

Abstract
Background
Obstructive sleep apnea is characterized by repetitive pharyngeal collapses during sleep, leading to intermittent hypoxia (IH), the main contributor of obstructive sleep apnea–related cardiovascular morbidity. In patients and rodents with obstructive sleep apnea exposed to IH, vascular inflammation and remodeling, endothelial dysfunction, and circulating inflammatory markers are linked with IH severity. The nonmuscle myosin light chain kinase (nmMLCK) isoform contributes to vascular inflammation and oxidative stress in different cardiovascular and inflammatory diseases. Thus, in the present study, we hypothesized that nmMLCK plays a key role in the IH‐induced vascular dysfunctions and inflammatory remodeling.
Methods and Results
Twelve‐week‐old nmMLCK +/+ or nmMLCK −/− mice were exposed to 14‐day IH or normoxia. IH was associated with functional alterations characterized by an elevation of arterial blood pressure and stiffness and perturbations of NO signaling. IH caused endothelial barrier dysfunction (ie, reduced transendothelial resistance in vitro) and induced vascular oxidative stress associated with an inflammatory remodeling, characterized by an increased intima‐media thickness and an increased expression and activity of inflammatory markers, such as interferon‐γ and nuclear factor‐κB, in the vascular wall. Interestingly, nmMLCK deletion prevented all IH‐induced functional and structural alterations, including the restoration of NO signaling, correction of endothelial barrier integrity, and reduction of both oxidative stress and associated inflammatory response.
Conclusions
nmMLCK is a key mechanism in IH‐induced vascular oxidative stress and inflammation and both functional and structural remodeling.
Keywords: high blood pressure, hypertension, hypoxia, inflammation, myosin light chain kinase, obstructive sleep apnea, oxidative stress, vascular remodeling
Subject Categories: High Blood Pressure
Clinical Perspective
What Is New?
Intermittent hypoxia induces functional and structural vascular alterations characterized by an elevation of arterial blood pressure and stiffness, perturbations of NO signaling, oxidative stress, and vascular inflammation, which were accompanied by endothelial barrier dysfunction in vitro.
Nonmuscle myosin light chain kinase deletion prevented these intermittent hypoxia–induced functional and structural alterations, restoring NO signaling, maintaining endothelial barrier integrity, and attenuating oxidative stress and inflammatory response. Nonmuscle myosin light chain kinase inhibition restored endothelial barrier function in vitro.
Nonmuscle myosin light chain kinase participates in intermittent hypoxia–induced vascular functional and structural disturbances.
What Are the Clinical Implications?
This study demonstrates that nonmuscle myosin light chain kinase is a hallmark mediator of intermittent hypoxia–induced functional and structural vascular dysfunctions, and related signaling pathways could be considered as potential targets for the management of hypoxia‐related vascular complications in obstructive sleep apnea.
Introduction
Obstructive sleep apnea (OSA) is recognized as a major worldwide health problem, affecting at least 10% of the general population. OSA is recognized as an important and independent risk factor for hypertension, coronary heart diseases, and stroke. The deleterious effects of OSA on cardiovascular outcomes are mainly triggered by intermittent hypoxia (IH) severity and the subsequent oxidative stress, leading to activation of the autonomic nervous system.1, 2, 3 Vascular endothelial dysfunction is a major intermediate mechanism of cardiovascular complications in patients with OSA,4, 5, 6 preceding and predicting late cardiovascular events, such as hypertension, atherosclerosis, and coronary diseases.1 The gold standard treatment for OSA remains continuous positive airway pressure during sleep. However, although continuous positive airway pressure exerts beneficial effects on psychocognitive functions (eg, excessive daytime sleepiness, mental depression, and quality of life), it has only a limited impact in terms of improvement of the cardiovascular risk.7 Therefore, understanding the pathophysiological characteristics of OSA‐related vascular disease would open new avenues for specific therapeutic strategies facilitating the management of cardiovascular complications in patients with OSA.
IH, by producing oxidative stress and inflammation, favors endothelial dysfunction, which has a causal role in promoting structural (ie, vascular remodeling) and functional (ie, decreased NO bioavailability) vascular alterations.8 In OSA, it has been shown that venous endothelial cells from apneic patients exhibited local oxidative stress and inflammation associated with decreased NO bioavailability and reduced flow‐mediated dilation.9 Experimental studies in rodents demonstrated that chronic IH per se represents a causal factor of vascular remodeling and hypertension,10, 11, 12, 13, 14 in part through vascular inflammation10, 11, 12 and oxidative stress.13, 14
A recently identified contributor to tissue inflammation and oxidative stress is the nonmuscular isoform of the Ca2+/calmodulin‐dependent enzyme myosin light chain kinase (nmMLCK). This kinase is mostly expressed in monocytes and platelets in comparison with muscular isoforms, and it is the only isoform of MLCK expressed in endothelial cells.15 This isoform (210 kDa) is, as the smooth muscle isoform (108 kDa), encoded by the mylk1 gene, although it differs both in its structure and function.16 nmMLCK phosphorylates the myosin light chain, leading to changes in cytoskeleton architecture, resulting in the retraction of the cells.17 In endothelial cells, this retraction is linked to endothelial permeability enhancement and vascular leakage under proinflammatory conditions. In this context, an activation of the nmMLCK has been linked to several pathophysiological conditions, such as vascular inflammation and atherosclerosis,18 acute lung injury,19 endotoxic shock,20 and severe burns.21
Therefore, in the present study, we hypothesized that nmMLCK plays a key role on IH‐related vascular dysfunction and that nmMLCK deletion could exert protective effects against IH‐induced vascular alterations (ie, vascular remodeling, arterial stiffness, and hypertension), through the modulation of oxidative stress, inflammation, and subsequent NO signaling disturbances.
Methods
The data, analytic methods, and study materials that support the findings of this study are available from the corresponding author on reasonable request.
Animals
The investigation conformed to the European Convention for the Protection of Vertebrate Animals Used for Experimental and Other Scientific Purposes (Council of Europe, European Treaties ETS 123, Strasbourg, March 18, 1986) and to the Guide for the Care and Use of Laboratory Animals (National Institutes of Health publication 85‐23, revised 1996). Our protocol was approved by the local committee and gained formal authorization (no. C3851610006).
Twelve‐week‐old male nmMLCK−/−, generated as previously described,19 and nmMLCK+/+ mice (C57Bl/6J background) (Janvier Labs, Le Genest‐Saint‐Isle, France) were used for this study. Animals were housed in local facilities and had free access to food and water.
Rodent IH
Mice were randomized to 14 days exposure to normoxia or IH, 8 h/d. Fraction of inspired oxygen in the hypoxic chambers was measured with a gas analyzer (ML206; ADInstruments) throughout the experiment. The IH stimulus consisted of a 60‐second cycle, with 30 seconds of hypoxia (hypoxic plateau at 5% fraction of inspired oxygen) and 30 seconds of normoxia (normoxic plateau at 21% fraction of inspired oxygen), as described.22 Normoxic mice were exposed to air flow turbulences related to gas circulation and noise stimuli similar to those undergone by animals submitted to IH. Ambient temperature was maintained at 20°C to 22°C.
In Vivo Investigations
Arterial blood pressure
Arterial blood pressure was measured weekly in conscious mice, using a noninvasive pressure recording system (CODA; Kent Scientific, Torrington, CT), which consists of placing a tail cuff, including a pressure sensor, while the mouse is restrained in a plexiglass tube. This noninvasive sphygmomanometry technique allows for measurements of blood pressure in the tail artery of awake mice, with values that are similar to those obtained separately by telemetry in the aorta.23, 24 Mice were habituated to the CODA restraining system for 4 consecutive days before the first measurement.
Carotid ultrasonography
All measurements were performed with the high‐resolution Vevo 770 system (VisualSonicsInc, Toronto, ON, Canada). Mice were anesthetized with 1% isoflurane and placed in the dorsal position; heart rate and body temperature were monitored throughout the experiment. Depilatory cream was used to remove fur from the throat, and ultrasound gel was used. Carotid pulsed‐wave Doppler ultrasound velocity measurements were performed as an estimation of arterial stiffness.25 Mean velocity, end‐diastolic velocity, and peak systolic velocity were determined, and the resistance index was calculated as follows: resistance index=(peak systolic velocity−end‐diastolic velocity)/peak systolic velocity.
Ex Vivo Investigations
The day after the last exposure to IH, animals were anesthetized by IP injection of ketamine (100 mg/kg) and xylazine (10 mg/kg). Blood was collected from cardiac puncture for hematocrit assessment. Aortas were harvested and either immediately frozen in liquid nitrogen and stored at −80°C until analysis (Western blot and quantitative reverse transcription–polymerase chain reaction) or fixed in 4% paraformaldehyde and processed for cryoembedding sectioning (Tissue‐Tek OCT; Fischer).
Aorta Mechanical Properties
Mice were anesthetized by IP injection of pentobarbital (60 mg/kg) before 100 IU heparin in 0.9% NaCl saline was injected in the saphenous vein. The ascending aortas were harvested and placed in HEPES‐buffered physiological saline solution (mmol/L): 135 NaCl, 5 KCl, 1.6 CaCl2, 1.17 MgSO4, 0.44 KH2 PO4, 2.6 NaHCO3, 0.34 Na2HPO4, 5.5 d‐glucose, 0.025 EDTA, and 10 HEPES (pH 7.4). Vessels were carefully cleaned of adipose tissue, cannulated, and mounted onto a pressure arteriograph, placed onto a microscope with video monitoring. Mechanistic study was performed, as previously described.26, 27, 28 Circumferential wall stress (representative of all forces that are circumferentially applied on each small portion of the vessel wall) and incremental elastic modulus (Einc) (representative of wall stiffness) were calculated from the inner and outer diameters at transluminal pressures ranging from 0 to 175 mm Hg, according to the formulas given by Gibbons and Shadwick.29
Vascular Reactivity
Variations of the ascending aorta diameters in response to 10 μmol/L phenylephrine (PE), a vasoconstrictor mainly acting through the α1‐adrenoceptors, then to 10 μmol/L acetylcholine (Ach), an endothelium‐dependent vasodilator, were measured at 75 mm Hg. Responses to PE are presented as the percentage decrease in inner diameter (ID), compared with the control diameter at 75 mm Hg before PE application: PE‐induced contraction (%)=100×(IDcontrol−IDPE)/IDcontrol. Responses to Ach are presented as the percentage restoration of the ID after PE‐induced constriction: Ach‐induced vasodilation (%)=100×(IDAch+PE−IDPE)/(IDcontrol−IDPE).27, 28
Intima‐Media Thickness, Fibrosis, and Elastic Fiber Network Analysis
Ascending aortas from normoxic and IH mice were sectioned (8 μm thick), and hematoxylin‐eosin staining was performed to assess global tissue morphological characteristics. Picrosirius red and Weigert stainings were performed to assess collagen deposition and elastic network in the aortic wall, respectively. Picrosirius red–stained sections were examined under bright‐field illumination for general collagen or polarized light for fibrillar collagen observations,30 in which collagen I appears in red‐orange and collagen III in green‐yellow.31, 32
Morphometric analysis was performed with a light microscope (Eclipse 80i; NIKON France, S.A., VILLE, France) and LUCIA‐G software Version 5.0 (Laboratory Imaging Ltd, Prague, Czech Republic) to determine intima‐media thickness, number of smooth muscle cells in vascular wall sections, and elastic fiber network (fiber thickness, ruptures, and distance between fibers). For each animal (n=6–10 mice per group), 4 noncontinuous sections were analyzed and a mean value was obtained by averaging 20 to 30 measurements spanning the entire cross section of the vessel at 10×10 and 10×40 magnification.
Western Blot Analysis
Cytoplasmic proteins from aortas were separated on polyacrylamide‐SDS gels and transferred onto polyvinylidene difluoride membranes. Membranes were then incubated overnight with rabbit anti–endothelial NO synthase (eNOS; 1:500; Cell Signaling, Danvers, MA), anti–PSer1177‐eNOS (phosphorylation on serine 1177 [1:500; Cell Signaling]), anti–inducible NOS (1:500; Cell Signaling), anti‐nitrotyrosine (1:2000; Thermo Fisher, Waltham, MA), and anti‐mouse actin (1:5000; Sigma‐Aldrich, Saint‐Quentin‐Fallavier, France) antibodies. Blots were then incubated for 1 hour with horseradish peroxidase–conjugated secondary antibodies at room temperature. Bands were visualized by enhanced chemiluminescence and quantified using the software ImageJ (National Institutes of Health, Bethesda, MD). All protein contents were normalized to the actin content.
Nuclear Factor‐κB Activity
Nuclear factor‐κB (NF‐κB) p50 activity was quantified on nuclear protein extracts from mice mesenteries (Nuclear Extract Kit; Active Motif Europe, Rixensart, Belgium) using ELISA DNA‐binding assay kit (TransAM; Active Motif Europe).
Immunohistochemistry
For immunostainings, aorta and carotid sections were fixed in 4% paraformaldehyde. Nonspecific binding was blocked with 5% nonfat dried milk in PBS, pH 7.4. Sections were incubated overnight at 4°C with monoclonal anti–inducible NOS (1:100; BD Biosciences, Le Pont de Claix, France), anti–NF‐κB (1:100; Abcam, Cambridge, UK), and anti‐F4/80 (1:100; Biolegend, San Diego, CA) antibodies. After 3 washes, the primary antibodies were detected after 1‐hour incubation with a secondary mouse or rabbit fluorescence‐labeled antibody Fluoprobes FP 546 (1:100; Interchim, San Diego, CA).
Dihydroethidium Staining
Dihydroethidium (DHE) staining was used to detect superoxide anion production in isolated aortas and carotid arteries. Frozen tissue samples were cryosectioned (14 μm thick) and collected onto Superfrost plus slides (Dutscher, Brumath, France). Slides were stained with 10 μmol/L dihydroethydine for 5 minutes (0.1% Tween PBS was used as control) in a dark moist chamber. After washing, the reaction was stopped with acetone (−20°C), cover slips were placed onto the slides, and fluorescent dihydroethydine signal was recorded using the confocal microscopy (LSM510 Meta confocal microscope; Zeiss, Oberkochen, Germany) and analyzed with ImageJ. The fluorescent dihydroethydine signal was expressed as a percentage of total vessel wall area.
Reactive Oxygen Species Production Assessment by Electronic Paramagnetic Resonance
Detection of reactive oxygen species production in femoral arteries was performed using a previously described technique with ferrous diethyldithiocarbamate (Sigma‐Aldrich) as a spin trap. Briefly, mice were euthanized, and femoral arteries were isolated and immediately incubated with 250 μL of Fe(diethyldithiocarbamate) for 45 minutes, at 37°C. Samples were then frozen in liquid nitrogen and stored at −80°C until analysis. The samples were analyzed using a tabletop x‐band spectrometer Miniscope (Magnettech, Berlin, Germany).
Vascular Barrier Integrity
Evans blue assay
In vivo vascular barrier integrity was examined with Evans blue assay. Briefly, after 14 days of normoxic or IH exposure, nmMLCK+/+ and nmMLCK−/− mice were injected with 20 mg/kg Evans blue dye. The day after the injection, mice were euthanized and perfused through the left ventricle with heparinized 0.1% lactate Ringer solution. Aorta and lung were excised. Organs were weighed, and Evans blue dye was extracted by incubation in formamide overnight at 62°C. Absorbance of the supernatants was measured at 620 nm and normalized to organ weights.
Transendothelial electrical resistance
Endothelial barrier property was also assessed in cell culture. Mouse microvascular bEnd.3 endothelial cell line (ATCC, Manassas, VA) was seeded in 6.5‐mm Transwells (Corning, Corning, NY) with 0.4‐μm‐diameter pores at a density of 400 000 cells/Transwell (area, 0.33 cm2). After 3 days, the Transwells were placed in gas‐permeable 24‐well plates (Zell‐kontakt, Nörten‐Hardenberg, Germany) and preincubated for 30 minutes in culture medium with 5 μmol/L ML‐7, a selective MLCK inhibitor (Sigma‐Aldrich), diluted in dimethyl sulfoxide at a final concentration of 0.12% dimethyl sulfoxide in the well or with 0.12% dimethyl sulfoxide as vehicle. They were then exposed to 8 hours of normoxia (constant 16% O2 and 5% CO2) or to IH consisting of 5 minutes at 16% and 5 minutes at 2% O2 with constant 5% CO2, as described.33 Oxygen concentration in the cell environment (ie, inside the Transwell) was monitored with an oxygen probe (Oxford Optronics, Abingdon, UK) and was found to cycle between 70 and 125 mm Hg. Transendothelial electrical resistance was measured before and after exposure to normoxia or IH with an epithelial voltometer (EVOM‐2; World Precision Instruments).
RNA Purification and Quantitative Reverse Transcription–Polymerase Chain Reaction
Total mRNA was extracted from snap‐frozen aortas (intima and media) from IH and normoxic mice using Trireagent RNA isolation reagent (Sigma Aldrich). Ptprc (CD45) and Ifng (interferon‐γ) gene expression was assessed by quantitative reverse transcription–polymerase chain reaction, normalized to RPLP0 (60S acidic ribosomal protein P0) gene expression and 18S rRNA content as internal controls, and expressed as fold change compared with control mice using the ∆∆CT method. Primer sequences are listed in the Table.
Table 1.
Primers Used for RT‐qPCR
| Gene | Forward primer 5′‐3′ | Reverse primer 5′‐3′ |
|---|---|---|
| Rplp0 | CCCTGCACTCTCGCTTTCTGGA | AGGGGCAGCAGCCGCAAATG |
| Rn18S | GGAAGAATAACTTCAGACCGCCC | TGCAGCGGACAGTGTCTTGTTT |
| Ptprc (CD45) | CAATGGAGTGTACGAGGGAGATTCA | TCATCCCTTTCAACGAGTTCCTGT |
| Ifng (interferon‐γ) | GGATATCTGGAGGAACTGGCAAAAG | TGACGCTTATGTTGTTGCTGATGG |
RT‐qPCR indicates quantitative reverse transcription–polymerase chain reaction.
Statistical Analysis
Experimental data are expressed as means±SEM. For each comparison group, normality and equal variance were tested using Shapiro‐Wilk and Bartlett tests, respectively. When these assumptions were met, results were analyzed using 2‐way ANOVA, followed by post hoc Sidak's multiple‐comparison tests. In other conditions and when sample size was too small, Kruskal‐Wallis test was used, followed by Dunn's multiple comparison test (Prism; GraphPad Software, La Jolla, CA). A 2‐sided P<0.05 was considered statistically significant.
Results
Vascular Physiological Parameters, Mechanics, Reactivity, and Structure
Although systolic arterial blood pressure was increased in nmMLCK+/+ mice after 7 days of IH and remained elevated at day 14 (Figure 1A), no change in response to IH was observed in nmMLCK−/− animals (Figure 1B). Similar to arterial blood pressure, mean blood flow velocity was significantly increased in nmMLCK+/+ mice exposed to IH, whereas it was unchanged in nmMLCK−/− animals (Figure 1C‐D); end‐diastolic blood flow velocity followed the same pattern (data not shown). By contrast, the carotid artery diameter and resistance index were similar in all experimental groups (data not shown).
Figure 1.
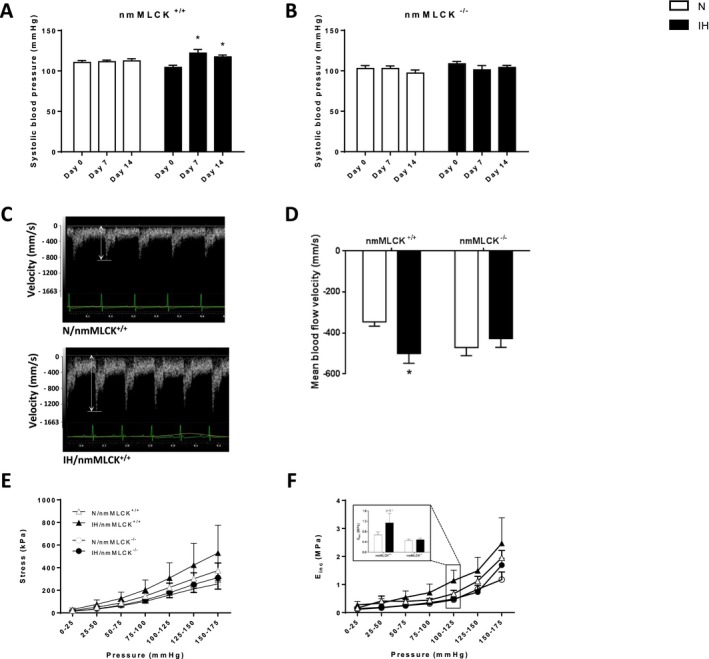
Arterial function. Systolic arterial blood pressure measured in nonmuscle myosin light chain kinase (nmMLCK)+/+ (A) and nmMLCK −/− (B) mice exposed to intermittent hypoxia (IH) or normoxia (N) (n=16–17 per group). *P<0.05 vs day 0. C, Representative carotid ultrasonography recordings of carotid artery blood flow velocity from N and IH nmMLCK +/+ mice. D, Mean blood flow velocity recorded by carotid ultrasonography in nmMLCK +/+ and nmMLCK −/− mice exposed to IH and N (n=7–9 per group). *P<0.05 vs N. Mechanical properties of aortas from nmMLCK +/+ and nmMLCK −/− mice exposed to IH and N: circumferential wall stress of the ascending aorta, representative of all forces that are circumferentially applied on each small portion of the vessel wall (E); and incremental elasticity modulus (EI nc) of the ascending aorta, representative of wall stiffness (F). The inset histogram represents EI nc at physiological pressure (100–125 mm Hg) (n=6–9 per group).
To complete the in vivo physiological vascular characteristics, the impact of IH was investigated on the reactivity and mechanical parameters of the excised ascending aorta from mice of the 2 genotypes. Although IH did not affect aortic reactivity (ie, contraction to 10 μmol/L PE and dilatation to 10 μmol/L Ach) in the animals from the 2 strains (data not shown), aortic stress and Einc were generally higher in nmMLCK+/+ than in nmMLCK−/− mice. Also, aortic stress and Einc seemed to be increased by IH only in nmMLCK+/+ mice. In particular, Einc appeared clearly increased by IH in the whole range of the tested pressures in nmMLCK+/+ mice, whereas a substantial augmentation of Einc was detected at the highest pressures only (150–175 mm Hg) in nmMLCK−/− mice (Figure 1E and 1F). IH triggered a 22% increase in the intima‐media thickness of nmMLCK+/+ mice, whereas it had no effect in nmMLCK−/− animals (Figure 2B). The IH‐induced aorta wall thickening in nmMLCK+/+ mice did not result from intima‐media cell proliferation because no change in cell number per area unit could be observed between normoxic and IH animals in both genotypes (Figure 2C). The overall collagen quantity and distribution did not seem to be different in all 4 groups. However, for the proportions of the different fibrillar collagen types, IH induced a switch from collagen I to collagen III, in the adventitia of nmMLCK+/+ mice, whereas it had no effect in nmMLCK−/− animals (Figure 2A, picrosirius red staining, polarized light analysis). Finally, IH induced a disorganization of the elastic network (Figure 2A, Weigert staining) in nmMLCK+/+ mice (Figure 2A through 2D).
Figure 2.
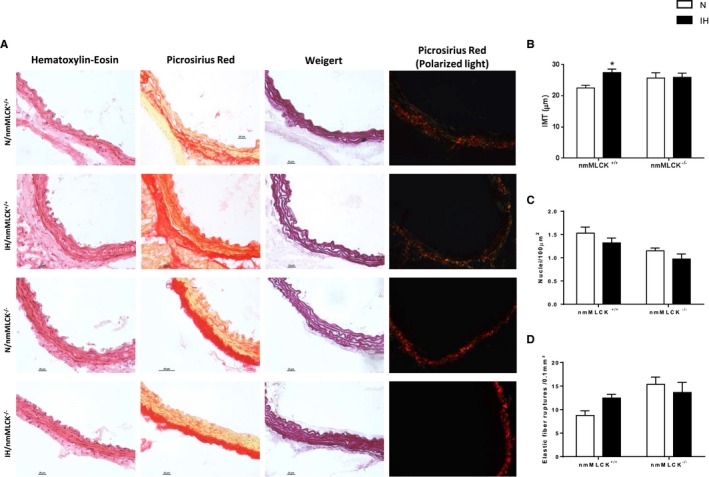
Vascular remodeling. A, Hematoxylin‐eosin, Sirius red, and Weigert stainings of aorta from nonmuscle myosin light chain kinase (nmMLCK)+/+ and nmMLCK −/− exposed to intermittent hypoxia (IH) and normoxia (N). To determine interstitial collagen content, Sirius red was also observed under polarized light (collagen I appears in red‐orange, and collagen III appears in green‐yellow) in each of the 4 groups. B, Aortic intima‐media thickness (IMT) measurements (n=9–10 per group). C, Smooth muscle cell nuclei count in the intima‐media. D, Quantification of the number of elastic fiber ruptures per 0.1 mm2 in intima from nmMLCK +/+ and nmMLCK −/− mice exposed to IH and N (n=5–10 per group). *P<0.05 vs N.
nmMLCK Deletion Abolished the IH‐Induced Nitrative and Oxidative Stress
Although total eNOS expression was not modified by IH in mice of both genotypes (Figure 3A), we demonstrated that IH induced a significant reduction in phosphorylated eNOS (Ser1177), the active form of eNOS, in aortas from nmMLCK+/+ mice (Figure 3B and 3C) and an increase in inducible NOS expression in the 2 strains, as evaluated by Western blot analysis (Figure 3D). The latter result was confirmed in another vascular bed (carotid artery) by immunostaining, which exhibited the same pattern (Figure 3E). This was accompanied by a trend towards reduced NO bioavailability, indicated by a slight elevation of nitrotyrosine, in the aorta of IH‐exposed nmMLCK+/+ mice only (Figure 3F). Oxidative stress and inflammation are key mechanisms of endothelial dysfunction, with direct effects on NO bioavailability.8 Experiments using DHE staining and electron paramagnetic resonance evidenced an IH‐induced increase in superoxide anion productions in aorta, carotid, and femoral arteries from nmMLCK+/+ mice (Figure 4A through 4E), which could corroborate elevation in nitrotyrosine. Interestingly, nmMLCK deletion prevented this IH‐induced vascular oxidative stress (Figure 4A through 4E). Taken together, these results suggest that nmMLCK deletion abolished the IH‐induced alterations of the NO signaling pathway and oxidative stress, leading to the correction of the nitrative stress.
Figure 3.
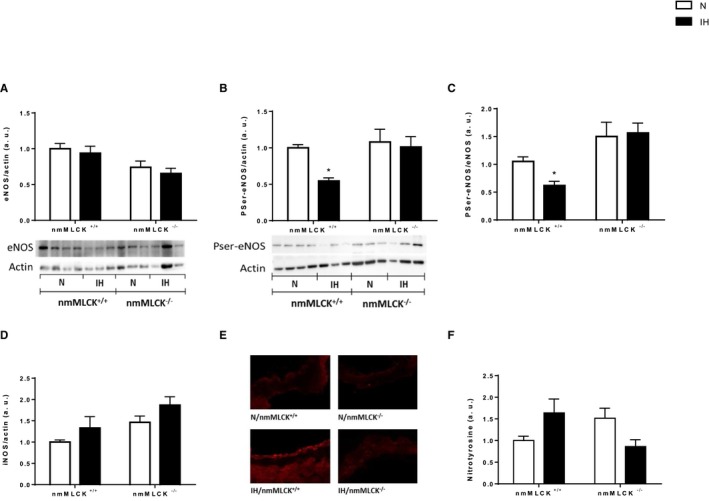
NO signaling. Aortic endothelial NO synthase (eNOS) expression (A) and phosphorylation at serine 1177 (PSer1177) (B) evaluated by Western blot analysis. C, Histogram of the PSer‐eNOS/eNOS ratio. Inducible NOS (iNOS) expression evaluated by Western blot analysis in aorta (Kruskal‐Wallis, P<0.05, general significant difference between intermittent hypoxia [IH] and normoxia [N]) (D) and immunohistochemistry of the carotid artery (E). F, Quantification of tyrosine‐nitrated proteins in aorta by Western blot analysis. All these results were obtained from nonmuscle myosin light chain kinase (nmMLCK)+/+ and nmMLCK −/− mice exposed to IH and N (n=6–8 per group). *P<0.05 vs N.
Figure 4.
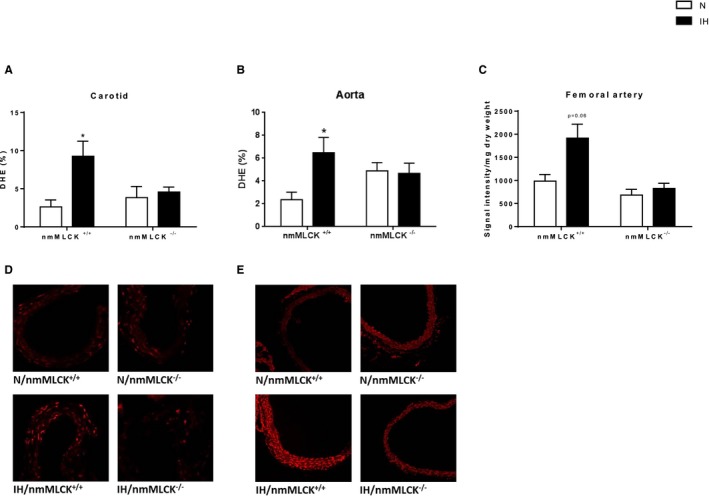
Vascular oxidative stress evaluated by Dihydroethidium (DHE) staining in carotid arteries (A through D) and aortas (B through E) and by electron paramagnetic resonance (EPR) in femoral arteries (C) from nonmuscle myosin light chain kinase (nmMLCK)+/+ and nmMLCK −/− mice exposed to intermittent hypoxia (IH) or normoxia (N) (n=6–8 per group). *P<0.05 vs N.
nmMLCK Deficiency Attenuates IH‐Induced Vascular Barrier Dysfunction and Vascular Inflammation
In vivo Evans blue assay exhibited a trend towards an IH‐induced increase in vascular permeability in the 2 strains (Figure 5A). This was supported in vitro by the demonstration, in cultured endothelial cell monolayers, that 8 hours of IH significantly decreased the transendothelial electrical resistance by 28%, whereas incubation with ML‐7, the MLCK inhibitor, abolished this effect (Figure 5B). This suggests that the alteration of the endothelial barrier by IH is, at least in part, mediated by nmMLCK. In addition, through analysis of several vascular beds, we showed that IH induces a low‐grade vascular inflammation in nmMLCK+/+ mice, which was partially prevented in nmMLCK−/− animals. First, mRNA expression of the leukocyte marker CD45 tended to increase only in intima from nmMLCK+/+ aorta mice exposed to IH (Figure 5C), whereas mRNA expression of the proinflammatory cytokine interferon‐γ was increased in the media from both strains exposed to IH (Figure 5D). As evidenced by carotid immunostainings, IH also seemed to increase the expression of the macrophage marker F4/80 and the proinflammatory transcription factor NF‐κB in carotids from wild‐type mice, which were abrogated in nmMLCK−/− animals (Figure 5E and 5F). Finally, NF‐κB activity was significantly increased by IH in mesenteric arteries from nmMLCK+/+ mice only. Altogether, these data strongly suggest that nmMLCK is involved in the IH‐induced vascular inflammation.
Figure 5.
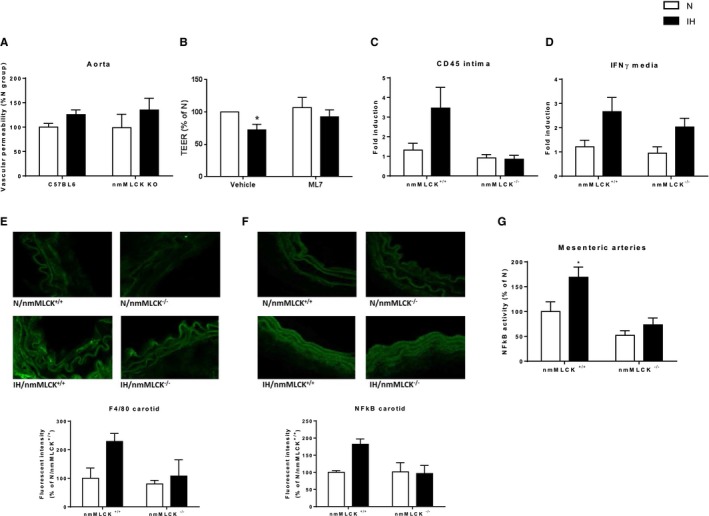
Endothelial barrier dysfunction and vascular inflammation. A, Assessment of endothelial barrier integrity by Evans blue assay in aorta from nonmuscle myosin light chain kinase (nmMLCK)+/+ and nmMLCK −/− mice exposed to normoxia (N) or intermittent hypoxia (IH) (ANOVA P=0.07, IH vs N). B, Transendothelial electrical resistance (TEER) of endothelial cell monolayers exposed to N or to IH for 8 hours in presence of ML‐7 (5 μmol/L) or vehicle. Ptprc (CD45) (C) and Ifng (interferon [IFN]‐γ) (D) gene expression in aorta from nmMLCK +/+ and nmMLCK −/− mice exposed to IH or N (n=6–9 per group, ANOVA P<0.05, general significant difference between IH and N for IFN‐γ expression). Immunostainings of F4/80 (E) and nuclear factor‐κB (NF‐κB) (F) in carotid arteries from nmMLCK +/+ and nmMLCK −/− mice exposed to IH or N (n=3–4 per group). G, NF‐κB activity in mesenteric arteries from nmMLCK +/+ and nmMLCK −/− mice exposed to IH or N (n=7–12 per group). *P<0.05 vs N.
Discussion
Our results demonstrate the implication of the multifunctional enzyme nmMLCK in the IH‐induced increases in arterial blood pressure, flow velocity, vascular stiffening, and remodeling. nmMLCK deletion prevented these IH‐induced functional alterations, allowing the preservation of endothelial NO signaling and preventing oxidative stress, endothelial barrier alteration, and vascular inflammation (Figure 6).
Figure 6.
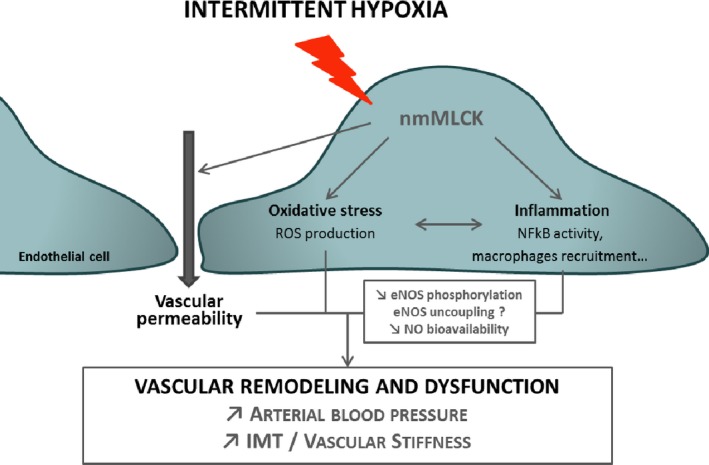
Role of nonmuscle myosin light chain kinase (nmMLCK) in the intermittent hypoxia (IH)–induced vascular dysfunctions. Our study demonstrates that the multifunctional enzyme nmMLCK plays a key role in the IH‐induced structural and functional dysfunctions, mediating disturbances in endothelial NO signaling, oxidative stress, endothelial barrier alteration, and vascular inflammation. eNOS indicates endothelial NO synthase; IMT, intima‐media thickness; NF‐κB, nuclear factor‐κB; and ROS, reactive oxygen species.
First, we observed in wild‐type mice an elevation of arterial blood pressure in response to IH. Our data are consistent with the literature, which reports the following: (1) an independent association between OSA and hypertension in humans34 and (2) a determinant role of chronic IH in inducing elevation of arterial and blood pressure in healthy humans and animals exposed to IH.35, 36, 37 We further demonstrated that, in addition to the elevation of arterial blood pressure, IH also induces an arterial inflammatory remodeling with subsequent stiffening. These results are consistent with the following: (1) clinical observations demonstrating a significant increase in arterial stiffness in patients with OSA, which correlates with the burden of the disease38, 39; and (2) results in a murine model, in which we previously demonstrated the presence of IH‐induced alterations of vascular mechanical properties (ie, reduced carotid compliance) in rats exposed to 28 days of IH.37 Hypertension and arterial stiffening are closely linked because, although it has been conventionally accepted that hypertension induces arterial stiffening, recent studies also demonstrate that arterial stiffening could precede and contribute to hypertension, therefore establishing a bidirectional relationship. Furthermore, these 2 phenomena are known to share similar pathophysiological pathways.40
Arterial stiffening might be explained by the IH‐induced arterial remodeling, characterized by intima‐media thickening, disruption of the elastic fiber network, and modification of production and/or distribution of fibrillar collagens I and III in adventice. These data are in accordance with clinical observations, demonstrating a significant increase in the carotid artery intima‐media thickness in patients with OSA that correlates to the severity of nocturnal hypoxemia.38, 39, 41 In rodent models, exposure to at least 14 days of IH resulted in enlarged intima‐media thickness and/or disruption of the elastic network.10, 11, 12, 37, 42, 43 In addition to structural remodeling, arterial stiffness and hypertension are also under the control of endothelial‐derived mediators, such as NO,40 and reduced NO bioavailability contributes to endothelial dysfunction, which represents a hallmark of hypertension and arterial stiffening. In the present study, IH‐induced endothelial dysfunction was evidenced by impaired NO signaling, because we observed a significant reduction of the phosphorylated (Ser1177) form of eNOS (ie, the active form of eNOS) in mice exposed to IH. These results are consistent with a recent study showing the same alterations of NO signaling in both the aorta of rats exposed to 6 weeks of IH and endothelial cells exposed to in vitro IH.44 Oxidative stress and inflammation also represent well‐known mechanisms interfering with endothelial function and playing an important role in the pathogenesis of hypertension and/or arterial stiffening through both structural and functional vascular alterations.45 Numerous lines of evidence indicate that oxidative stress affects the vascular tone, on one hand through decreased NO bioavailability contributing to endothelial dysfunction and, on the other hand, by promoting vascular cell proliferation, inflammation, and extracellular matrix remodeling.8 Our results are in accordance with previous studies demonstrating that long‐term exposure to IH induces vascular oxidative stress13, 14 (ie, superoxide anion production) and inflammation10, 11, 12, 44, 46 (ie, NF‐κB, F4/80, CD45, and interferon‐γ expression). In addition, using in vitro transendothelial electrical resistance measurement, we demonstrated that IH also induces endothelial barrier dysfunctions, confirming recent data from Prabhakar's group that demonstrates in endothelial cell cultures a central role of reactive oxygen species in promoting IH‐associated endothelial barrier dysfunctions.47 Therefore, our results strongly suggest that IH, triggering vascular oxidative stress and inflammation, leads to a reduced NO bioavailability and an increase in arterial blood pressure and stiffness.
Our study focused on the potential role of the nmMLCK in these IH‐induced vascular alterations, and we observed that nmMLCK gene deletion induces substantial changes in the mouse pathophysiological responses to IH. Unlike nmMLCK+/+ animals, nmMLCK−/− mice did not exhibit elevation in arterial blood pressure, mean blood flow velocity, and arterial wall remodeling in response to IH. Also, the IH‐induced increases in aorta wall stress and stiffness were attenuated in nmMLCK−/− mice, compared with nmMLCK+/+ animals. The abrogation or the correction of the IH‐induced vascular alterations previously described in nmMLCK−/− mice could result from several factors. After exposure of these mice to IH, the NO pathway is maintained functional, and we did not observe any induction of oxidative stress or inflammation as in nmMLCK+/+ animals. Actually, stability of eNOS expression and lack of nitrotyrosine increase strongly suggest that NO levels in the arteries of nmMLCK−/− mice are unaffected by IH, which is essential for vascular physiology and homeostasis.8, 45 Thus, as opposed to the response of their wild‐type counterparts, the absence of oxidative stress under IH exposure in nmMLCK−/− mice could contribute to the conservation of vascular properties. Interestingly, this result is in accordance with a previous study describing the same protective effect of nmMLCK deletion on vascular oxidative stress in a model of lipopolysaccharide‐induced sepsis.20 We further demonstrated that nmMLCK inhibition abolishes the IH‐induced endothelial barrier dysfunctions, which could directly result from the prevention of oxidative stress47 and could explain the limitation of inflammation in the vascular wall of nmMLCK−/− compared with nmMLCK+/+ mice. These results indicate that nmMLCK plays a key role in the IH‐induced vascular inflammation, which is consistent with previous studies showing a participation of nmMLCK in other inflammatory conditions. In particular, in lipopolysaccharide‐induced lung injuries, a beneficial effect of nmMLCK deletion has been demonstrated on endothelial barrier integrity and prevention of neutrophil migration.19, 48 More recently, we demonstrated that the protective effect of nmMLCK deletion on the vascular wall could be mediated by circulating microvesicles, which were able to activate endothelial proresolving anti‐inflammatory pathways, allowing prevention of endotoxic shock‐induced oxidative and nitrative stresses.49
Taken together, this study demonstrates that nmMLCK is a hallmark mediator of IH‐induced functional and structural vascular dysfunctions (ie, hypertension, arterial stiffening, and remodeling), through the inhibition of most of their determinants (ie, endothelial dysfunction, oxidative stress, and inflammation). Thus, nmMLCK and related signaling pathways could be considered as potential targets for the management of hypoxia‐related vascular complications in OSA.
Sources of Funding
This work was supported by grants from University Grenoble Alpes and Institut National de la Santé et de la Recherche Médicale (France), the French National Research Agency (ANR‐12‐BSV1‐0024‐01, France), the Fond de dotation Agir Pour les Maladies Chroniques (France), and Université d'Angers. This work was also supported by the French National Research Agency in the framework of the “Investissements d'avenir” program (ANR‐15‐IDEX‐02, France). Recoquillon is recipient of a PhD fellowship from Société Française de Cardiologie and Groupe de Réflexion sur la Recherche Cardiovasculaire.
Disclosures
None.
(J Am Heart Assoc. 2018;7:e007893 DOI: 10.1161/JAHA.117.007893.)29371201
References
- 1. Baguet JP, Barone‐Rochette G, Tamisier R, Levy P, Pepin JL. Mechanisms of cardiac dysfunction in obstructive sleep apnea. Nat Rev Cardiol. 2012;9:679–688. [DOI] [PubMed] [Google Scholar]
- 2. Lévy P, Kohler M, McNicholas WT, Barbé F, McEvoy RD, Somers VK, Lavie L, Pépin J‐L. Obstructive sleep apnoea syndrome. Nat Rev Dis Primers. 2015;1:15015. [DOI] [PubMed] [Google Scholar]
- 3. Javaheri S, Barbe F, Campos‐Rodriguez F, Dempsey JA, Khayat R, Malhotra A, Martinez‐Garcia MA, Mehra R, Pack AI, Polotsky VY, Redline S, Somers VK. Sleep apnea: types, mechanisms, and clinical cardiovascular consequences. J Am Coll Cardiol. 2017;69:841–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kato M, Roberts‐Thomson P, Phillips BG, Haynes WG, Winnicki M, Accurso V, Somers VK. Impairment of endothelium‐dependent vasodilation of resistance vessels in patients with obstructive sleep apnea. Circulation. 2000;102:2607–2610. [DOI] [PubMed] [Google Scholar]
- 5. Dyugovskaya L, Lavie P, Lavie L. Increased adhesion molecules expression and production of reactive oxygen species in leukocytes of sleep apnea patients. Am J Respir Crit Care Med. 2002;165:934–939. [DOI] [PubMed] [Google Scholar]
- 6. Hoyos CM, Melehan KL, Liu PY, Grunstein RR, Phillips CL. Does obstructive sleep apnea cause endothelial dysfunction? A critical review of the literature. Sleep Med Rev. 2015;20:15–26. [DOI] [PubMed] [Google Scholar]
- 7. McEvoy RD, Antic NA, Heeley E, Luo Y, Ou Q, Zhang X, Mediano O, Chen R, Drager LF, Liu Z, Chen G, Du B, McArdle N, Mukherjee S, Tripathi M, Billot L, Li Q, Lorenzi‐Filho G, Barbe F, Redline S, Wang J, Arima H, Neal B, White DP, Grunstein RR, Zhong N, Anderson CS. CPAP for prevention of cardiovascular events in obstructive sleep apnea. N Engl J Med. 2016;375:919–931. [DOI] [PubMed] [Google Scholar]
- 8. Schulz E, Gori T, Munzel T. Oxidative stress and endothelial dysfunction in hypertension. Hypertens Res. 2011;34:665–673. [DOI] [PubMed] [Google Scholar]
- 9. Jelic S, Padeletti M, Kawut SM, Higgins C, Canfield SM, Onat D, Colombo PC, Basner RC, Factor P, LeJemtel TH. Inflammation, oxidative stress, and repair capacity of the vascular endothelium in obstructive sleep apnea. Circulation. 2008;117:2270–2278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Arnaud C, Beguin PC, Lantuejoul S, Pepin JL, Guillermet C, Pelli G, Burger F, Buatois V, Ribuot C, Baguet JP, Mach F, Levy P, Dematteis M. The inflammatory preatherosclerotic remodeling induced by intermittent hypoxia is attenuated by RANTES/CCL5 inhibition. Am J Respir Crit Care Med. 2011;184:724–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Poulain L, Richard V, Levy P, Dematteis M, Arnaud C. Toll‐like receptor‐4 mediated inflammation is involved in the cardiometabolic alterations induced by intermittent hypoxia. Mediators Inflamm. 2015;2015:620258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gras E, Belaidi E, Briancon‐Marjollet A, Pepin JL, Arnaud C, Godin‐Ribuot D. Endothelin‐1 mediates intermittent hypoxia‐induced inflammatory vascular remodeling through HIF‐1 activation. J Appl Physiol (1985). 2016;120:437–443. [DOI] [PubMed] [Google Scholar]
- 13. Troncoso Brindeiro CM, da Silva AQ, Allahdadi KJ, Youngblood V, Kanagy NL. Reactive oxygen species contribute to sleep apnea‐induced hypertension in rats. Am J Physiol Heart Circ Physiol. 2007;293:H2971–H2976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Friedman JK, Nitta CH, Henderson KM, Codianni SJ, Sanchez L, Ramiro‐Diaz JM, Howard TA, Giermakowska W, Kanagy NL, Gonzalez Bosc LV. Intermittent hypoxia‐induced increases in reactive oxygen species activate NFATc3 increasing endothelin‐1 vasoconstrictor reactivity. Vascul Pharmacol. 2014;60:17–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Verin AD, Lazar V, Torry RJ, Labarrere CA, Patterson CE, Garcia JG. Expression of a novel high molecular‐weight myosin light chain kinase in endothelium. Am J Respir Cell Mol Biol. 1998;19:758–766. [DOI] [PubMed] [Google Scholar]
- 16. Garcia JG, Lazar V, Gilbert‐McClain LI, Gallagher PJ, Verin AD. Myosin light chain kinase in endothelium: molecular cloning and regulation. Am J Respir Cell Mol Biol. 1997;16:489–494. [DOI] [PubMed] [Google Scholar]
- 17. Goeckeler ZM, Wysolmerski RB. Myosin light chain kinase‐regulated endothelial cell contraction: the relationship between isometric tension, actin polymerization, and myosin phosphorylation. J Cell Biol. 1995;130:613–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sun C, Wu MH, Yuan SY. Nonmuscle myosin light‐chain kinase deficiency attenuates atherosclerosis in apolipoprotein E‐deficient mice via reduced endothelial barrier dysfunction and monocyte migration. Circulation. 2011;124:48–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wainwright MS, Rossi J, Schavocky J, Crawford S, Steinhorn D, Velentza AV, Zasadzki M, Shirinsky V, Jia Y, Haiech J, Van Eldik LJ, Watterson DM. Protein kinase involved in lung injury susceptibility: evidence from enzyme isoform genetic knockout and in vivo inhibitor treatment. Proc Natl Acad Sci U S A. 2003;100:6233–6238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ralay Ranaivo H, Carusio N, Wangensteen R, Ohlmann P, Loichot C, Tesse A, Chalupsky K, Lobysheva I, Haiech J, Watterson DM, Andriantsitohaina R. Protection against endotoxic shock as a consequence of reduced nitrosative stress in MLCK210‐null mice. Am J Pathol. 2007;170:439–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Reynoso R, Perrin RM, Breslin JW, Daines DA, Watson KD, Watterson DM, Wu MH, Yuan S. A role for long chain myosin light chain kinase (MLCK‐210) in microvascular hyperpermeability during severe burns. Shock. 2007;28:589–595. [DOI] [PubMed] [Google Scholar]
- 22. Dematteis M, Godin‐Ribuot D, Arnaud C, Ribuot C, Stanke‐Labesque F, Pepin JL, Levy P. Cardiovascular consequences of sleep‐disordered breathing: contribution of animal models to understanding the human disease. ILAR J. 2009;50:262–281. [DOI] [PubMed] [Google Scholar]
- 23. Daugherty A, Rateri D, Hong L, Balakrishnan A. Measuring blood pressure in mice using volume pressure recording, a tail‐cuff method. J Vis Exp. 2009:e1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wilde E, Aubdool AA, Thakore P, Baldissera L Jr, Alawi KM, Keeble J, Nandi M, Brain SD. Tail‐cuff technique and its influence on central blood pressure in the mouse. J Am Heart Assoc. 2017;6:e005204 DOI: 10.1161/JAHA.116.005204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Williams R, Needles A, Cherin E, Zhou YQ, Henkelman RM, Adamson SL, Foster FS. Noninvasive ultrasonic measurement of regional and local pulse‐wave velocity in mice. Ultrasound Med Biol. 2007;33:1368–1375. [DOI] [PubMed] [Google Scholar]
- 26. Faury G, Maher GM, Li DY, Keating MT, Mecham RP, Boyle WA. Relation between outer and luminal diameter in cannulated arteries. Am J Physiol. 1999;277:H1745–H1753. [DOI] [PubMed] [Google Scholar]
- 27. Faury G, Pezet M, Knutsen RH, Boyle WA, Heximer SP, McLean SE, Minkes RK, Blumer KJ, Kovacs A, Kelly DP, Li DY, Starcher B, Mecham RP. Developmental adaptation of the mouse cardiovascular system to elastin haploinsufficiency. J Clin Invest. 2003;112:1419–1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Pezet M, Jacob MP, Escoubet B, Gheduzzi D, Tillet E, Perret P, Huber P, Quaglino D, Vranckx R, Li DY, Starcher B, Boyle WA, Mecham RP, Faury G. Elastin haploinsufficiency induces alternative aging processes in the aorta. Rejuvenation Res. 2008;11:97–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Gibbons CA, Shadwick RE. Functional similarities in the mechanical design of the aorta in lower vertebrates and mammals. Experientia. 1989;45:1083–1088. [DOI] [PubMed] [Google Scholar]
- 30. McConnell JC, O'Connell OV, Brennan K, Weiping L, Howe M, Joseph L, Knight D, O'Cualain R, Lim Y, Leek A, Waddington R, Rogan J, Astley SM, Gandhi A, Kirwan CC, Sherratt MJ, Streuli CH. Increased peri‐ductal collagen micro‐organization may contribute to raised mammographic density. Breast Cancer Res. 2016;18:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Rodriguez M, Pascual G, Cifuentes A, Perez‐Kohler B, Bellon JM, Bujan J. Role of lysyl oxidases in neointima development in vascular allografts. J Vasc Res. 2011;48:43–51. [DOI] [PubMed] [Google Scholar]
- 32. Coquand‐Gandit M, Jacob MP, Fhayli W, Romero B, Georgieva M, Bouillot S, Esteve E, Andrieu JP, Brasseur S, Bouyon S, Garcia‐Honduvilla N, Huber P, Bujan J, Atanasova M, Faury G. Chronic treatment with minoxidil induces elastic fiber neosynthesis and functional improvement in the aorta of aged mice. Rejuvenation Res. 2017;20:218–230. [DOI] [PubMed] [Google Scholar]
- 33. Minoves M, Morand J, Perriot F, Chatard M, Gonthier B, Lemarie E, Menut JB, Polak J, Pepin JL, Godin‐Ribuot D, Briancon‐Marjollet A. An innovative intermittent hypoxia model for cell cultures allowing fast Po2 oscillations with minimal gas consumption. Am J Physiol Cell Physiol. 2017;313:C460–C468. [DOI] [PubMed] [Google Scholar]
- 34. Nieto FJ, Young TB, Lind BK, Shahar E, Samet JM, Redline S, D'Agostino RB, Newman AB, Lebowitz MD, Pickering TG. Association of sleep‐disordered breathing, sleep apnea, and hypertension in a large community‐based study: Sleep Heart Health Study. JAMA. 2000;283:1829–1836. [DOI] [PubMed] [Google Scholar]
- 35. Tamisier R, Pepin JL, Remy J, Baguet JP, Taylor JA, Weiss JW, Levy P. 14 Nights of intermittent hypoxia elevate daytime blood pressure and sympathetic activity in healthy humans. Eur Respir J. 2011;37:119–128. [DOI] [PubMed] [Google Scholar]
- 36. Belaidi E, Joyeux‐Faure M, Ribuot C, Launois SH, Levy P, Godin‐Ribuot D. Major role for hypoxia inducible factor‐1 and the endothelin system in promoting myocardial infarction and hypertension in an animal model of obstructive sleep apnea. J Am Coll Cardiol. 2009;53:1309–1317. [DOI] [PubMed] [Google Scholar]
- 37. Totoson P, Fhayli W, Faury G, Korichneva I, Cachot S, Baldazza M, Ribuot C, Pepin JL, Levy P, Joyeux‐Faure M. Atorvastatin protects against deleterious cardiovascular consequences induced by chronic intermittent hypoxia. Exp Biol Med (Maywood). 2013;238:223–232. [DOI] [PubMed] [Google Scholar]
- 38. Doonan RJ, Scheffler P, Lalli M, Kimoff RJ, Petridou ET, Daskalopoulos ME, Daskalopoulou SS. Increased arterial stiffness in obstructive sleep apnea: a systematic review. Hypertens Res. 2011;34:23–32. [DOI] [PubMed] [Google Scholar]
- 39. Drager LF, Bortolotto LA, Lorenzi MC, Figueiredo AC, Krieger EM, Lorenzi‐Filho G. Early signs of atherosclerosis in obstructive sleep apnea. Am J Respir Crit Care Med. 2005;172:613–618. [DOI] [PubMed] [Google Scholar]
- 40. Mitchell GF. Arterial stiffness and hypertension. Hypertension. 2014;64:13–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Baguet JP, Hammer L, Levy P, Pierre H, Launois S, Mallion JM, Pepin JL. The severity of oxygen desaturation is predictive of carotid wall thickening and plaque occurrence. Chest. 2005;128:3407–3412. [DOI] [PubMed] [Google Scholar]
- 42. Cortese R, Gileles‐Hillel A, Khalyfa A, Almendros I, Akbarpour M, Khalyfa AA, Qiao Z, Garcia T, Andrade J, Gozal D. Aorta macrophage inflammatory and epigenetic changes in a murine model of obstructive sleep apnea: potential role of CD36. Sci Rep. 2017;7:43648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Castro‐Grattoni AL, Alvarez‐Buve R, Torres M, Farre R, Montserrat JM, Dalmases M, Almendros I, Barbe F, Sanchez‐de‐la‐Torre M. Intermittent hypoxia‐induced cardiovascular remodeling is reversed by normoxia in a mouse model of sleep apnea. Chest. 2016;149:1400–1408. [DOI] [PubMed] [Google Scholar]
- 44. Lee MY, Wang Y, Mak JC, Ip MS. Intermittent hypoxia induces NF‐kappaB‐dependent endothelial activation via adipocyte‐derived mediators. Am J Physiol Cell Physiol. 2016;310:C446–C455. [DOI] [PubMed] [Google Scholar]
- 45. Yannoutsos A, Levy BI, Safar ME, Slama G, Blacher J. Pathophysiology of hypertension: interactions between macro and microvascular alterations through endothelial dysfunction. J Hypertens. 2014;32:216–224. [DOI] [PubMed] [Google Scholar]
- 46. Greenberg H, Ye X, Wilson D, Htoo AK, Hendersen T, Liu SF. Chronic intermittent hypoxia activates nuclear factor‐kappaB in cardiovascular tissues in vivo. Biochem Biophys Res Commun. 2006;343:591–596. [DOI] [PubMed] [Google Scholar]
- 47. Makarenko VV, Usatyuk PV, Yuan G, Lee MM, Nanduri J, Natarajan V, Kumar GK, Prabhakar NR. Intermittent hypoxia‐induced endothelial barrier dysfunction requires ROS‐dependent MAP kinase activation. Am J Physiol Cell Physiol. 2014;306:C745–C752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Eutamene H, Theodorou V, Schmidlin F, Tondereau V, Garcia‐Villar R, Salvador‐Cartier C, Chovet M, Bertrand C, Bueno L. LPS‐induced lung inflammation is linked to increased epithelial permeability: role of MLCK. Eur Respir J. 2005;25:789–796. [DOI] [PubMed] [Google Scholar]
- 49. Gaceb A, Vergori L, Martinez MC, Andriantsitohaina R. Activation of endothelial pro‐resolving anti‐inflammatory pathways by circulating microvesicles from non‐muscular myosin light chain kinase‐deficient mice. Front Pharmacol. 2016;7:322. [DOI] [PMC free article] [PubMed] [Google Scholar]