

ABSTRACT
The Mo- and V-nitrogenases are two homologous members of the nitrogenase family that are distinguished mainly by the presence of different heterometals (Mo or V) at their respective cofactor sites (M- or V-cluster). However, the V-nitrogenase is ~600-fold more active than its Mo counterpart in reducing CO to hydrocarbons at ambient conditions. Here, we expressed an M-cluster-containing, hybrid V-nitrogenase in Azotobacter vinelandii and compared it to its native, V-cluster-containing counterpart in order to assess the impact of protein scaffold and cofactor species on the differential reactivities of Mo- and V-nitrogenases toward CO. Housed in the VFe protein component of V-nitrogenase, the M-cluster displayed electron paramagnetic resonance (EPR) features similar to those of the V-cluster and demonstrated an ~100-fold increase in hydrocarbon formation activity from CO reduction, suggesting a significant impact of protein environment on the overall CO-reducing activity of nitrogenase. On the other hand, the M-cluster was still ~6-fold less active than the V-cluster in the same protein scaffold, and it retained its inability to form detectable amounts of methane from CO reduction, illustrating a fine-tuning effect of the cofactor properties on this nitrogenase-catalyzed reaction. Together, these results provided important insights into the two major determinants for the enzymatic activity of CO reduction while establishing a useful framework for further elucidation of the essential catalytic elements for the CO reactivity of nitrogenase.
KEYWORDS: carbon monoxide, cofactor, hydrocarbons, molybdenum, nitrogenase, vanadium
IMPORTANCE
This is the first report on the in vivo generation and in vitro characterization of an M-cluster-containing V-nitrogenase hybrid. The “normalization” of the protein scaffold to that of the V-nitrogenase permits a direct comparison between the cofactor species of the Mo- and V-nitrogenases (M- and V-clusters) in CO reduction, whereas the discrepancy between the protein scaffolds of the Mo- and V-nitrogenases (MoFe and VFe proteins) housing the same cofactor (M-cluster) allows for an effective assessment of the impact of the protein environment on the CO reactivity of nitrogenase. The results of this study provide a first look into the “weighted” contributions of protein environment and cofactor properties to the overall activity of CO reduction; more importantly, they establish a useful platform for further investigation of the structural elements attributing to the CO-reducing activity of nitrogenase.
INTRODUCTION
Nitrogenase is an important metalloenzyme that catalyzes certain remarkable chemical transformations under ambient conditions (1). Catalysis by nitrogenase is enabled by ATP-dependent transfer of electrons from a reductase component to a catalytic component of the enzyme, followed by the subsequent reduction of substrates at the cofactor site of the catalytic component upon accumulation of sufficient electrons (2, 3). Using this two-component mechanism, the nitrogenase is capable of reducing nitrogen (N2) to ammonia (NH3), as well as carbon monoxide (CO) to hydrocarbons (e.g., propane [C3H8] and butane [C4H10]) (4, 5) at ambient conditions. Interestingly, these two reactions parallel the industrial Haber-Bosch and Fischer-Tropsch processes, respectively, which are used for large-scale production of ammonia and carbon fuels. However, in contrast to the energy-demanding industrial processes, the enzymatic reactions occur under ambient temperatures and pressures (6, 7). The unique features of the nitrogenase-catalyzed reactions make them fascinating subjects of study from a perspective of chemical energy while suggesting the potential of using these systems as prototypes for future development of biomimetic catalysts for energy- and cost-efficient production of useful chemical compounds.
The molybdenum (Mo)- and vanadium (V)-dependent nitrogenases are two homologous members of the nitrogenase family (8). Mainly distinguished by the presence of a different heterometal (i.e., Mo or V) at the cofactor site, the two nitrogenases comprise a pair of homologous component proteins: a homodimeric reductase component (nifH- or vnfH-encoded Fe protein), which contains a subunit-bridging [Fe4S4] cluster and an MgATP-binding site within each subunit; and a multimeric catalytic component (nifDK-encoded MoFe protein or vnfDGK-encoded VFe protein), which contains an 8Fe P-cluster species (P- or P*-cluster) at each α/β-subunit interface and a 7Fe/1Mo or 7Fe/1V cofactor species (M- or V-cluster) within each α subunit (9). Intriguingly, while biochemical, spectroscopic, and structural analyses reveal a striking resemblance between the Mo- and V-nitrogenases in terms of protein structure and cluster species (9, 10), the two nitrogenase systems are clearly distinct in their catalytic behaviors. Most notably, the Mo- and V-nitrogenases display significantly different reactivities toward the substrate CO, with the former showing a marginal activity of ~0.02 nmol of reduced carbon/nmol of protein/min, and the latter demonstrating a significantly increased activity at ~16 nmol of reduced carbon/nmol of protein/min—substantially higher than its Mo counterpart (4, 5). The observation of highly differential CO-reducing activities of two homologous nitrogenases has prompted us to define key features of these systems that contribute to this discrepancy in activity; in particular, the question of whether the protein environment or the cofactor species determines the reactivity of nitrogenase toward CO needs to be addressed, as knowledge in this regard represents the first step toward understanding the CO-reducing activity of nitrogenase for the potential applications of this reactivity in the future.
RESULTS
To tackle the question in hand, the M- and V-clusters must be placed in the same protein environment for direct comparison. Using a genetically altered Azotobacter vinelandii strain, a pair of VFe proteins containing either the V- or M-cluster can be generated in vivo for this line of investigation. This A. vinelandii strain expresses a His-tagged form of VFe protein in a genetic background that contains deletions of (i) the nifDK genes, which encode the MoFe protein, and (ii) the mod genes, which encode the Mo uptake system (locus tag Avin_50650-Avin_50730 of the A. vinelandii DJ strain) (11–14). Using this A. vinelandii strain, a V-cluster-containing native form of the VFe protein (designated VnfDGKV) was produced in vivo when V was supplemented in the growth medium (Fig. 1A), where deletion of the Mo transporter prevented incorporation of trace Mo into the cofactor (12–14), whereas an M-cluster-containing hybrid form of the VFe protein (designated VnfDGKM) was produced in vivo when Mo was added in excess to the growth medium (Fig. 1A), where the uptake of Mo was accomplished by other transporter systems, such as those involving siderophores (15, 16).
FIG 1 .

Subunit and metal compositions of VnfDGKV and VnfDGKM. (A) SDS-PAGE analysis of VnfDGKV and VnfDGKM. The molecular masses (in kilodaltons) of the protein standards are shown to the left of the gel. (B) Metal contents of NifDKM, VnfDGKV, and VnfDGKM.
Like the native VnfDGKV protein, the VnfDGKM hybrid consists of α, β, and δ subunits, although the δ subunit is present in a much reduced quantity in VnfDGKM than in VnfDGKV (Fig. 1A). Metal analysis reveals a metal content of 0.9 nmol Mo and less than 0.07 nmol V per nmol protein (Fig. 1B), suggesting that VnfDGKM houses an Mo-containing cofactor in place of a V-containing species. Not too surprisingly, the heterologous incorporation of the Mo-containing cofactor into the VnfDGK scaffold is less efficient than the homologous incorporation of the V-containing cofactor, as the Mo content of VnfDGKM supports the assignment of one M-cluster per protein, which is lower than the assignment of ~1.5 V-clusters per protein in the case of VnfDGKV. The identity of the cofactor species in VnfDGKM is confirmed by extracting the cofactor from VnfDGKM into an organic solvent, N-methylformamide (NMF), and subsequently inserting it into the cofactor-deficient apo-NifDK (designated NifDKapo). As shown in Fig. 2A, the apo-NifDK protein reconstituted with the cofactor extracted from VnfDGKM (designated MVnfDGK) exhibits EPR features (g = 4.31, 3.67, 2.01, and 1.91) identical to those of the NifDKapo protein reconstituted with the cofactor extracted from the wild-type NifDK (designated MNifDK). Moreover, when combined with the reductase component (designated NifH), the MVnfDGK- and MNifDK-reconstituted NifDKapo proteins demonstrate nearly indistinguishable substrate-reducing activities when N2, proton (H+) or acetylene (C2H2) is supplied as the substrate (Fig. 2B). Together, these observations establish VnfDGKM as an M-cluster-containing counterpart of VnfDGKV.
FIG 2 .
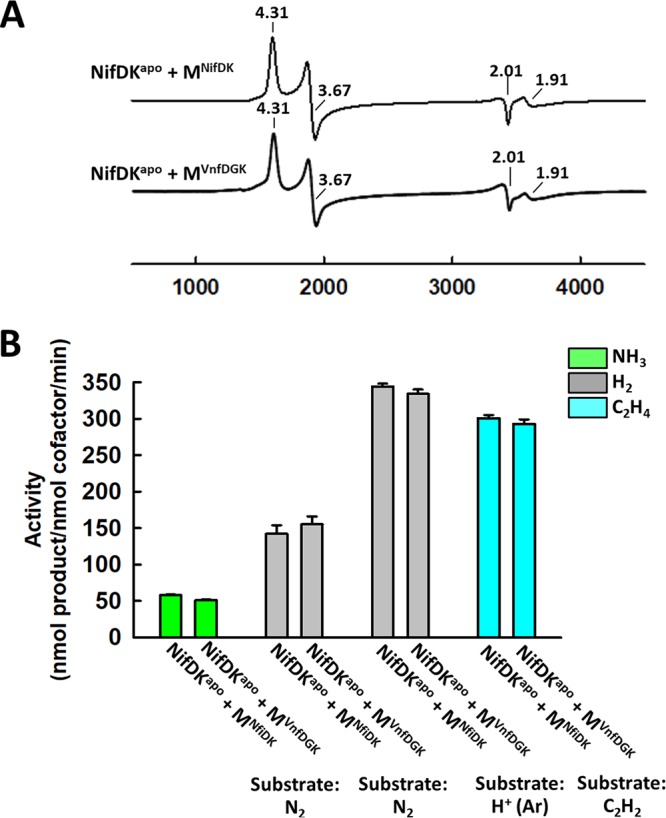
Spectroscopic and catalytic properties of the M-cluster extracted from VnfDGKM. (A and B) EPR spectra (A) and activity profiles (B) of the cofactor-deficient NifDKapo protein reconstituted with the M-cluster extracted from NifDK (NifDKapo + MNifDK) or VnfDGKM (NifDKapo + MVnfDGK). The g values are indicated in panel A. Activities are expressed as nanomoles of product per nanomole of cofactor per min in panel B.
Interestingly, the M-cluster in VnfDGKM displays EPR features (g = 5.36, 4.48, and 3.46) similar to those of the native V-cluster in VnfDGKV (g = 5.50, 4.32, and 3.77), both of which are clearly distinct from the EPR features of the native M-cluster in NifDKM (g = 4.31 and 3.67) (Fig. 3A). This observation is interesting, as it highlights a strong impact of protein environment on the properties of the cofactor. Consistent with the observed similarity between their EPR features, VnfDGKM seems to follow its native VnfDGKV counterpart in terms of the overall product distribution patterns, demonstrating decreased NH3/H2 and C2H4/H2 ratios relative to those generated by NifDKM, the ability to generate C2H6 from C2H2 reduction that is absent from NifDKM, and higher activity than NifDKM in producing hydrocarbons from CO reduction (Fig. 3B) (4, 5, 8, 11). The similarity between the CO reactivities of VnfDGKM and VnfDGKV is particularly striking. Both VnfDGKM and VnfDGKV reduce CO to hydrocarbons of up to C4 in length, whereas in comparison, NifDKM has a narrower product profile comprising up to C3 hydrocarbons (Fig. 4A). Moreover, the product distribution profiles of VnfDGKM and VnfDGKV are remarkably similar, with C2H4/C2H6 comprising 96.3%/2.5% and 94.6%/3.1%, respectively, of the total amounts of hydrocarbons generated by these proteins, displaying a clear tendency toward formation of the unsaturated C2 product (C2H4); in contrast, NifDKM generates C2H4/C2H6 at 56.9%/28.4% of the total amounts of hydrocarbons, showing a preference for formation of the saturated C2 product (C2H6) (Fig. 4B). The protein environment, therefore, appears to “normalize” the product profiles of the M- and V-clusters in CO reduction once they are inserted into the same protein scaffold, VnfDGK. Further, the fact that VnfDGKM is considerably more active than NifDKM (by ~100-fold) in CO reduction illustrates the higher efficiency of the VnfDGK scaffold in catalyzing this reaction (Fig. 4C).
FIG 3 .

Spectroscopic and catalytic properties of NifDKM, VnfDGKV, and VnfDGKM. (A and B) EPR spectra (A) and activity profiles (B) of NifDKM, VnfDGKV, and VnfDGKM. Note the presence of the same S = 1/2 signal in the spectra of VnfDGKV and VnfDGKM (see panel A), which was assigned to the P*-cluster (i.e., a pair of [Fe4S4]-like clusters) in the case of VnfDGKV (11). The g values are indicated in panel A, and the products are color coded in panel B. The substrates are indicated at the bottom of the bar chart, and the ratios of N2/H2 and C2H4/H2 generated in the reactions of N2 and C2H2 reduction are indictedabove the respective bars in panel B. HCs, hydrocarbons.
FIG 4 .

CO-reducing activities and product profiles of NifDKM, VnfDGKV, and VnfDGKM. (A to C) Individual activities of hydrocarbon formation (A), distributions of hydrocarbon products (B), and total activities of hydrocarbon product formation (C) by NifDKM, VnfDGKV, and VnfDGKM when CO is supplied as a substrate. Activities are expressed as nanomoles of product per nanomole of cofactor per minute in panels A and C. The percentage activities of proteins are shown in red in panel C, with the total activity of VnfDGKV set at 100% and those of NifDKM and VnfDGKM calculated accordingly. Note that there is a contribution of V to the activity of VnfDGKM due to the presence of <0.07 nmol of V per nmol of VnfDGKM. However, the percentage contribution of V to the overall activity of VnfDGKM cannot be conclusively determined due to the inaccuracy of V determination in this low concentration range.
There is, however, a clear contribution of the cofactor properties to the CO-reducing activity, as VnfDGKV is ~6-fold more active than VnfDGKM in hydrocarbon formation, which demonstrates that the V-cluster is better tuned toward CO reduction than the M-cluster (Fig. 4C). Moreover, despite the “normalization” of the protein environment, the ability of VnfDGKV to form detectable amounts of CH4 is not observed in the case of VnfDGKM under the same experimental conditions (Fig. 4B). Given the absence of CH4 from the product profile of NifDKM (Fig. 4B), this trait seems to be carried over to VnfDGKM by the M-cluster, further highlighting the characteristics of the unique properties of the M-cluster in the reaction of CO reduction. Taken together, these results suggest a combined effect of protein environment and cofactor properties on the reactivity of nitrogenase toward CO: the protein scaffold has a significant impact on the overall activity of CO reduction (Fig. 4C, VnfDGKM versus NifDKM), whereas the cofactor species fine-tunes the product profile of CO reduction while exerting a moderate impact on the overall activity (Fig. 4C, VnfDGKV versus VnfDGKM). It is interesting to note that a “weighted” contribution of protein environment and cofactor properties to the CO-reducing activity can be derived from these comparisons, with (i) the ~100-fold difference that arises from the difference in protein scaffold and (ii) the ~6-fold difference that arises from the difference in cofactor species contributing collectively to an ~600-fold difference between the CO-reducing activities of Mo- and V-nitrogenases.
DISCUSSION
The impact of protein environment on the CO reactivity of nitrogenase is intimately associated with the immediate surroundings of the cofactor that could play a significant role in the interactions between the cofactor and the substrate CO. The cofactor “pocket” in the recently reported crystal structure of VnfDGK is slightly more polar than its counterpart in NifDK, which may influence the electrochemical properties of the cofactor (17). Moreover, the cofactor captured in the crystal structure of VnfDGK has a “belt” sulfur substituted by a carbonate moiety (17). A comparison between the cofactor-binding sites in the crystal structures of VnfDGK and NifDK reveals comparable hydrogen bonding networks around the homocitrate moieties of the two cofactors but markedly different hydrogen bonding at the position where carbonate is bound to the V-cluster, which could contribute to the differences in the catalytic activities of the two proteins (17). Other than the cofactor environment, the P-cluster species, which mediates electron transfer to the cofactor, could also impact the CO reactivity of nitrogenase. While the P-cluster in the crystal structure of VnfDGK is determined to have the same [Fe8S7] structure as its counterpart in NifDK, there are additional electron densities at the P-cluster site that suggest the possible existence of an additional P-cluster conformation(s) that is not populated or captured in the specific redox state of the VnfDGK crystal (17). This observation is in line with the X-ray absorption spectroscopy (XAS)/extended X-ray absorption fine structure (EXAFS)-derived structure of the P-cluster of a cofactor-deficient VnfDGK, which suggests that this cluster assumes the conformation of a [Fe4S4]-like cluster pair in the solution state. It is likely, therefore, that the P-cluster of VnfDGK is capable of undergoing different conformational changes than those of its counterpart in NifDK upon redox changes. In this context, it is interesting to note that the P-cluster of the reduced, resting-state VnfDGK exhibits analogous S = 1/2 and S = 5/2 electron paramagnetic resonance (EPR) signals to the one-electron-oxidized, P1+ state of the P-cluster of NifDK (8, 18), which has been implicated in substrate turnover. Such a difference in redox states, likely associated with the conformational differences between the two P-cluster species, could very well impact the ability of the respective proteins to transfer electrons to their cofactor sites and, consequently, the catalytic activities of these proteins.
The impact of cofactor properties on the CO reactivity of nitrogenase, on the other hand, could stem from the presence of different heterometals in the M- and V-clusters. Interestingly, differential abilities of synthetic V- and Mo-containing compounds to reductively couple two CO moieties into functionalized acetylene ligands have been observed previously (19), which suggests a higher capacity of V (a first-row transition metal) than Mo (a second-row transition metal) in this type of reactions. While this observation may be used to account for the differential reactivities of V- and M-clusters toward CO, it remains unclear whether the heterometal directly participates in substrate reduction or exerts an indirect effect on the electronic/catalytic properties of the cofactor. Apart from the differential heterometal compositions of the V- and M-clusters, the presence of a carbonate moiety at the belt region of the V-cluster—a feature that is absent from any M-cluster structure reported so far—may also impact the nitrogenase reactivity (17). The observed substitution of a belt sulfide of the V-cluster by carbonate is interesting, as carbonate is a potential carbon substrate of this cofactor. However, the sulfide displaced by carbonate in the structure of the V-cluster is different than the sulfide equivalent displaced by CO in the structure of the CO-bound M-cluster (20). Moreover, a catalysis-dependent migration of belt sulfide has been suggested recently for the M-cluster, which could very well enable displacement of carbonate by a sulfide during substrate reduction in the case of the V-cluster (21). This proposal is also consistent with our XAS/EXAFS-derived structure of the isolated V-cluster, where a sulfide is modeled in place of carbonate in the belt region of this cofactor (22). The unlikely scenario that carbonate, a very weak ligand, has survived the cluster extraction procedure, along with the observation that the isolated V-cluster can be used to reconstitute cofactor-deficient proteins, suggests that a carbonate-free conformation of the V-cluster is likely the competent form in substrate reduction (22). Clearly, the origin and catalytic relevance of the carbonate moiety needs to be clarified before mechanistic interpretations can be made based on this finding.
The in vitro formation of an M-cluster containing the VnfDGK hybrid and analysis of its N2-reducing activity was reported earlier (23). However, the in vivo generation of this hybrid, which permits a direct comparison of the activities of hydrocarbon formation by the M- and V-cluster-containing VnfDGK proteins generated under cell growth conditions, has not been accomplished prior to the current study. Other than facilitating a direct assessment of the contributions of protein scaffold and cofactor species to the CO-reducing activity of nitrogenase, our in vivo generation of a heterologous form of VnfDGK that contains an M-cluster at its cofactor-binding site also sheds light on the regulation of nitrogenase expression and the biosynthesis of the “alternative” nitrogenase. It is interesting to note that, despite the deletion of the mod-encoded Mo uptake system (11–14) in A. vinelandii, the cells still manage to acquire sufficient Mo from a growth medium supplemented with excess Mo for the synthesis of M-clusters. In contrast to earlier suggestions (24), the expression of vnf genes in a nifDK deletion background is not suppressed by the amount of Mo taken up by this mechanism. Moreover, unlike NifEN that is specific for M-cluster synthesis (25, 26), VnfEN is apparently capable of synthesizing both M- and V-clusters for VnfDGK, further facilitating the formation of VnfDGKM via this approach. Finally, there is an obvious reduction in the amount of the vnfG-encoded δ subunit in the VnfDGKM protein (Fig. 1A). This observation coincides with results derived from the characterization of a cofactor-deficient form of VnfDGK, which reveals the absence of the δ subunit and an incomplete, αβ2-trimeric composition of this cofactor-less protein (27). The positive correlation between the decreased amount of δ subunit and the absence of V-cluster suggests a possible role of the δ subunit in specifically delivering the V-cluster to the cofactor-binding site and maintaining the stability at the α/β subunit interface once its delivery job is finished.
While many aspects related to the expression and assembly of the alternative nitrogenase await investigation, the outcome of this work provides a useful framework for further investigation of the two major determinants—the protein environment and the cofactor species—in order to narrow down the key elements attributing to the CO reactivity of nitrogenase. Moreover, the strategy used in this work for the successful generation of VnfDGKM in vivo could potentially be employed for generation of other heterologous forms of nitrogenase, which may facilitate further exploration of this unique reactivity of nitrogenase for potential applications in the future.
MATERIALS AND METHODS
Strain construction and cell growth.
Azotobacter vinelandii strains YM68A and YM13A (expressing His-tagged VnfDGK and NifDK, respectively) were constructed as described earlier (11, 28). Both strains were grown in 180-liter batches in a 200-liter New Brunswick fermentor (New Brunswick Scientific) in Burke’s minimal medium supplemented with 2 mM ammonium acetate (11, 28). The molybdate in Burke’s medium was replaced by an equal amount of vanadate for the expression of the native VnfDGKV protein in strain YM68A. In preparation for the expression of the VnfDGKM hybrid in strain YM68A, the fermentor was scrubbed with acid and water, followed by growth of two consecutive 180-liter batches of YM68A in Burke’s medium that contained no Mo or V, which permitted removal of trace amounts of V in the vessel. Subsequently, strain YM68A was grown in Burke’s medium supplemented with 2.5-fold molybdate, and cell growth was monitored by measuring the cell density at 436 nm using a Spectronic 20 Genesys spectrophotometer. Cells were harvested in the late exponential phase by a flowthrough centrifugal harvester (Cepa), and the cell paste was washed with a buffer containing 50 mM Tris-HCl (pH 8.0). Published methods were then used for the purification of His-tagged NifDK and VnfDGK and nontagged NifH and VnfH (11, 28).
Protein characterization and activity assays.
VnfDGK proteins were subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) analysis on a 4 to 20% precast Tris-glycine gel (Bio-Rad). The metal contents of the proteins were determined by inductively coupled plasma-optical emission spectroscopy (ICP-OES) based on previously established protocols (29). All nitrogenase activity assays were carried out as described earlier (30, 31). The hydrocarbon products were analyzed as described elsewhere (4, 5, 29). Ammonium was determined by a high-performance liquid chromatography fluorescence method (32), and hydrogen was analyzed as described previously (33).
Cofactor extraction and reconstitution of NifDKapo.
The NifDK- and VnfDGKM-bound M-clusters were extracted into N-methylformamide (NMF) using a previously established method (22). The extracted cofactor was then incubated with the M-cluster-deficient, apo-NifDK protein (NifDKapo) for 20 min prior to removal of excess metal cluster by passing the reconstituted protein through a G25 column.
EPR spectroscopy.
EPR samples were prepared in a Vacuum Atmospheres dry box at an oxygen level of <4 ppm. All samples contained 25 mM Tris-HCl (pH 8.0), 10% glycerol, and 2 mM sodium dithionite (Na2S2O4). The EPR spectra were taken in perpendicular mode using a Bruker ESP 300 Ez spectrophotometer (Bruker) interfaced with an Oxford Instruments ESR-9002 liquid helium continuous flow cryostat. All spectra were recorded at 10 K, using a microwave power of 20 mW, a gain of 5 × 104, a modulation frequency of 100 kHz, and a modulation amplitude of 5 G. A microwave frequency of 9.62 GHz was used to collect five scans for each sample.
ACKNOWLEDGMENT
This work is supported by DOE (BES) grant DE-SC0014470 (to M.W.R. and Y.H.).
Footnotes
Citation Rebelein JG, Lee CC, Newcomb M, Hu Y, Ribbe MW. 2018. Characterization of an M-cluster-substituted nitrogenase VFe protein. mBio 9:e00310-18. https://doi.org/10.1128/mBio.00310-18.
REFERENCES
- 1.Burgess BK, Lowe DJ. 1996. Mechanism of molybdenum nitrogenase. Chem Rev 96:2983–3012. doi: 10.1021/cr950055x. [DOI] [PubMed] [Google Scholar]
- 2.Rees DC, Akif Tezcan F, Haynes CA, Walton MY, Andrade S, Einsle O, Howard JB. 2005. Structural basis of biological nitrogen fixation. Philos Trans A Math Phys Eng Sci 363:971–984, 1035–1040. doi: 10.1098/rsta.2004.1539. [DOI] [PubMed] [Google Scholar]
- 3.Hoffman BM, Lukoyanov D, Yang ZY, Dean DR, Seefeldt LC. 2014. Mechanism of nitrogen fixation by nitrogenase: the next stage. Chem Rev 114:4041–4062. doi: 10.1021/cr400641x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lee CC, Hu Y, Ribbe MW. 2010. Vanadium nitrogenase reduces CO. Science 329:642. doi: 10.1126/science.1191455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hu Y, Lee CC, Ribbe MW. 2011. Extending the carbon chain: hydrocarbon formation catalyzed by vanadium/molybdenum nitrogenases. Science 333:753–755. doi: 10.1126/science.1206883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rofer-DePoorter CK. 1981. A comprehensive mechanism for the Fischer-Tropsch synthesis. Chem Rev 81:447–474. doi: 10.1021/cr00045a002. [DOI] [Google Scholar]
- 7.Lee CC, Hu Y, Ribbe MW. 2011. Tracing the hydrogen source of hydrocarbons formed by vanadium nitrogenase. Angew Chem Int Ed Engl 50:5545–5547. doi: 10.1002/anie.201100869. [DOI] [PubMed] [Google Scholar]
- 8.Eady RR. 1996. Structure-function relationships of alternative nitrogenases. Chem Rev 96:3013–3030. doi: 10.1021/cr950057h. [DOI] [PubMed] [Google Scholar]
- 9.Hu Y, Ribbe MW. 2015. Nitrogenase and homologs. J Biol Inorg Chem 20:435–445. doi: 10.1007/s00775-014-1225-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hu Y, Ribbe MW. 2016. Biosynthesis of the metalloclusters of nitrogenases. Annu Rev Biochem 85:455–483. doi: 10.1146/annurev-biochem-060614-034108. [DOI] [PubMed] [Google Scholar]
- 11.Lee CC, Hu Y, Ribbe MW. 2009. Unique features of the nitrogenase VFe protein from Azotobacter vinelandii. Proc Natl Acad Sci U S A 106:9209–9214. doi: 10.1073/pnas.0904408106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bishop PE, Jarlenski DM, Hetherington DR. 1980. Evidence for an alternative nitrogen fixation system in Azotobacter vinelandii. Proc Natl Acad Sci U S A 77:7342–7346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bishop PE, Jarlenski DM, Hetherington DR. 1982. Expression of an alternative nitrogen fixation system in Azotobacter vinelandii. J Bacteriol 150:1244–1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Noar JD, Bruno-Bárcena JM. 2013. Complete genome sequences of Azotobacter vinelandii wild-type strain CA and tungsten-tolerant mutant strain CA6. Genome Announc 1:e00313-13. doi: 10.1128/genomeA.00313-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kraepiel AM, Bellenger JP, Wichard T, Morel FM. 2009. Multiple roles of siderophores in free-living nitrogen-fixing bacteria. Biometals 22:573–581. doi: 10.1007/s10534-009-9222-7. [DOI] [PubMed] [Google Scholar]
- 16.Wichard T, Bellenger JP, Morel FM, Kraepiel AM. 2009. Role of the siderophore azotobactin in the bacterial acquisition of nitrogenase metal cofactors. Environ Sci Technol 43:7218–7224. doi: 10.1021/es8037214. [DOI] [PubMed] [Google Scholar]
- 17.Sippel D, Einsle O. 2017. The structure of vanadium nitrogenase reveals an unusual bridging ligand. Nat Chem Biol 13:956–960. doi: 10.1038/nchembio.2428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rupnik K, Hu Y, Lee CC, Wiig JA, Ribbe MW, Hales BJ. 2012. P+ state of nitrogenase P-cluster exhibits electronic structure of a [Fe4S4]+ cluster. J Am Chem Soc 134:13749–13754. doi: 10.1021/ja304077h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Carnahan EM, Protasiewicz JD, Lippard SJ. 1993. The 15 years of reductive coupling: what have we learned? Acc Chem Res 26:90–97. doi: 10.1021/ar00027a003. [DOI] [Google Scholar]
- 20.Spatzal T, Perez KA, Einsle O, Howard JB, Rees DC. 2014. Ligand binding to the FeMo-cofactor: structures of CO-bound and reactivated nitrogenase. Science 345:1620–1623. doi: 10.1126/science.1256679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Spatzal T, Perez KA, Howard JB, Rees DC. 2015. Catalysis-dependent selenium incorporation and migration in the nitrogenase active site iron-molybdenum cofactor. eLife 4:e11620. doi: 10.7554/eLife.11620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fay AW, Blank MA, Lee CC, Hu Y, Hodgson KO, Hedman B, Ribbe MW. 2010. Characterization of isolated nitrogenase FeVco. J Am Chem Soc 132:12612–12618. doi: 10.1021/ja1019657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Moore VG, Tittsworth RC, Hales BJ. 1994. Construction and characterization of hybrid component 1 from V-nitrogenase containing FeMo cofactor. J Am Chem Soc 116:12101–12102. doi: 10.1021/ja00105a080. [DOI] [Google Scholar]
- 24.Burns RC, Fuchsman WH, Hardy RW. 1971. Nitrogenase from vanadium-grown Azotobacter: isolation, characteristics, and mechanistic implications. Biochem Biophys Res Commun 42:353–358. doi: 10.1016/0006-291X(71)90377-9. [DOI] [PubMed] [Google Scholar]
- 25.Hu Y, Corbett MC, Fay AW, Webber JA, Hodgson KO, Hedman B, Ribbe MW. 2006. FeMo cofactor maturation on NifEN. Proc Natl Acad Sci U S A 103:17119–17124. doi: 10.1073/pnas.0602647103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yoshizawa JM, Fay AW, Lee CC, Hu Y, Ribbe MW. 2010. Insertion of heterometals into the NifEN-associated iron-molybdenum cofactor precursor. J Biol Inorg Chem 15:421–428. doi: 10.1007/s00775-009-0614-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Blanchard CZ, Hales BJ. 1996. Isolation of two forms of the nitrogenase VFe protein from Azotobacter vinelandii. Biochemistry 35:472–478. doi: 10.1021/bi951429j. [DOI] [PubMed] [Google Scholar]
- 28.Hu Y, Fay AW, Schmid B, Makar B, Ribbe MW. 2006. Molecular insights into nitrogenase FeMoco insertion: Trp-444 of MoFe protein α-subunit locks FeMoco in its binding site. J Biol Chem 281:30534–30541. doi: 10.1074/jbc.M605527200. [DOI] [PubMed] [Google Scholar]
- 29.Sickerman NS, Tanifuji K, Lee CC, Ohki Y, Tatsumi K, Ribbe MW, Hu Y. 2017. Reduction of C1 substrates to hydrocarbons by the homometallic precursor and synthetic mimic of the nitrogenase cofactor. J Am Chem Soc 139:603–606. doi: 10.1021/jacs.6b11633. [DOI] [PubMed] [Google Scholar]
- 30.Hu Y, Fay AW, Dos Santos PC, Naderi F, Ribbe MW. 2004. Characterization of Azotobacter vinelandii nifZ deletion strains. Indication of stepwise MoFe protein assembly. J Biol Chem 279:54963–54971. doi: 10.1074/jbc.M408983200. [DOI] [PubMed] [Google Scholar]
- 31.Burgess BK, Jacobs DB, Stiefel EI. 1980. Large-scale purification of high activity Azotobacter vinelandii nitrogenase. Biochim Biophys Acta 614:196–209. doi: 10.1016/0005-2744(80)90180-1. [DOI] [PubMed] [Google Scholar]
- 32.Corbin JL. 1984. Liquid chromatographic-fluorescence determination of ammonia from nitrogenase reactions: a 2-min assay. Appl Environ Microbiol 47:1027–1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gavini N, Burgess BK. 1992. FeMo cofactor synthesis by a nifH mutant with altered MgATP reactivity. J Biol Chem 267:21179–21186. [PubMed] [Google Scholar]