during low‐grade inflammation through experimental endotoxemia in humans. Sci Rep. 2016;6:30162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Danis VA, Millington M, Hyland VJ, Grennan D. Cytokine production by normal human monocytes: inter‐subject variation and relationship to an IL‐1 receptor antagonist (IL‐1RA) gene polymorphism. Clin Exp Immunol. 1995;99:303–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Rossol M, Heine H, Meusch U, Quandt D, Klein C, Sweet MJ, Hauschildt S. LPS‐induced cytokine production in human monocytes and macrophages. Crit Rev Immunol. 2011;31:379–446. [DOI] [PubMed] [Google Scholar]
- 91. Smedman C, Ernemar T, Gudmundsdotter L, Gille‐Johnson P, Somell A, Nihlmark K, Gardlund B, Andersson J, Paulie S. Fluorospot analysis of TLR‐activated monocytes reveals several distinct cytokine‐secreting subpopulations. Scand J Immunol. 2012;75:249–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Patel H, Davidson D. Control of pro‐inflammatory cytokine release from human monocytes with the use of an interleukin‐10 monoclonal antibody. Methods Mol Biol. 2014;1172:99–106. [DOI] [PubMed] [Google Scholar]
- 93. Tsou CL, Peters W, Si Y, Slaymaker S, Aslanian AM, Weisberg SP, Mack M, Charo IF. Critical roles for CCR2 and MCP‐3 in monocyte mobilization from bone marrow and recruitment to inflammatory sites. J Clin Invest. 2007;117:902–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Belge KU, Dayyani F, Horelt A, Siedlar M, Frankenberger M, Frankenberger B, Espevik T, Ziegler‐Heitbrock L. The proinflammatory CD14+CD16+DR++ monocytes are a major source of TNF. J Immunol. 2002;168:3536–3542. [DOI] [PubMed] [Google Scholar]
- 95. Majmudar MD, Keliher EJ, Heidt T, Leuschner F, Truelove J, Sena BF, Gorbatov R, Iwamoto Y, Dutta P, Wojtkiewicz G, Courties G, Sebas M, Borodovsky A, Fitzgerald K, Nolte MW, Dickneite G, Chen JW, Anderson DG, Swirski FK, Weissleder R, Nahrendorf M. Monocyte‐directed RNAI targeting CCR2 improves infarct healing in atherosclerosis‐prone mice. Circulation. 2013;127:2038–2046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Leuschner F, Courties G, Dutta P, Mortensen LJ, Gorbatov R, Sena B, Novobrantseva TI, Borodovsky A, Fitzgerald K, Koteliansky V, Iwamoto Y, Bohlender M, Meyer S, Lasitschka F, Meder B, Katus HA, Lin C, Libby P, Swirski FK, Anderson DG, Weissleder R, Nahrendorf M. Silencing of CCR2 in myocarditis. Eur Heart J. 2015;36:1478–1488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Nahrendorf M, Swirski FK. Monocyte and macrophage heterogeneity in the heart. Circ Res. 2013;112:1624–1633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Panizzi P, Swirski FK, Figueiredo JL, Waterman P, Sosnovik DE, Aikawa E, Libby P, Pittet M, Weissleder R, Nahrendorf M. Impaired infarct healing in atherosclerotic mice with Ly‐6C(hi) monocytosis. J Am Coll Cardiol. 2010;55:1629–1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Meyer IS, Jungmann A, Dieterich C, Zhang M, Lasitschka F, Werkmeister S, Haas J, Muller OJ, Boutros M, Nahrendorf M, Katus HA, Hardt SE, Leuschner F. The cardiac microenvironment uses non‐canonical WNT signaling to activate monocytes after myocardial infarction. EMBO Mol Med. 2017;9:1279–1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Auffray C, Fogg D, Garfa M, Elain G, Join‐Lambert O, Kayal S, Sarnacki S, Cumano A, Lauvau G, Geissmann F. Monitoring of blood vessels and tissues by a population of monocytes with patrolling behavior. Science. 2007;317:666–670. [DOI] [PubMed] [Google Scholar]
- 101. Frantz S, Hofmann U, Fraccarollo D, Schafer A, Kranepuhl S, Hagedorn I, Nieswandt B, Nahrendorf M, Wagner H, Bayer B, Pachel C, Schon MP, Kneitz S, Bobinger T, Weidemann F, Ertl G, Bauersachs J. Monocytes/macrophages prevent healing defects and left ventricular thrombus formation after myocardial infarction. FASEB J. 2013;27:871–881. [DOI] [PubMed] [Google Scholar]
- 102. Swirski FK, Libby P, Aikawa E, Alcaide P, Luscinskas FW, Weissleder R, Pittet MJ. Ly‐6C(hi) monocytes dominate hypercholesterolemia‐associated monocytosis and give rise to macrophages in atheromata. J Clin Invest. 2007;117:195–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Tacke F, Alvarez D, Kaplan TJ, Jakubzick C, Spanbroek R, Llodra J, Garin A, Liu J, Mack M, van Rooijen N, Lira SA, Habenicht AJ, Randolph GJ. Monocyte subsets differentially employ CCR2, CCR5, and CX3CR1 to accumulate within atherosclerotic plaques. J Clin Invest. 2007;117:185–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Arnold L, Henry A, Poron F, Baba‐Amer Y, van Rooijen N, Plonquet A, Gherardi RK, Chazaud B. Inflammatory monocytes recruited after skeletal muscle injury switch into anti inflammatory macrophages to support myogenesis. J Exp Med. 2007;204:1057–1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Ramachandran P, Pellicoro A, Vernon MA, Boulter L, Aucott RL, Ali A, Hartland SN, Snowdon VK, Cappon A, Gordon‐Walker TT, Williams MJ, Dunbar DR, Manning JR, Van Rooijen N, Fallowfield JA, Forbes SJ, Iredale JP. Differential Ly‐6C expression identifies the recruited macrophage phenotype, which orchestrates the regression of murine liver fibrosis. Proc Natl Acad Sci USA. 2012;109:E3186–E3195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Patel AA, Zhang Y, Fullerton JN, Boelen L, Rongvaux A, Maini AA, Bigley V, Flavell RA, Gilroy DW, Asquith B, Macallan D, Yona S. The fate and lifespan of human monocyte subsets in steady state and systemic inflammation. J Exp Med. 2017;214:1913–1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Mandl M, Schmitz S, Weber C, Hristov M. Characterization of the CD14++CD16+ monocyte population in human bone marrow. PLoS One. 2014;9:e112140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Thompson WL, Van Eldik LJ. Inflammatory cytokines stimulate the chemokines CCL2/MCP‐1 and CCL7/MCP‐7 through NFκB and MAPK dependent pathways in rat astrocytes. Brain Res. 2009;1287:47–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Nguyen KD, Fentress SJ, Qiu Y, Yun K, Cox JS, Chawla A. Circadian gene Bmal1 regulates diurnal oscillations of Ly6C(hi) inflammatory monocytes. Science. 2013;341:1483–1488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Meuret G, Bammert J, Hoffmann G. Kinetics of human monocytopoiesis. Blood. 1974;44:801–816. [PubMed] [Google Scholar]
- 111. Soehnlein O, Drechsler M, Döring Y, Lievens D, Hartwig H, Kemmerich K, Ortega‐Gómez A, Mandl M, Vijayan S, Projahn D, Garlichs CD, Koenen RR, Hristov M, Lutgens E, Zernecke A, Weber C. Distinct functions of chemokine receptor axes in the atherogenic mobilization and recruitment of classical monocytes. EMBO Mol Med. 2013;5:471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Deshmane SL, Kremlev S, Amini S, Sawaya BE. Monocyte chemoattractant protein‐1 (MCP‐1): an overview. J Interferon Cytokine Res. 2009;29:313–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Hirota H, Izumi M, Hamaguchi T, Sugiyama S, Murakami E, Kunisada K, Fujio Y, Oshima Y, Nakaoka Y, Yamauchi‐Takihara K. Circulating interleukin‐6 family cytokines and their receptors in patients with congestive heart failure. Heart Vessels. 2004;19:237–241. [DOI] [PubMed] [Google Scholar]
- 114. Kleinbongard P, Schulz R, Heusch G. TNF alpha in myocardial ischemia/reperfusion, remodeling and heart failure. Heart Fail Rev. 2011;16:49–69. [DOI] [PubMed] [Google Scholar]
- 115. Caruso R, De Chiara B, Campolo J, Verde A, Musca F, Belli O, Parolini M, Cozzi L, Moreo A, Frigerio M, Parodi O. Neopterin levels are independently associated with cardiac remodeling in patients with chronic heart failure. Clin Biochem. 2013;46:94–98. [DOI] [PubMed] [Google Scholar]
- 116. Swirski FK, Weissleder R, Pittet MJ. Heterogeneous in vivo behavior of monocyte subsets in atherosclerosis. Arterioscler Thromb Vasc Biol. 2009;29:1424–1432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Apostolakis S, Vogiatzi K, Krambovitis E, Spandidos DA. IL‐1 cytokines in cardiovascular disease: diagnostic, prognostic and therapeutic implications. Cardiovasc Hematol Agents Med Chem. 2008;6:150–158. [DOI] [PubMed] [Google Scholar]
- 118. Heidt T, Courties G, Dutta P, Sager HB, Sebas M, Iwamoto Y, Sun Y, Da Silva N, Panizzi P, Van der Laan AM, Swirski FK, Weissleder R, Nahrendorf M. Differential contribution of monocytes to heart macrophages in steady‐state and after myocardial infarction. Circ Res. 2014;115:284–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Lu W, Zhang Z, Fu C, Ma G. Intermediate monocytes lead to enhanced myocardial remodelling in STEMI patients with diabetes. Int Heart J. 2015;56:22–28. [DOI] [PubMed] [Google Scholar]
- 120. Barisione C, Garibaldi S, Ghigliotti G, Fabbi P, Altieri P, Casale MC, Spallarossa P, Bertero G, Balbi M, Corsiglia L, Brunelli C. CD14CD16 monocyte subset levels in heart failure patients. Dis Markers. 2010;28:115–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Shioi T, Matsumori A, Kihara Y, Inoko M, Ono K, Iwanaga Y, Yamada T, Iwasaki A, Matsushima K, Sasayama S. Increased expression of interleukin‐1 beta and monocyte chemotactic and activating factor/monocyte chemoattractant protein‐1 in the hypertrophied and failing heart with pressure overload. Circ Res. 1997;81:664–671. [DOI] [PubMed] [Google Scholar]
- 122. Bradham WS, Moe G, Wendt KA, Scott AA, Konig A, Romanova M, Naik G, Spinale FG. TNF‐α and myocardial matrix metalloproteinases in heart failure: relationship to LV remodeling. Am J Physiol Heart Circ Physiol. 2002;282:H1288–H1295. [DOI] [PubMed] [Google Scholar]
- 123. Bosco MC, Puppo M, Blengio F, Fraone T, Cappello P, Giovarelli M, Varesio L. Monocytes and dendritic cells in a hypoxic environment: spotlights on chemotaxis and migration. Immunobiology. 2008;213:733–749. [DOI] [PubMed] [Google Scholar]
- 124. Hristov M, Heine GH. Monocyte subsets in atherosclerosis. Hamostaseologie. 2015;35:105–112. [DOI] [PubMed] [Google Scholar]
- 125. Jaipersad AS, Lip GY, Silverman S, Shantsila E. The role of monocytes in angiogenesis and atherosclerosis. J Am Coll Cardiol. 2014;63:1–11. [DOI] [PubMed] [Google Scholar]
- 126. Tapp LD, Shantsila E, Wrigley BJ, Pamukcu B, Lip GY. The CD14++CD16+ monocyte subset and monocyte‐platelet interactions in patients with ST‐elevation myocardial infarction. J Thromb Haemost. 2012;10:1231–1241. [DOI] [PubMed] [Google Scholar]
- 127. Zouggari Y, Ait‐Oufella H, Bonnin P, Simon T, Sage AP, Guerin C, Vilar J, Caligiuri G, Tsiantoulas D, Laurans L, Dumeau E, Kotti S, Bruneval P, Charo IF, Binder CJ, Danchin N, Tedgui A, Tedder TF, Silvestre JS, Mallat Z. B lymphocytes trigger monocyte mobilization and impair heart function after acute myocardial infarction. Nat Med. 2013;19:1273–1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Hilgendorf I, Gerhardt LM, Tan TC, Winter C, Holderried TA, Chousterman BG, Iwamoto Y, Liao R, Zirlik A, Scherer‐Crosbie M, Hedrick CC, Libby P, Nahrendorf M, Weissleder R, Swirski FK. Ly‐6Chigh monocytes depend on Nr4a1 to balance both inflammatory and reparative phases in the infarcted myocardium. Circ Res. 2014;114:1611–1622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Zeng S, Zhou X, Ge L, Ji WJ, Shi R, Lu RY, Sun HY, Guo ZZ, Zhao JH, Jiang TM, Li YM. Monocyte subsets and monocyte‐platelet aggregates in patients with unstable angina. J Thromb Thrombolysis. 2014;38:439–446. [DOI] [PubMed] [Google Scholar]
- 130. Dobreanu M, Dobreanu D, Fodor A, Bacarea A. Integrin expression on monocytes and lymphocytes in unstable angina short term effects of atorvastatin. Rom J Intern Med. 2007;45:193–199. [PubMed] [Google Scholar]
- 131. Hojo Y, Ikeda U, Takahashi M, Shimada K. Increased levels of monocyte‐related cytokines in patients with unstable angina. Atherosclerosis. 2002;161:403–408. [DOI] [PubMed] [Google Scholar]
- 132. Goonewardena SN, Stein AB, Tsuchida RE, Rattan R, Shah D, Hummel SL. Monocyte subsets and inflammatory cytokines in acute decompensated heart failure. J Card Fail. 2016;22:358–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Apostolakis S, Lip GY, Shantsila E. Monocytes in heart failure: relationship to a deteriorating immune overreaction or a desperate attempt for tissue repair? Cardiovasc Res. 2010;85:649–660. [DOI] [PubMed] [Google Scholar]
- 134. Subimerb C, Pinlaor S, Lulitanond V, Khuntikeo N, Okada S, McGrath MS, Wongkham S. Circulating CD4(+) CD16(+) monocyte levels predict tissue invasive character of cholangiocarcinoma. Clin Exp Immunol. 2010;161:471–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Amir O, Spivak I, Lavi I, Rahat MA. Changes in the monocytic subsets CD14(dim)CD16(+) and CD14(++)CD16(‐) in chronic systolic heart failure patients. Mediators Inflamm. 2012;2012:616384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Ghigliotti G, Barisione C, Garibaldi S, Brunelli C, Palmieri D, Spinella G, Pane B, Spallarossa P, Altieri P, Fabbi P, Sambuceti G, Palombo D. CD16(+) monocyte subsets are increased in large abdominal aortic aneurysms and are differentially related with circulating and cell‐associated biochemical and inflammatory biomarkers. Dis Markers. 2013;34:131–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Kawaguchi M, Takahashi M, Hata T, Kashima Y, Usui F, Morimoto H, Izawa A, Takahashi Y, Masumoto J, Koyama J, Hongo M, Noda T, Nakayama J, Sagara J, Taniguchi S, Ikeda U. Inflammasome activation of cardiac fibroblasts is essential for myocardial ischemia/reperfusion injury. Circulation. 2011;123:594–604. [DOI] [PubMed] [Google Scholar]
- 138. Frangogiannis NG, Youker KA, Rossen RD, Gwechenberger M, Lindsey MH, Mendoza LH, Michael LH, Ballantyne CM, Smith CW, Entman ML. Cytokines and the microcirculation in ischemia and reperfusion. J Mol Cell Cardiol. 1998;30:2567–2576. [DOI] [PubMed] [Google Scholar]
- 139. Frangogiannis NG, Lindsey ML, Michael LH, Youker KA, Bressler RB, Mendoza LH, Spengler RN, Smith CW, Entman ML. Resident cardiac mast cells degranulate and release preformed TNF‐alpha, initiating the cytokine cascade in experimental canine myocardial ischemia/reperfusion. Circulation. 1998;98:699–710. [DOI] [PubMed] [Google Scholar]
- 140. Zarember KA, Godowski PJ. Tissue expression of human Toll‐like receptors and differential regulation of Toll‐like receptor mRNAs in leukocytes in response to microbes, their products, and cytokines. J Immunol. 2002;168:554–561. [DOI] [PubMed] [Google Scholar]
- 141. Anker SD, Egerer KR, Volk HD, Kox WJ, Poole‐Wilson PA, Coats AJ. Elevated soluble CD14 receptors and altered cytokines in chronic heart failure. Am J Cardiol. 1997;79:1426–1430. [DOI] [PubMed] [Google Scholar]
- 142. Niebauer J, Volk HD, Kemp M, Dominguez M, Schumann RR, Rauchhaus M, Poole‐Wilson PA, Coats AJ, Anker SD. Endotoxin and immune activation in chronic heart failure: a prospective cohort study. Lancet. 1999;353:1838–1842. [DOI] [PubMed] [Google Scholar]
- 143. Sarafoff N, Ndrepepa G, Mehilli J, Dorrler K, Schulz S, Iijima R, Byrne R, Schomig A, Kastrati A. Aspirin and clopidogrel with or without phenprocoumon after drug eluting coronary stent placement in patients on chronic oral anticoagulation. J Intern Med. 2008;264:472–480. [DOI] [PubMed] [Google Scholar]
- 144. Satoh M, Shimoda Y, Maesawa C, Akatsu T, Ishikawa Y, Minami Y, Hiramori K, Nakamura M. Activated Toll‐like receptor 4 in monocytes is associated with heart failure after acute myocardial infarction. Int J Cardiol. 2006;109:226–234. [DOI] [PubMed] [Google Scholar]
- 145. Földes G, von Haehling S, Okonko DO, Jankowska EA, Poole‐Wilson PA, Anker SD. Fluvastatin reduces increased blood monocyte Toll‐like receptor 4 expression in whole blood from patients with chronic heart failure. Int J Cardiol. 2008;124:80–85. [DOI] [PubMed] [Google Scholar]
- 146. Frantz S, Kobzik L, Kim YD, Fukazawa R, Medzhitov R, Lee RT, Kelly RA. Toll4 (TLR4) expression in cardiac myocytes in normal and failing myocardium. J Clin Invest. 1999;104:271–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Hoshino K, Takeuchi O, Kawai T, Sanjo H, Ogawa T, Takeda Y, Takeda K, Akira S. Cutting edge: Toll‐like receptor 4 (TLR4)‐deficient mice are hyporesponsive to lipopolysaccharide: evidence for TLR4 as the LPS gene product. J Immunol. 1999;162:3749–3752. [PubMed] [Google Scholar]
- 148. Krüger S, Kunz D, Graf J, Stickel T, Merx MW, Koch KC, Janssens U, Hanrath P. Endotoxin hypersensitivity in chronic heart failure. Int J Cardiol. 2007;115:159–163. [DOI] [PubMed] [Google Scholar]
- 149. Sandek A, Swidsinski A, Schroedl W, Watson A, Valentova M, Herrmann R, Scherbakov N, Cramer L, Rauchhaus M, Grosse‐Herrenthey A, Krueger M, Von Haehling S, Doehner W, Anker SD, Bauditz J. Intestinal blood flow in patients with chronic heart failure: a link with bacterial growth, gastrointestinal symptoms, and cachexia. J Am Coll Cardiol. 2014;64:1092–1102. [DOI] [PubMed] [Google Scholar]
- 150. Prasad K, Kalra J, Bharadwaj B. Increased chemiluminescence of polymorphonuclear leucocytes in dogs with volume overload heart failure. Br J Exp Pathol. 1989;70:463–468. [PMC free article] [PubMed] [Google Scholar]
- 151. Uthamalingam S, Patvardhan E, Subramanian S, Ahmed W, Martin W, Daley M, Capodilupo R. Utility of the neutrophil to lymphocyte ratio in predicting long‐term outcomes in acute decompensated heart failure. Am J Cardiol. 2011;107:433–438. [DOI] [PubMed] [Google Scholar]
- 152. McMurray JJ, Adamopoulos S, Anker SD, Auricchio A, Böhm M, Dickstein K, Falk V, Filippatos G, Fonseca C, Gomez‐Sanchez MA, Jaarsma T, Køber L, Lip GY, Maggioni AP, Parkhomenko A, Pieske BM, Popescu BA, Ronnevik PK, Rutten FH, Schwitter J, Seferovic P, Stepinska J, Trindade PT, Voors AA, Zannad F, Zeiher A; ESC Committee for Practice Guidelines . ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure 2012: the Task Force for the Diagnosis and Treatment of Acute and Chronic Heart Failure 2012 of the European Society of Cardiology. Developed in collaboration with the Heart Failure Association (HFA) of the ESC. Eur Heart J. 2012;33:1787–1847. [DOI] [PubMed] [Google Scholar]
- 153. Van Heerebeek L, Franssen CP, Hamdani N, Verheugt FW, Somsen GA, Paulus WJ. Molecular and cellular basis for diastolic dysfunction. Curr Heart Fail Rep. 2012;9:293–302. [DOI] [PubMed] [Google Scholar]
- 154. Glezeva N, Voon V, Watson C, Horgan S, McDonald K, Ledwidge M, Baugh J. Exaggerated inflammation and monocytosis associate with diastolic dysfunction in heart failure with preserved ejection fraction: evidence of M2 macrophage activation in disease pathogenesis. J Card Fail. 2015;21:167–177. [DOI] [PubMed] [Google Scholar]
- 155. Glezeva N, Collier P, Watson C, Ledwidge M, McDonald K, Baugh J. Increased inflammatory and alternative monocyte/macrophage markers in patients with hypertension and heart failure with preserved ejection fraction. Heart. 2012;98:A6–A7. [Google Scholar]
- 156. Armstrong EJ, Morrow DA, Sabatine MS. Inflammatory biomarkers in acute coronary syndromes: part I: introduction and cytokines. Circulation. 2006;113:e72–e75. [DOI] [PubMed] [Google Scholar]
- 157. Seta Y, Shan K, Bozkurt B, Oral H, Mann DL. Basic mechanisms in heart failure: the cytokine hypothesis. J Card Fail. 1996;2:243–249. [DOI] [PubMed] [Google Scholar]
- 158. Gu L, Okada Y, Clinton SK, Gerard C, Sukhova GK, Libby P, Rollins BJ. Absence of monocyte chemoattractant protein‐1 reduces atherosclerosis in low density lipoprotein receptor‐deficient mice. Mol Cell. 1998;2:275–281. [DOI] [PubMed] [Google Scholar]
- 159. Mann DL. Inflammatory mediators and the failing heart: past, present, and the foreseeable future. Circ Res. 2002;91:988–998. [DOI] [PubMed] [Google Scholar]
- 160. Pagani FD, Baker LS, Hsi C, Knox M, Fink MP, Visner MS. Left ventricular systolic and diastolic dysfunction after infusion of tumor necrosis factor‐α in conscious dogs. J Clin Invest. 1992;90:389–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Janssen SP, Gayan‐Ramirez G, Van den Bergh A, Herijgers P, Maes K, Verbeken E, Decramer M. Interleukin‐6 causes myocardial failure and skeletal muscle atrophy in rats. Circulation. 2005;111:996–1005. [DOI] [PubMed] [Google Scholar]
- 162. Niethammer M, Sieber M, Von Haehling S, Anker SD, Munzel T, Horstick G, Genth‐Zotz S. Inflammatory pathways in patients with heart failure and preserved ejection fraction. Int J Cardiol. 2008;129:111–117. [DOI] [PubMed] [Google Scholar]
- 163. Mewhort HE, Lipon BD, Svystonyuk DA, Teng G, Guzzardi DG, Silva C, Yong VW, Fedak PW. Monocytes increase human cardiac myofibroblast‐mediated extracellular matrix remodeling through TGF β1. Am J Physiol Heart Circ Physiol. 2016;310:H716–H724. [DOI] [PubMed] [Google Scholar]
- 164. Westermann D, Lindner D, Kasner M, Zietsch C, Savvatis K, Escher F, Von Schlippenbach J, Skurk C, Steendijk P, Riad A, Poller W, Schultheiss HP, Tschöpe C. Cardiac inflammation contributes to changes in the extracellular matrix in patients with heart failure and normal ejection fraction. Circ Heart Fail. 2011;4:44–52. [DOI] [PubMed] [Google Scholar]
- 165. Paulus WJ, Vantrimpont PJ, Shah AM. Paracrine coronary endothelial control of left ventricular function in humans. Circulation. 1995;92:2119–2126. [DOI] [PubMed] [Google Scholar]
- 166. Bolego C, Cignarella A, Staels B, Chinetti‐Gbaguidi G. Macrophage function and polarization in cardiovascular disease: a role of estrogen signaling? Arterioscler Thromb Vasc Biol. 2013;33:1127–1134. [DOI] [PubMed] [Google Scholar]
- 167. Moore JP, Vinh A, Tuck KL, Sakkal S, Krishnan SM, Chan CT, Lieu M, Samuel CS, Diep H, Kemp‐Harper BK, Tare M, Ricardo SD, Guzik TJ, Sobey CG, Drummond GR. M2 macrophage accumulation in the aortic wall during angiotensin II infusion in mice is associated with fibrosis, elastin loss, and elevated blood pressure. Am J Physiol Heart Circ Physiol. 2015;309:H906–H917. [DOI] [PubMed] [Google Scholar]
- 168. Ben‐Mordechai T, Holbova R, Landa‐Rouben N, Harel‐Adar T, Feinberg MS, Abd Elrahman I, Blum G, Epstein FH, Silman Z, Cohen S, Leor J. Macrophage subpopulations are essential for infarct repair with and without stem cell therapy. J Am Coll Cardiol. 2013;62:1890–1901. [DOI] [PubMed] [Google Scholar]
- 169. Dzeshka MS, Lip GY, Snezhitskiy V, Shantsila E. Cardiac fibrosis in patients with atrial fibrillation: mechanisms and clinical implications. J Am Coll Cardiol. 2015;66:943–959. [DOI] [PubMed] [Google Scholar]
- 170. Anne W, Willems R, Roskams T, Sergeant P, Herijgers P, Holemans P, Ector H, Heidbuchel H. Matrix metalloproteinases and atrial remodeling in patients with mitral valve disease and atrial fibrillation. Cardiovasc Res. 2005;67:655–666. [DOI] [PubMed] [Google Scholar]
- 171. Ohtani K, Yutani C, Nagata S, Koretsune Y, Hori M, Kamada T. High prevalence of atrial fibrosis in patients with dilated cardiomyopathy. J Am Coll Cardiol. 1995;25:1162–1169. [DOI] [PubMed] [Google Scholar]
- 172. Lie JT, Hammond PI. Pathology of the senescent heart: anatomic observations on 237 autopsy studies of patients 90 to 105 years old. Mayo Clin Proc. 1988;63:552–564. [DOI] [PubMed] [Google Scholar]
- 173. Pellett AA, Myers L, Welsch M, Jazwinski SM, Welsh DA. Left atrial enlargement and reduced physical function during aging. J Aging Phys Act. 2013;21:417–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Koura T, Hara M, Takeuchi S, Ota K, Okada Y, Miyoshi S, Watanabe A, Shiraiwa K, Mitamura H, Kodama I, Ogawa S. Anisotropic conduction properties in canine atria analyzed by high‐resolution optical mapping: preferential direction of conduction block changes from longitudinal to transverse with increasing age. Circulation. 2002;105:2092–2098. [DOI] [PubMed] [Google Scholar]
- 175. Verheule S, Sato T, Everett T IV, Engle SK, Otten D, Rubart‐Von der Lohe M, Nakajima HO, Nakajima H, Field LJ, Olgin JE. Increased vulnerability to atrial fibrillation in transgenic mice with selective atrial fibrosis caused by overexpression of TGF‐β1. Circ Res. 2004;94:1458–1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Li H, Li S, Yu B, Liu S. Expression of miR‐133 and miR‐30 in chronic atrial fibrillation in canines. Mol Med Rep. 2012;5:1457–1460. [DOI] [PubMed] [Google Scholar]
- 177. Morillo CA, Klein GJ, Jones DL, Guiraudon CM. Chronic rapid atrial pacing. Structural, functional, and electrophysiological characteristics of a new model of sustained atrial fibrillation. Circulation. 1995;91:1588–1595. [DOI] [PubMed] [Google Scholar]
- 178. De Jong AM, Van Gelder IC, Vreeswijk‐Baudoin I, Cannon MV, Van Gilst WH, Maass AH. Atrial remodeling is directly related to end‐diastolic left ventricular pressure in a mouse model of ventricular pressure overload. PLoS One. 2013;8:e72651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179. Ren M, Li X, Hao L, Zhong J. Role of tumor necrosis factor alpha in the pathogenesis of atrial fibrillation: a novel potential therapeutic target? Ann Med. 2015;47:316–324. [DOI] [PubMed] [Google Scholar]
- 180. Boldt A, Wetzel U, Lauschke J, Weigl J, Gummert J, Hindricks G, Kottkamp H, Dhein S. Fibrosis in left atrial tissue of patients with atrial fibrillation with and without underlying mitral valve disease. Heart. 2004;90:400–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181. Saito T, Tamura K, Uchida D, Saito T, Togashi M, Nitta T, Sugisaki Y. Histopathological features of the resected left atrial appendage as predictors of recurrence after surgery for atrial fibrillation in valvular heart disease. Circ J. 2007;71:70–78. [DOI] [PubMed] [Google Scholar]
- 182. Gramley F, Lorenzen J, Plisiene J, Rakauskas M, Benetis R, Schmid M, Autschbach R, Knackstedt C, Schimpf T, Mischke K, Gressner A, Hanrath P, Kelm M, Schauerte P. Decreased plasminogen activator inhibitor and tissue metalloproteinase inhibitor expression may promote increased metalloproteinase activity with increasing duration of human atrial fibrillation. J Cardiovasc Electrophysiol. 2007;18:1076–1082. [DOI] [PubMed] [Google Scholar]
- 183. Verheule S, Wilson E, Banthia S, Everett TH, Shanbhag S, Sih HJ, Olgin J. Direction‐dependent conduction abnormalities in a canine model of atrial fibrillation due to chronic atrial dilatation. Am J Physiol Heart Circ Physiol. 2004;287:H634. [DOI] [PubMed] [Google Scholar]
- 184. Kong CW, Yu WC, Chen SA, Lin YJ, Huang CY, Chung SL. Development of atrial fibrillation in patients with atrioventricular block after atrioventricular synchronized pacing. Pacing Clin Electrophysiol. 2004;27:352–357. [DOI] [PubMed] [Google Scholar]
- 185. Goette A, Juenemann G, Peters B, Klein HU, Roessner A, Huth C, Rocken C. Determinants and consequences of atrial fibrosis in patients undergoing open heart surgery. Cardiovasc Res. 2002;54:390–396. [DOI] [PubMed] [Google Scholar]
- 186. Xie W, Santulli G, Reiken SR, Yuan Q, Osborne BW, Chen BX, Marks AR. Mitochondrial oxidative stress promotes atrial fibrillation. Sci Rep. 2015;5:11427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187. Youn JY, Zhang J, Zhang Y, Chen H, Liu D, Ping P, Weiss JN, Cai H. Oxidative stress in atrial fibrillation: an emerging role of NADPH oxidase. J Mol Cell Cardiol. 2013;62:72–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188. Chung MK, Martin DO, Sprecher D, Wazni O, Kanderian A, Carnes CA, Bauer JA, Tchou PJ, Niebauer MJ, Natale A, Van Wagoner DR. C‐reactive protein elevation in patients with atrial arrhythmias: inflammatory mechanisms and persistence of atrial fibrillation. Circulation. 2001;104:2886–2891. [DOI] [PubMed] [Google Scholar]
- 189. Jiang Z, Dai L, Song Z, Li H, Shu M. Association between C‐reactive protein and atrial fibrillation recurrence after catheter ablation: a meta‐analysis. Clin Cardiol. 2013;36:548–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190. Kumagai K, Nakashima H, Urata H, Gondo N, Arakawa K, Saku K. Effects of angiotensin II type 1 receptor antagonist on electrical and structural remodeling in atrial fibrillation. J Am Coll Cardiol. 2003;41:2197–2204. [DOI] [PubMed] [Google Scholar]
- 191. Lee KW, Everett TH IV, Rahmutula D, Guerra JM, Wilson E, Ding C, Olgin JE. Pirfenidone prevents the development of a vulnerable substrate for atrial fibrillation in a canine model of heart failure. Circulation. 2006;114:1703–1712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192. Li D, Shinagawa K, Pang L, Leung TK, Cardin S, Wang Z, Nattel S. Effects of angiotensin‐converting enzyme inhibition on the development of the atrial fibrillation substrate in dogs with ventricular tachypacing‐induced congestive heart failure. Circulation. 2001;104:2608–2614. [DOI] [PubMed] [Google Scholar]
- 193. Milliez P, Deangelis N, Rucker‐Martin C, Leenhardt A, Vicaut E, Robidel E, Beaufils P, Delcayre C, Hatem SN. Spironolactone reduces fibrosis of dilated atria during heart failure in rats with myocardial infarction. Eur Heart J. 2005;26:2193–2199. [DOI] [PubMed] [Google Scholar]
- 194. Orso F, Fabbri G, Maggioni AP. Upstream treatment of atrial fibrillation with n‐3 polyunsaturated fatty acids: myth or reality? Arrhythm Electrophysiol Rev. 2015;4:163–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195. Shiroshita‐Takeshita A, Brundel BJ, Burstein B, Leung TK, Mitamura H, Ogawa S, Nattel S. Effects of simvastatin on the development of the atrial fibrillation substrate in dogs with congestive heart failure. Cardiovasc Res. 2007;74:75–84. [DOI] [PubMed] [Google Scholar]
- 196. Pellman J, Lyon RC, Sheikh F. Extracellular matrix remodeling in atrial fibrosis: mechanisms and implications in atrial fibrillation. J Mol Cell Cardiol. 2010;48:461–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197. Ling LH, Kistler PM, Ellims AH, Iles LM, Lee G, Hughes GL, Kalman JM, Kaye DM, Taylor AJ. Diffuse ventricular fibrosis in atrial fibrillation: noninvasive evaluation and relationships with aging and systolic dysfunction. J Am Coll Cardiol. 2012;60:2402–2408. [DOI] [PubMed] [Google Scholar]
- 198. Rahmutula D, Marcus GM, Wilson EE, Ding CH, Xiao Y, Paquet AC, Barbeau R, Barczak AJ, Erle DJ, Olgin JE. Molecular basis of selective atrial fibrosis due to overexpression of transforming growth factor‐beta1. Cardiovasc Res. 2013;99:769–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199. Burstein B, Libby E, Calderone A, Nattel S. Differential behaviors of atrial versus ventricular fibroblasts: a potential role for platelet‐derived growth factor in atrial‐ventricular remodeling differences. Circulation. 2008;117:1630–1641. [DOI] [PubMed] [Google Scholar]