

In cardiomyocytes, excitation–contraction (E‐C) coupling is the fundamental mechanism governing the contractile function of the heart, by which working ventricular myocytes generate sufficient contractile force to pump blood throughout the body.1 In the past several decades, numerous studies have greatly increased our understanding of the structure and function of cardiac E‐C coupling.2 During E‐C coupling, an action potential drives membrane depolarization to initiate a cascade of Ca2+‐mediated processes, among which Ca2+‐induced Ca2+ release is the central process controlling the efficacy and fidelity of E‐C coupling. During the process of Ca2+‐induced Ca2+ release, Ca2+ influx through the opening of voltage‐gated L‐type Ca2+ channels (LTCCs) located on T‐tubule membrane triggers a much larger release of Ca2+ from the sarcoplasmic reticulum (SR) via Ca2+ release channels or ryanodine receptors residing on the terminal cisternae of the SR or junctional SR. In ventricular cardiomyocytes, T‐tubules and the junctional SR closely appose each other to form tight junctional couplings with a gap of 12 to 15 nm, dubbed cardiac dyads, which provide nanoscale ultrastructural microdomains for highly efficient and precisely controlled Ca2+‐induced Ca2+ release (Figure).3 It is also appreciated that the structural protein junctophilin‐2 is responsible for the formation of the junctional coupling between T‐tubules and the terminal cisternae of SR membranes, as well as for maintaining T‐tubule integrity in a regularly arrayed organization.4, 5, 6
Figure 1.
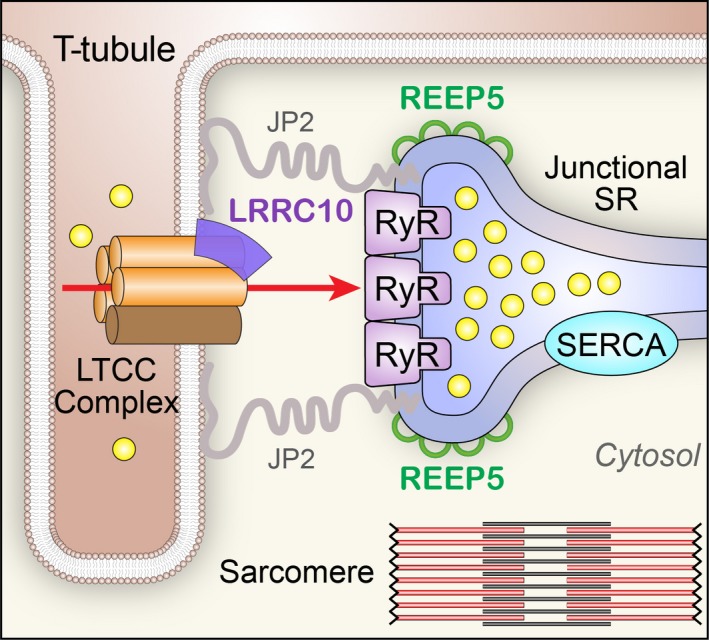
Novel E‐C coupling partners uncovered. New findings indicate that LRRC10 is a novel LTCC accessory protein, and REEP5 plays an important role in shaping the morphology of junctional SR. JP2 indicates junctophilin type 2; LRRC10, leucine‐rich repeat‐containing protein 10; LTCC, L‐type Ca2+ channel; REEP5, receptor expression‐enhancing protein 5; RyR, ryanodine receptor; SERCA, sarco/endoplasmic reticulum Ca2+‐ATPase; SR, sarco/endoplasmic reticulum; T‐tubule, transverse tubule.
Novel Proteins Responsible for Shaping the Junctional SR: The Receptor Expression‐Enhancing Proteins Family Protein
The SR system is a network of tubules running throughout the cardiomyocytes that uptakes and stores Ca2+ ions. The SR contains 3 distinct but continuous regions: the network SR, the corbular SR, and the junctional SR.7 A majority of the total SR system is the network SR that is organized in an anastomosing system surrounding the myofibrils. The junctional and corbular SR are structurally specialized domains extending from the network SR. Although both contain ryanodine receptors, the junctional SR is physically connected to T‐tubules or sarcolemma (as mentioned above), whereas the corbular SR is free of sarcolemmal contact. Morphologically, the junctional SR, as the terminal cisternae of the continuous SR network, changes shape drastically, from a long, randomly running tubular‐like structure into a flat SR cisternae that closely apposes to the surface sarcolemma or associates along with T‐tubules.8 Even though much is known regarding the SR network, what remains unclear is how the junctional SR is shaped in cardiomyocytes.
In this issue of Journal of the American Heart Association (JAHA), Chen and colleagues elegantly describe a novel role for REEP5 (receptor accessory protein 5) in the formation of the junctional SR.9 REEP5 (also known as DP1) belongs to a family of receptor expression‐enhancing proteins (REEPs) that are part of a superfamily of “morphogens” that establish the curvature of the endoplasmic reticulum.10 While mutations in REEP1 and REEP6 have been associated with neurological and retinal disorders, respectively, the function of REEP proteins in the heart is completely unknown. In the article by Chen and colleagues, the authors demonstrate that REEP5 is the only REEP family protein enriched in the heart and colocalizes with ryanodine receptors in the junctional SR region. Using a novel CRISPR/Cas9‐based REEP5 knockout rat model, focused‐ion‐beam scanning electron microscopy was used to reconstruct the ultrastructure of cardiac SR, with a focus on the junctional SR. These sophisticated and state‐of‐the‐art techniques revealed that REEP5 deficiency alters the morphology of the junctional SR, resulting in a defect in the connections between the SR and T‐tubules. At the cellular level, REEP5 deficiency reduces the triggering efficiency of L‐type Ca2+ current and decreases Ca2+ release from the SR. This defect in Ca2+‐induced Ca2+ release under REEP5 deficiency culminates in compromised cardiac contractility.
Novel Auxiliary Subunit of Cardiac LTCC and Its Roles in Dilated Cardiomyopathy
Leucine‐rich repeats (LRRs) are 20 to 29 amino acid motifs that are found in many proteins of diverse biological functions and contain a conserved 11‐residue segment with the consensus sequence LxxLxLxxN/CxL (x represents any amino acid and L positions can be occupied by valine, isoleucine, or phenylalanine).11 Recent structural analyses have provided insights into the role of leucine‐rich repeats as versatile structural domains critical for protein–protein interactions, including a curved overall shape with mostly helical elements on the convex side and a parallel β sheet on the concave side where the protein–protein interactions occur.11 LRR‐containing (LRRC) protein 10 (LRRC10) is a cardiac‐specific member of the LRRC superfamily with critical roles in cardiac function.12 It contains 7 LRR motifs with no other known functional domains. LRRC10 has been shown to be highly conserved and expressed exclusively in cardiomyocytes where it localizes to the dyad region. Previous studies in zebrafish demonstrated that Lrrc10 plays important roles in cardiac function and that knockdown of Lrrc10 in zebrafish results in cardiac developmental abnormalities with reduced cardiac function and death.13 Subsequent studies in mice supported these earlier findings where homozygous knockout of Lrrc10 results in dilated cardiomyopathy (DCM).14 Critical roles of LRRC10 in cardiac function were further solidified where mutations in LRRC10 have recently been identified in human DCM.15 However, the exact mechanisms of how LRRC10 may regulate cardiac function remain unknown.
In an intriguing and comprehensive study by Woon and colleagues in the current issue of JAHA,16 the authors used whole exome sequencing to identify a homozygous recessive variant (I195T) in the LRR region of LRRC10 in a sporadic case of pediatric DCM. Comprehensive mechanistic study reveals the unique roles of LRRC10 in the modulation of cardiac LTCC. Specifically, it was established that LRRC10 forms multiprotein complexes with Cav1.2 LTCC. Co‐expression of LRRC10 with Cav1.2 in a heterologous expression system results in a significant increase in L‐type Ca2+ current density while the opposite effect is observed when the mutant LRRC10 (I195T) was used for the co‐expression. More importantly, co‐expression of LRRC10 or mutant LRRC10 with Cav1.2 significantly impacts the voltage‐dependent gating of the channels with hyperpolarization and depolarization shift in the steady‐state activation, respectively. There is no alteration in channel trafficking to the membrane. These data strongly support the critical roles of LRRC10 as an auxiliary subunit of Cav1.2 Ca2+ channel. Quantitative analyses show that co‐expression of LRRC10 results in a decrease in the late Ca2+ current, while co‐expression of the mutant LRRC10 increases the late Ca2+ current. Indeed, the increase in the late Ca2+ current may lead to increased Ca2+ loading and DCM. Finally, the authors corroborate their findings by taking advantage of the Lrrc10 knockout mice.
New Insights Into the Regulatory Mechanisms for Cardiac E‐C Coupling, Cardiomyopathy, and Heart Failure
DCM and hypertrophic cardiomyopathy are the most common primary myocardial diseases with a prevalence of 1 in 2500 and 1 in 500 individuals, respectively.17 Patients with either form of cardiomyopathy have increased propensity for fatal arrhythmias and sudden cardiac death. Over the past several decades, a large number of mutations in sarcomeric proteins have been shown to be linked to hypertrophic cardiomyopathy. In contrast, DCM has been associated with mutations in a wide range of genes encoding for proteins of the Z‐disc, costamere, sarcolemma, nuclear lamina, and cytoskeletal proteins.17 In addition, up to 50% of cases of DCM are caused by acquired pathogeneses including ischemia and viral myocarditis. However, the underlying causes in a significant number of patients with DCM remain unknown. Therefore, there is a critical need to determine new genetic determinants underlying DCM. Moreover, insights into novel proteins mechanistically linked to DCM help in further understanding the underlying pathophysiology of acquired causes of DCM.
The 2 complementary articles in this issue of JAHA reveal novel regulatory mechanisms for cardiac E‐C coupling. Importantly, these findings help to fill our knowledge gaps about the exquisite and intricate control of cardiac E‐C coupling and how the defects may result in DCM and cardiac dysfunction. Specifically, Yao et al demonstrate that REEP5 plays a critical role in shaping the SR at the junctions and that defect in REEP5 deforms the architecture of junctional SR, resulting in a decrease in cardiac contractility.9 Moreover, REEP5 is downregulated in a chronic pressure‐induced model of heart failure, supporting its possible contribution to cardiac dysfunction. Nonetheless, additional studies are needed to further determine the specific mechanisms of how REEP5 maintains the integrity of the junctional SR membrane. In addition, the mechanistic underpinnings of the downregulation of REEP5 in heart failure require further investigations.
Complementary findings by Woon et al provide new mechanistic insights into regulation of LTCC gating by identifying LRRC10 as a novel auxiliary subunit.16 In a parallel study, LRRC26 proteins have been shown to modulate the gating of a large conductance Ca2+‐activated K+ (BK) channel by enhancing the allosteric coupling between voltage‐sensor activation and the channel's closed–open transition. This results in a large hyperpolarization shift in voltage dependence and plays important roles for activation of BK channels in nonexcitable cells.18 Here, Woon and coworkers demonstrated with compelling evidence that defects in LRRC10 result in a decrease in peak Ca2+ current but an increase in delayed current, which can predispose patients to Ca2+ overload as a possible pathophysiologic mechanism for DCM. Further substantiating the role of LRRC10 in normal cardiac function, the same group of investigators has previously shown that Lrrc10 knockout mice develop severe cardiac dysfunction and ventricular dilation following mild pressure overload.19 However, additional studies are required to fully decipher the interacting partners of LRRC10 within the macromolecular complex of LTCCs. LRRC10 proteins are localized within the dyad regions and may interact and regulate other component(s) of the molecular complexes involved in cardiac E‐C coupling. LRRC10 was shown to interact with α‐actinin and α‐sarcomeric actin at the Z‐disc in cardiomyocytes.20 The biophysical effects of LRRC10 on the voltage‐dependent gating of Cav1.2 channels require additional study to fully dissect the molecular mechanisms for the observed changes. Since LRRC10 also induces a hyperpolarization shift in the voltage‐dependent activation of LTCCs similar to the effects of LRRC26 on BK channels, it is tempting to hypothesize that LRRC10 may also enhance the allosteric coupling between the voltage sensor and the channel's closed–open transition similar to BK channels. However, this remains to be investigated. Structural information on the LRRC protein family may help to further determine the mechanistic interactions of LRRC10 with Cav1.2. It is also appealing to hypothesize that there may be a cohort of LRRC family members that facilitate the fine tuning of the voltage dependence of other ion channels. Finally, possible changes of LRRC10 in the acquired type of DCM need further investigations.
Thus, the 2 articles in this issue of JAHA have further peeled back and revealed the multiple layers of the intricate and precise regulation of cardiac E‐C coupling. Importantly, these studies help to raise new unanswered questions: Are there additional interacting partners within the macromolecular complex of LTCCs? Do changes in the expression of LRRC10 and REEP5 lead to cardiomyopathies from different causes? How can we leverage this new information to design mechanism‐based treatment for DCM and heart failure? With emerging tools such as high‐resolution imaging and protein structure information as well as patient‐specific pluripotent stem cell–derived cardiomyocytes, the answers to these questions may be closer than we think (perhaps even within 12–15 nm).
Sources of Funding
This work is supported by NIH R01s (HL137228, HL085844, HL085727, HL090905 & HL130346), and VA Merit Awards (I01‐BX000576, I01‐CX001490, and I01‐BX002344).
Disclosures
None.
Acknowledgments
The authors would like to acknowledge artistic help from Rafael Shimkunas and Shawn Roach for the figure in the editorial.
(J Am Heart Assoc. 2018;7:e008260 DOI: 10.1161/JAHA.117.008260.)29431105
The opinions expressed in this article are not necessarily those of the editors or of the American Heart Association.
Contributor Information
Nipavan Chiamvimonvat, Email: nchiamvimonvat@ucdavis.edu.
Long‐Sheng Song, Email: long-sheng-song@uiowa.edu.
References
- 1. Bers DM. Cardiac excitation‐contraction coupling. Nature. 2002;415:198–205. [DOI] [PubMed] [Google Scholar]
- 2. Eisner DA, Caldwell JL, Kistamas K, Trafford AW. Calcium and excitation‐contraction coupling in the heart. Circ Res. 2017;121:181–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Wang SQ, Song LS, Lakatta EG, Cheng H. Ca2+ signalling between single L‐type Ca2+ channels and ryanodine receptors in heart cells. Nature. 2001;410:592–596. [DOI] [PubMed] [Google Scholar]
- 4. Takeshima H, Komazaki S, Nishi M, Iino M, Kangawa K. Junctophilins: a novel family of junctional membrane complex proteins. Mol Cell. 2000;6:11–22. [DOI] [PubMed] [Google Scholar]
- 5. Wei S, Guo A, Chen B, Kutschke W, Xie YP, Zimmerman K, Weiss RM, Anderson ME, Cheng H, Song LS. T‐tubule remodeling during transition from hypertrophy to heart failure. Circ Res. 2010;107:520–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Guo A, Zhang X, Iyer VR, Chen B, Zhang C, Kutschke WJ, Weiss RM, Franzini‐Armstrong C and Song LS. Overexpression of junctophilin‐2 does not enhance baseline function but attenuates heart failure development after cardiac stress. Proc Natl Acad Sci U S A. 2014;111:12240–12245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Jorgensen AO, Shen AC, Arnold W, McPherson PS, Campbell KP. The Ca2+‐release channel/ryanodine receptor is localized in junctional and corbular sarcoplasmic reticulum in cardiac muscle. J Cell Biol. 1993;120:969–980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Franzini‐Armstrong C, Protasi F, Tijskens P. The assembly of calcium release units in cardiac muscle. Ann N Y Acad Sci. 2005;1047:76–85. [DOI] [PubMed] [Google Scholar]
- 9. Yao L, Xie D, Geng L, Shi D, Huang J, Wu Y, Lv F, Liang D, Li L, Liu Y, Li J, Chen YH. REEP5 (Receptor Accessory Protein 5) acts as a sarcoplasmic reticulum membrane sculptor to modulate cardiac function. J Am Heart Assoc. 2018;7:e007205 DOI: 10.1161/JAHA.117.007205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Goyal U, Blackstone C. Untangling the web: mechanisms underlying ER network formation. Biochim Biophys Acta. 2013;1833:2492–2498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kobe B, Kajava AV. The leucine‐rich repeat as a protein recognition motif. Curr Opin Struct Biol. 2001;11:725–732. [DOI] [PubMed] [Google Scholar]
- 12. Brody MJ, Lee Y. The role of leucine‐rich repeat containing protein 10 (LRRC10) in dilated cardiomyopathy. Front Physiol. 2016;7:337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kim KH, Antkiewicz DS, Yan L, Eliceiri KW, Heideman W, Peterson RE, Lee Y. Lrrc10 is required for early heart development and function in zebrafish. Dev Biol. 2007;308:494–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Manuylov NL, Manuylova E, Avdoshina V, Tevosian S. Serdin1/Lrrc10 is dispensable for mouse development. Genesis. 2008;46:441–446. [DOI] [PubMed] [Google Scholar]
- 15. Qu XK, Yuan F, Li RG, Xu L, Jing WF, Liu H, Xu YJ, Zhang M, Liu X, Fang WY, Yang YQ, Qiu XB. Prevalence and spectrum of LRRC10 mutations associated with idiopathic dilated cardiomyopathy. Mol Med Rep. 2015;12:3718–3724. [DOI] [PubMed] [Google Scholar]
- 16. Woon MT, Long PA, Reilly L, Evans JM, Keefe AM, Lea MR, Beglinger CJ, Balijepalli RC, Lee Y, Olson TM, Kamp TJ. Pediatric dilated cardiomyopathy‐associated LRRC10 (Leucine‐Rich Repeat‐Containing 10) variant reveals LRRC10 as an auxiliary subunit of cardiac L‐type Ca2+ channels. J Am Heart Assoc. 2018;7:e006428 DOI: 10.1161/JAHA.117.006428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. McNally EM, Barefield DY, Puckelwartz MJ. The genetic landscape of cardiomyopathy and its role in heart failure. Cell Metab. 2015;21:174–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Yan J, Aldrich RW. LRRC26 auxiliary protein allows BK channel activation at resting voltage without calcium. Nature. 2010;466:513–516. [DOI] [PubMed] [Google Scholar]
- 19. Brody MJ, Feng L, Grimes AC, Hacker TA, Olson TM, Kamp TJ, Balijepalli RC, Lee Y. LRRC10 is required to maintain cardiac function in response to pressure overload. Am J Physiol Heart Circ Physiol. 2016;310:H269–H278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Brody MJ, Hacker TA, Patel JR, Feng L, Sadoshima J, Tevosian SG, Balijepalli RC, Moss RL, Lee Y. Ablation of the cardiac‐specific gene leucine‐rich repeat containing 10 (Lrrc10) results in dilated cardiomyopathy. PLoS One. 2012;7:e51621. [DOI] [PMC free article] [PubMed] [Google Scholar]