

Abstract
Hybridization is a natural or anthropogenic process that can deeply affect the genetic make-up of populations, possibly decreasing individual fitness but sometimes favoring local adaptations. The population of Italian wolves (Canis lupus), after protracted demographic declines and isolation, is currently expanding in anthropic areas, with documented cases of hybridization with stray domestic dogs. However, identifying admixture patterns in deeply introgressed populations is far from trivial. In this study, we used a panel of 170,000 SNPs analyzed with multivariate, Bayesian and local ancestry reconstruction methods to identify hybrids, estimate their ancestry proportions and timing since admixture. Moreover, we carried out preliminary genotype–phenotype association analyses to identify the genetic bases of three phenotypic traits (black coat, white claws, and spur on the hind legs) putative indicators of hybridization. Results showed no sharp subdivisions between nonadmixed wolves and hybrids, indicating that recurrent hybridization and deep introgression might have started mostly at the beginning of the population reexpansion. In hybrids, we identified a number of genomic regions with excess of ancestry in one of the parental populations, and regions with excess or resistance to introgression compared with neutral expectations. The three morphological traits showed significant genotype–phenotype associations, with a single genomic region for black coats and white claws, and with multiple genomic regions for the spur. In all cases the associated haplotypes were likely derived from dogs. In conclusion, we show that the use of multiple genome-wide ancestry reconstructions allows clarifying the admixture dynamics even in highly introgressed populations, and supports their conservation management.
Keywords: admixture history, Canis lupus, genotype–phenotype association, hybridization, introgression
Introduction
Hybridization is a biological process that, through the cross-breeding between individuals from distinct but closely related taxa, or between discrete entities that exchange genes, can deeply affect their genetic make-up, long-term survival and evolution (Parham et al. 2013; Saetre 2013; Gompert and Buerkle 2016). Natural hybridization is no longer viewed as a sporadic and undesirable evolutionary dead-end, but rather as a relatively frequent and potentially creative process (Mallet 2008; Larsen et al. 2010; Bailey et al. 2013; Stern 2013; Rius and Darling 2014). Natural hybrid zones represent hot-spots of genetic diversity, where natural selection can expose novel gene assemblages to the adaptive process, possibly leading to hybrid speciation (Lavrenchenko and Bulatova 2016). Episodic hybridization may introduce genetic variation into isolated populations, contrasting the deleterious consequences of small effective size and inbreeding (genetic rescue; Brennan et al. 2015), and cases of adaptive introgression have been recently documented in diverse animal and plant species (Grossen et al. 2014; Hovick and Whitney 2014; Fulgione et al. 2016). Conversely, anthropogenic hybridization, caused by habitat changes and by the spread of alien or domestic species, is mostly viewed as a risk factor in conservation biology, especially for the deleterious consequences of gene introgression into wild populations through backcrossing (Allendorf et al. 2001; Laikre et al. 2010). Introgression may spread maladaptive variants causing fitness declines and outbreeding depression, swamp genetic diversity, or disrupt epistatic equilibria, and local adaptations, driving local populations, or entire species to extinction (genomic extinctions; Rhymer and Simberloff 1996; Todesco et al. 2016). However, the long-term evolutionary consequences of genetic admixtures remain largely unpredictable and can sometimes be beneficial (Coulson et al. 2011; Hedrick et al. 2016).
Hybridized populations may have complex histories. Hybrids can be generated either by discrete events (e.g., from single migrations waves) or by recurrent gene flow and admixture (e.g., in stable areas of sympatry; Allendorf et al. 2001; Currat et al. 2008; Moorjani et al. 2011; Muhlfeld et al. 2014). Whereas modeling the former case is relatively simple, since the relative proportions of ancestral components will be fairly homogeneous across all the hybrid individuals (Rhymer and Simberloff 1996; Rieseberg et al. 1999; Rosenberg et al. 2010), identifying the patterns and timing of recurrent hybridization events is far more complex (Currat and Excoffier 2011; Twyford and Ennos 2012) and is conditioned by the availability of efficient ancestry-informative markers (AIMs). Moreover, hybridization is expected to affect the phenotypes of admixed individuals, depending on the dominance and quantitative nature of the considered traits (Mallet 2008; Patterson et al. 2010). Therefore, the study of hybridization and introgression in natural populations also provides the opportunity to identify loci associated with phenotypic features, adaptation and selection, which might underlie the genetic bases of fitness in hybrid individuals (Arnold and Martin 2010; Gompert and Buerkle 2012).
Canids (genus Canis) represent interesting examples of hybridizing taxa, sharing related and recently divergent genomes. In Eurasia, wolves (C. lupus) can hybridize in the wild with the closely related golden jackals (C. aureus; Freedman et al. 2014; Moura et al. 2014). In North America, historical wolf x coyote (C. latrans) hybridization originated complex patterns of admixed taxa, such as the red wolf (C. rufus), although their origin and taxonomic status is highly debated (Fredrickson and Hedrick 2006; Koblmuller et al. 2009; VonHoldt et al. 2011; Gese et al. 2015; Rutledge et al. 2015; VonHoldt et al. 2016a, 2016b). Wolves, coyotes and golden jackals can successfully reproduce also with domestic dogs (C. l. familiaris), principally in human-dominated regions where the widespread diffusion of stray dogs threatens the gene pool of several wild canid populations, constituting a serious conservation concern (Stronen and Paquet 2013; Monzón et al. 2014; Randi et al. 2014; Galov et al. 2015).
A particular case-study is offered by the Italian wolf population, which is sharply genetically differentiated from any other wolf population due to protracted isolation south of the Alps and to demographic declines that led it to shrink down to <100 individuals in the 1970s (Zimen and Boitani 1975; Lucchini et al. 2004; Pilot et al. 2014; Randi et al. 2014). Thanks to the increased availability of prey and legal protection, Italian wolves are now recovering (Galaverni et al. 2016), but they are still threatened by heavy poaching (Caniglia et al. 2010; Imbert et al. 2016) and hybridization with stray dogs, as documented by genetic evidences (Lucchini et al. 2004; Caniglia et al. 2013; Randi et al. 2014).
To date, except for a few studies where SNP arrays were applied, but not to assess hybridization patterns in this population (Vonholdt et al. 2011; Stronen and Paquet 2013; Pilot et al. 2014), Italian wolves have been genotyped using small panels of microsatellites (STRs) and uniparental markers (mitochondrial DNA and Y-chromosome sequences), which allow the detection of hybridization only up to 2–3 generations in the past (Vähä and Primmer 2006; Randi 2008), but not to understand the temporal patterns of older hybridization events. In particular, it is unknown whether the Italian wolves prevalently hybridized during or before the bottleneck of the 1970s, or if they recurrently admixed during the reexpansion along the Apennine ridge in the last 40 years. To overcome these limitations, in this study we genotyped 118 wolves, 31 village dogs, and 72 putative genetic and/or phenotypical wolf x dog hybrids, in addition to 456 publicly available dog genotypes, at a panel of 170,000 genome-wide single nucleotide polymorphisms (SNPs) and we exploited the additional information inferred from their haplotype blocks, aiming to identify admixed genotypes older than the first few generations of backcrossing and to estimate their timing of admixture (Lindblad-Toh et al. 2005; Vonholdt et al. 2010; Patterson et al. 2012). Furthermore, we applied local ancestry reconstruction methods to identify regions of the genomes of hybrids that were significantly deviating from random inheritance patterns, likely indicating selective forces locally acting on such regions. Moreover, wolf x dog hybridization may be reflected at the phenotypic level. The causative mutations of some phenotypic variants are well known, such as the one for the black coat color (Candille et al. 2007). This trait is coded by a 3-bp deletion at the β-defensin gene CDB103 that was likely introduced into North American wolves by ancient hybridization with dogs (Anderson et al. 2009). Other traits have also been suggested as potential indicators of hybridization, such as white claws (compared with the usual black or dark-grey wild phenotype) or the spur on the hind legs (a relict fifth finger, also known as dewclaw or preaxial polydactyly; Deng et al. 2015), but their genetic determinants are unknown or uncertain (Ciucci et al. 2003; Caniglia et al. 2013; Randi et al. 2014). Therefore, we used local genome ancestry (Gompert and Buerkle 2012) and genotype–phenotype association procedures (Gorlova et al. 2011) to identify putative dog-derived causal mutations associated with phenotypic variants that might be introgressed into the Italian wolves.
Results
Data Filtering, Sample and Marker Selection
Following genotyping and quality cleaning steps performed in SVS, both per sample and per locus, we retained 213 unrelated samples with high call rate (96.4%) that were successfully genotyped at 156,132 autosomal SNPs (90%, hereafter referred to as the “156k data set”): these samples included 114 wolves, 68 putative hybrids (identified based on previous STR assignments and/or anomalous phenotypic traits; Randi et al. 2014) and 31 village dogs, which were added to 456 dog samples from 30 breeds publicly available from the LUPA project data set, created for the genetic mapping of a number of canine diseases (Lequarré et al. 2011; Vaysse et al. 2011). A subset of 25,061 SNPs (14.5%) was obtained after LD pruning performed in Plink (Purcell et al. 2007) at threshold r2 = 0.1 (the 25 k data set). A smaller set of 19,732 SNPs (11.4%) was obtained after discarding all sites with any missing data (19 k data set).
Assignment and Admixture Analyses
The first component of an exploratory PCA, performed in SVS using the 156 k data set, explained most of the genetic variability (72%; fig. 1) and clearly differentiated dogs from wolves. The putative genetic hybrids were scattered along the first axis between wolves and dogs, much closer to the former except one intermediate sample. Conversely, the putative hybrids identified only by atypical phenotypes were mostly included within the wolf cluster. The second axis, which explained a 10-time smaller portion of the genetic diversity, differentiated the most divergent dog breed, the Dobermanns, followed by Border Terriers and Weimaraners along the additional axes (supplementary fig. S1, Supplementary Material online).
Fig. 1.

PC1 versus PC2 results from an exploratory principal component analysis (PCA) computed in SVS on the 156 k SNP data set and including dogs (yellow dots), putatively nonadmixed wolves (blue squares), black wolves (black squares), genetically admixed wolves based on STR data (pink squares) and wolves with atypical phenotypes (gray squares). The two axes are not to scale, in order to better distinguish individuals along PC2. The shaded rectangle includes a zoom on the wolf/hybrid cluster. The dog cluster in the bottom-right corner of the figure is composed of Dobermann individuals.
Results from Admixture (Alexander et al. 2009), run with the 25k data set to reassign each sample to its population of origin, were concordant with the PCA analysis (fig. 2). At increasing values of K, the cross validation error steadily decreased up to K = 34 (supplementary fig. S2, Supplementary Material online). However, the first main decrease in CV error was observed at K = 2, that clearly separated wolves from dogs. Thus, we retained K = 2 to identify the proportions of admixture in wolf x dog hybrids. All dogs (DOG, n = 487) had an individual assignment value qw < 0.15 (fig. 2). Similarly, based on the distribution of individual values in the reference wolves (fig. 2), we selected a conservative threshold qw ≥ 0.999 to assign genotypes to the reference Italian wolf cluster (WIT, n = 90), and all those with an intermediate assignment value as possible hybrids (HYB, n = 92) in the downstream analyses, independently of phenotypic information. Since at K > 10 some level of substructuring was observed within wolves, we used K = 10 to identify the prevalent dog components in the admixed individuals, which turned out to correspond to German Shepherds (fig. 2). At K = 34, corresponding to the optimal number of genetic clusters (supplementary fig. S2, Supplementary Material online), all breeds clearly separated from one another (fig. 2), and also from wolves, which were split into five main groups (centred around the Northern Apennines, Eastern Coast—Adriatic, Western Coast—Maremma, Central Apennines, Southern Apennines) reflecting a rough geographical substructure along the Apennines. ThreePOP results for the f3 test (Pickrell and Pritchard 2012) confirmed a highly significant admixture between wolves and dogs in the HYB samples (z-score = −72.064). To better estimate the actual admixture proportions in the analyzed hybrids (Falush et al. 2016), we applied a PCA-based admixture deconvolution approach implemented in PCAdmix (Brisbin et al. 2012), which identified 1.2–51.6% dog blocks in the genome of the hybrid individuals. These proportions highly correlated with those estimated in Admixture at K = 2 (R2 = 0.969; P < 0.01), but highlighted larger dog components (supplementary fig. S3, Supplementary Material online).
Fig. 2.

Admixture results from the 25 k SNP data set at K = 2, K = 10, and K = 34. K = 2 clearly separates wolves from dogs, whereas at K = 10 it is possible to identify the prevalent dog component in the admixed wolves, namely from German Shepherds (in blue). At K = 34 all the dog breeds clearly separates from one another and wolves are split into five main groups, reflecting a rough geographical structure of the population along the Apennines (Northern Apennines in yellow, Eastern Coast—Adriatic in blue, Western Coast—Maremma in dark gray, Central Apennines in pink and Southern Apennines in light brown). The intermediate hybrid identified in the PCA is the third individual of the HIT subset. DIT = village dogs (31), BeT = Belgian Tervuren (12), Bgl = Beagle (10), BMD = Bernese Mountain Dog (12), BoC = Border Collie (16), BoT = Border Terrier (25), BrS = Brittany Spaniel (12), CoS = Cocker Spaniel (14), Dac = Dachshund (12), Dob = Doberman Pinscher (25), EBD = English Bulldog (13), Elk = Elkhound (12), ESt = English Setter (12), Eur = Eurasian (12), Finnish Spitz FSp (12), GoS = Gordon Setter (25), Gry = Golden Retriever (11), GRe = Greyhound (14), GSh = German Shepherd (12), GSl = Greenland Sledge Dog (12), IrW = Irish Wolfhound (11), JRT = Jack Russell Terrier (12), LRe = Labrador Retriever (14), NFd = Newfoundland (25), NSD = Nova Scotia Duck Tolling Retriever (23), Rtw = Rottweiler (12), Sci = Schipperke (25), ShP = Shar-Pei (11), StP = Standard Poodle (12), TYo = Yorkshire Terrier (12), Wei = Weimaraner (26), PIT = wolves with atypical phenotypes (10), BIT = black wolves (14), HIT = genetically admixed wolves based on STR data (45), WIT = putatively nonadmixed Italian wolves (118).
Timing of Admixture
The software Alder (Loh et al. 2013), by exploiting information derived from the haplotype structure, based on recombination rates and the extent of LD decay among neighboring loci, identified significant admixture in each generation cohort or pool of cohorts of putative hybrids (generations G1, G2, G3–4, and G5–9 from the present, all with P values < 1.4*10−6; supplementary table S1, Supplementary Material online), although it reported inconsistent decay rates (meaning that one of the parental populations could have been not fully sampled; Loh et al. 2013; supplementary fig. S4, Supplementary Material online). Hybridization was estimated to have occurred 3.5–5.0 generations before sampling and, assuming a wolf generation time of 3 years (Skoglund et al. 2011), this corresponded to four partially overlapping intervals (one per cohort) in which admixture possibly originated, centerd around years 1987, 1995, 1999, and 2000 (fig. 3).
Fig. 3.

Top: timing since the admixture event for each admixed individual, deduced from the empirical distribution of the number of chromosomal switches inferred from PCAdmix in relation to the individual assignment values (proportion of wolf blocks). The expected distributions at increasing generations since admixture are indicated by the colored lines. Bottom: temporal distribution of the admixture events deduced from PCAdmix (vertical bars) compared with the time intervals reconstructed by Alder (horizontal bars) from four generation cohorts or pools of cohorts, with G1 being the last sampled generation (years 2013–2015) and so on, considering a wolf generation time of 3 years (Skoglund et al. 2011). Square, triangle and diamond symbols represent mean values, and vertical sticks represent confidence intervals.
PCAdmix results, that we also used to estimate individual admixture times, indicated that admixture mostly occurred 3–4 generations before sampling, with the oldest events tracing back up to 19 generations before sampling (supplementary fig. S5, Supplementary Material online). Converted into years, this indicated that the hybridization events likely started in the 1940s, followed in the late 1970s, but peaked in the late 1990s (fig. 3), highly concordant with the results from Alder. Although our methods could be expected to be more efficient in tracing more recent hybridization events, the estimated timing of admixture through PCAdmix appeared robust, since estimates from different generation cohorts were highly concordant (supplementary fig. S5, Supplementary Material online). From a geographical point of view, the patterns of admixture showed an initial South-to-North trend, followed by more complex dynamics (fig. 4, supplementary file S1, Supplementary Material online) likely reflecting the range expansion of the species along the Apennines (Fabbri et al. 2007).
Fig. 4.
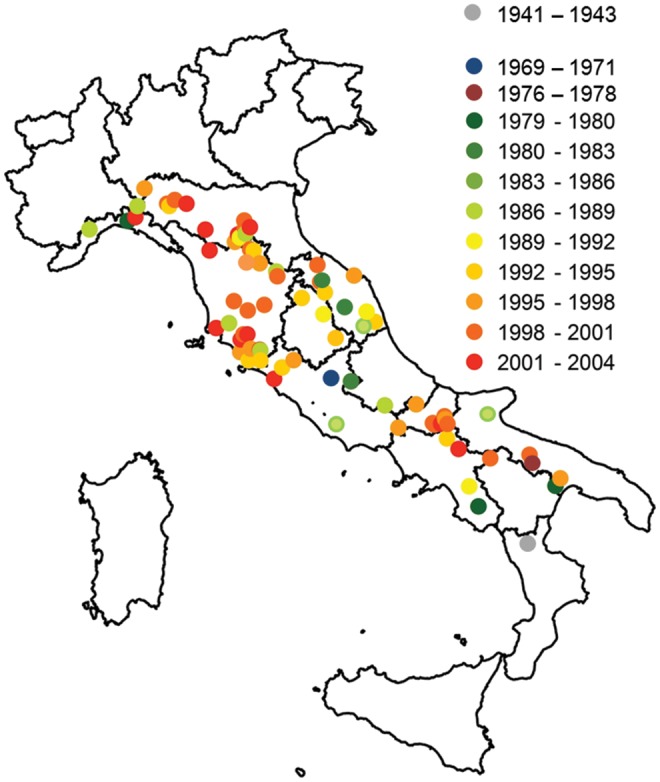
Map visualizing the geographical distribution of the wolf x dog admixture events in Italy through time, as reconstructed from PCAdmix results. Locations are plotted where the admixed individuals have been sampled and cannot fully reflect the potential movements from where the first parental hybrids were sampled. An animated version of the map is available in supplementary file S1, Supplementary Material online.
Deviations from Admixture Neutrality
In order to look at deviations from neutrality in the inheritance patterns of parental haplotypes in hybrids, all blocks analyzed by PCAdmix in the hybrids were ranked according to their relative proportion of “dog” or “wolf” assignment, and labeled as admixture outliers if falling into the top or bottom 1% of the genome-wide frequency distribution. In this way, we identified 73 regions with high frequency of dog alleles in hybrids (mostly mapping to chromosomes chr3, chr10, chr28, and chr32) and 106 blocks with high frequency of wolf alleles (mainly on chr7, chr8, and chr12), likely indicating outlier regions for excess of ancestry from one of the parental populations (fig. 5). The “dog-like” outlier regions included 179 annotated protein-coding genes, significantly enriched for Gene Ontology (GO) categories related to transmembrane transport, and a number of Human Phenotype (HP) categories, the most significant of which were linked to nasal and ear morphology (supplementary file S2, Supplementary Material online). The “wolf-like” outlier regions included 235 protein-coding genes, significantly enriched for immunity-related, metabolic and enzymatic biological processes (BP), and for HP categories related to abnormal ossification (supplementary file S2, Supplementary Material online).
Fig. 5.

Distribution of the introgressed regions along the genomes of admixed individuals. Top: PCAdmix outlier regions. Blue bars indicate regions with excess of wolf-derived alleles, yellow bars indicate excess of dog-derived alleles. Center: BGC (Bayesian Genomic Cline analysis; Gompert and Buerkle 2012) alpha parameter outlier SNPs. Values higher than 0 indicate excess of dog alleles, values lower than 0 indicate excess of wolf alleles. Bottom: outlier SNPs for the BGC beta parameter. Values higher than 0 indicate resistance to introgression, lower than 0 an excess of introgression, compared to random expectations. In both cases, BGC significant outliers are indicated by blue crosses (top or bottom 1% of the empirical distribution of values) and by red dots (95% credibility intervals of 10,000 iterations not including 0).
Local deviations from neutrality were also investigate through a Bayesian Genomic Cline analysis in BGC (Gompert and Buerkle 2012), which identifies regions with excess of ancestry in one of the parental populations compared with neutral expectations (α parameter), as well as those with excess of or resistance to introgression (ẞ parameter). BGC results for the α parameter indicated that 187 SNPs, distributed throughout most of the chromosomes, had an excess of dog ancestry (significantly positive values of α) and 132 SNPs an excess of wolf ancestry (α < 0), with overall higher absolute values in the former (fig. 5). A significant excess of introgression was observed only in six SNPs on chr19 and chr24 (ẞ < 0), whereas nine SNPs on chr17, chr21, chr27, and chr35 showed values of ẞ > 0, indicating a resistance to introgression or steeper genetic clines (fig. 5). The 50-kb regions surrounding the SNPs with excess of dog ancestry contained 210 protein-coding genes that were mostly enriched for a GO biological process related to inflammatory response and a HP category linked to short stature (supplementary file S2, Supplementary Material online). Conversely, the regions with excess of wolf ancestry contained 156 coding genes, enriched for membrane and protein-related cellular components (CC) and for skin-related HP categories (supplementary file S2, Supplementary Material online).
The 50-kb regions surrounding the outlier SNPs for the ẞ parameter included only eight genes with excess of introgression, enriched for GO categories related to telomere processing, and 18 genes with resistance to introgression, enriched for GO categories mainly linked to neural cells functions and glucose transport (supplementary file S2, Supplementary Material online). No SNP was significant for both parameters (supplementary fig. S2, Supplementary Material online), although a region located on chr24 around 46–47 Mb included both SNPs that were outlier for ẞ < 0, and SNPs either significantly positive or negative for α, indicating a possible excess of introgression in this region for both wolf and dog alleles. We found only a partial overlap between the outlier SNPs identified from the BGC α parameter and the blocks of ancestry-excess identified from PCAdmix, namely a region on chr25 for dog excess, and a region on chr27 for wolf excess (supplementary file S2, Supplementary Material online).
Ancestry-Informative Marker Selection
To verify their power to identify wolves, dogs and their hybrids, we selected three panels of the most ancestry-informative markers (192, 96 and 48 SNPs; supplementary file S2, Supplementary Material online) from the 25 k data set (FST = 0.16, HO-dog = 0.2312, HO-wolf = 0.1921) and used them to perform additional PCA analyses. Results were highly concordant and well differentiated dogs and wolves (192 top SNPs: FST = 0.58, HO-dog = 0.3051, HO-wolf = 0.0075; Top 96 SNPs: FST = 0.60, HO-dog = 0.2957, HO-wolf = 0.0087; Top 48 SNPs: FST = 0.61, HO-dog = 0.2843, HO-wolf = 0.0095). Most of the admixed individuals clustered in an intermediate position between the parental groups, although there was still no clear subdivision between some of the hybrids and the wolves (fig. 6). The assignment values from Admixture run at K = 2 on the 192, 96, and 48 SNPs were highly concordant with the 25k data set (R2 ≥ 0.87 in all cases) and all performed better than a set of 39 STRs (which had FST = 0.18, HO-dog = 0.5765, HO-wolf = 0.4582; supplementary fig. S9, Supplementary Material online) commonly used to identify wolf x dog hybrids in Europe (Randi et al. 2014). In particular, when fixing an operative individual assignment threshold of qw = 0.95 for the identification of hybrids (as currently used in the ongoing European LIFE projects for the management of wolf x dog hybrids in Italy), all the top-differentiating SNP sets were able to correctly recognize >85% of the hybrids identified with the 25 k data set. Conversely, although based on different Bayesian algorithms, assignment results from Structure on the 39 STRs identified only 37% of the hybrids detected with the 25 k SNP data set.
Fig. 6.

Principal component analysis (PCA) computed in SVS on the 25 k SNP data set and on the 192, 96, 48 SNPs with the highest wolf-dog FST values. For each data set, PC1 versus PC2 are indicated (axes are not to scale), and the overall FST distribution for the 25 k data set is represented in the histogram. Yellow dots indicate dogs, blue dots nonadmixed wolves, light blue dots admixed wolves (Admixtureqw < 0.999). The power of the top 48 SNPs is comparable to that of the 25 k data set, indicating that they can be used as reliable ancestry-informative-markers (AIMs), although no clear subdivision could be traced between some of the admixed and the nonadmixed wolves.
Genotype–Phenotype Associations
To identify the genetic bases of three atypical phenotypic traits described in the literature as possible indicators of hybridization (Ciucci et al. 2003; Anderson et al. 2009), we performed an exploratory genotype–phenotype association test. We contrasted the genotypes of nine cases showing black coat (BC) phenotypes to 102 wild-type controls. The presence of one or more white claws (WC) was described in 16 cases, whereas 98 controls had all claws with the typical dark grey color. The presence of the spur (SP) on the hind legs was identified in five cases, compared with 108 controls where the spur was absent. The genotype–phenotype analysis revealed a main peak associated to the black coat color, with 90 SNPs above the significance threshold after Bonferroni correction (fig. 7), in an interval of about 12 Mb at the end of chr16 that included the β-defensin gene CBD103, known to be responsible for such trait in North American and Italian wolves (Anderson et al. 2009; Caniglia et al. 2013; Randi et al. 2014). The alleles of the three most significant SNPs (chr16:60391793, chr16:61370693, chr16:61718721; table 1) were perfectly associated to the phenotypes of all the cases, plus another four samples for which the presence of a black coat was not known, but that carried the KB deletion (supplementary table S2, Supplementary Material online). To evaluate whether the regions containing the SNPs associated with the atypical phenotypes had a wolf or dog ancestry, we proceeded in two ways: first, we verified their assignment in PCAdmix for all the cases; second, we concatenated all the significantly associated SNPs and reconstructed their haplotype networks. The network reconstructed from the 90 SNPs significantly associated to the BC showed a single haplotype largely represented in wild-type wolves, whereas the six different haplotypes found in individuals with BC were split into two main groups, both rooted on dog-derived nodes (fig. 8). Concordantly, the significant SNPs fell into 41 PCAdmix blocks where all the BC cases had at least one haplotype of dog ancestry.
Fig. 7.

Genotype–phenotype association results for three phenotypic traits considered as potential indicators of hybridization: black coat (top), white claws (center) and spur on the hind legs (also known as dewclaw; bottom panel). Significance thresholds for the –log10 Chi-Squared P values are indicated, corresponding to a Bonferroni-corrected probability of 0.01. (Photo credits, from top to bottom: Renato Fabbri, Willy Reggioni, and Luigi Molinari).
Table 1.
Summary Statistics of the SNPs Most-Significantly Associated with Three Atypical Phenotypic Traits.
| Trait | Top SNPs | ||
|---|---|---|---|
| Black coat | 16:60391793(A/G) | 16:61370693(A/G) | 16:61718721(A/G) |
| Cases (n = 9) | 0.444 | 0.500 | 0.500 |
| Controls (n = 102) | 0.005 | 0.005 | 0.005 |
| Chi-squared –10 log P | 21.2133 | 21.5568 | 21.5568 |
| Chi-squared Bonferroni P | 8.53*10−17 | 3.86*10−17 | 3.86*10−17 |
| White claws | 20:27360052(A/G) | 20:31224886(A/T) | 20:37232759(A/G) |
| Cases (n = 16) | 0.188 | 0.188 | 0.188 |
| Controls (n = 98) | 0.000 | 0.000 | 0.000 |
| Chi-squared –10 log P | 9.0931 | 9.0931 | 9.0931 |
| Chi-squared Bonferroni P | 0.00011 | 0.00011 | 0.00011 |
| Spur | 11:24498328(A/G) | 11:27397738(A/C) | 11:35263745(A/C) |
| Cases (n = 5) | 0.400 | 0.300 | 0.400 |
| Controls (n = 113) | 0.000 | 0.000 | 0.000 |
| Chi-squared –10 log P | 20.1745 | 15.2735 | 20.1745 |
| Chi-squared Bonferroni P | 9.33*10−16 | 7.43*10−11 | 9.33*10−16 |
Note.—For each SNP (indicated as chr:position), the allele associated to the atypical phenotype is showed in italics, together with its frequency in “cases” and in “controls” and the association values detected by SVS.
Fig. 8.

Haplotype networks reconstructed from the SNPs significantly associated to the considered phenotypic traits. Top-left: black coat. Top-right: white claws. Bottom: spur (only referred to chr11). Node dimensions are proportional to the number of times each haplotype has been observed. Haplotypes found in wolves are indicated in blue, those found in hybrids (Admixtureqw < 0.999) in light blue, those found in dogs in yellow. Dashed ovals around nodes identify haplotypes reconstructed from SNPs significantly associated to each phenotypic trait.
The genotype–phenotype association results for the presence of WC revealed a main, large peak on chr20, with 74 significantly associated SNPs (fig. 7). These SNPs spanned an interval of about 20 Mb, which included 286 annotated genes, some of which known to be related to pigmentation (supplementary file S2, Supplementary Material online). However, none of the significant SNPs was associated to all the 16 cases (table 1), but the 22 most significant SNPs on chr20 (between chr20:27360052 and chr20:37651111) clearly explained six of them (supplementary table S2, Supplementary Material online). Another four cases were associated to a solitary SNP on chr16 (chr16:50237955), although with a lower genome-wide significance (fig. 7). The haplotypes reconstructed from the significant SNPs on chr20 formed a network centered around the haplotype most commonly observed in wild-type wolves (fig. 8). Two haplotypes were associated to the WC phenotype and formed a distinct branch rooted on a dog node, which included two additional admixed individuals for which no phenotypic information was available. In PCAdmix, the blocks that included the significant SNPs on chr20 showed that all the six explained cases had at least one dog-derived haplotype, as for the blocks of the other four cases possibly explained by the single significant SNP on chr16.
Several regions were associated to the presence of the spur, with a main peak of 145 significant SNPs on chr11, and other peaks on a number of other chromosomes, mainly chr7, chr12 and chr28 (fig. 7). In particular, the three most significant SNPs lied on chr11 (chr11:24498328, chr11:35263745, chr11:41596702; fig. 7) and their alleles were perfectly associated to the phenotypes of up to four of the five cases (table 1; supplementary table S2, Supplementary Material online). The 50-kb regions surrounding the significant SNPs included 467 genes (supplementary file S2, Supplementary Material online), enriched for the BP category GO:0048856 (anatomical structure development) and a number of HP categories related to phalangeal development, syndactyly and polydactyly, represented by 11 genes (supplementary file S2, Supplementary Material online). The haplotypes reconstructed from the significant SNPs in the main peak (chr11) formed a network where the four haplotypes found in the cases were rooted between the main wolf node and a wider panel of dog branches (fig. 8), not allowing a plain discrimination of their origin. However, PCAdmix clearly assigned as dog-derived all the blocks that included the SNPs significantly associated to the spur.
Discussion
Anthropogenic hybridization is considered a global threat to biodiversity, especially in human-dominated contexts where the growing diffusion of domestic species might increase the risks of hybridization and introgression in the gene pool of wild parental populations (Allendorf et al. 2001). Our genome-wide assessment of hybridization in the Italian wolf population, as highlighted by both multivariate and assignment procedures, showed that the wolf x dog putative hybrids we analyzed (selected by their low STR assignment or atypical phenotypic traits; Randi et al. 2014) ranged from complete wolf assignments to c. 20% dog-derived genomes. Only a few cases (8% of the hybrids) had a higher dog content and likely fell within the first three generations from the admixture.
This pattern indicates that recurrent admixture events occurred in the past, but their legacy has been mostly diluted through time, as expected in a selectively neutral perspective where the retained dog ancestry should be <1% after seven generations of backcrossing with wolves.
The estimated timing of the hybridization events suggests that most of the hybrids do not trace back to the last bottleneck of the Italian wolf population, but rather to its following reexpansion phase. In fact, even if the first cases were dated in the 1940s and in the 1970s (but not in between), the main peak occurred in the late 1990s, when the population was expanding in most of its current range (Lucchini et al. 2002; Fabbri et al. 2007). However, the frequency of hybridization events seems to decline in the following decade, possibly due to the increasing availability of potential wolf mates and to the stronger social structuring and stability of packs, which can lower the need for dog contributions to reproduction (parallel to what described by Rutledge et al. 2010 between eastern wolves and coyotes). Of course, given that 170 k SNPs offer only a moderately resolved snapshot on the whole Canis genome, we cannot exclude that the legacy of more ancient hybridization events could have been left in the whole population. PCAdmix is indeed agnostic on the number of admixture events that may have occurred as it simply identified the “dog” blocks within the genomes of hybrids. However, when applying Alder or similar dating methods, these models are constrained to pick up either the major admixture event (if a main one occurred), the midpoint (in case of continuous admixture events) or the latest event (if these were punctuated). The future comparison of multiple complete genomes from the Italian and other European populations will be needed to shed more light on this possibility.
From a geographical point of view, results show that the first hybridization events might have occurred in the main population refugia in central and southern Italy, followed by more frequent events in the northern Apennines and finally in human-dominated landscapes along the Tyrrhenian and Adriatic coasts, likely surfing the main population expansion wave around the 1990s. In particular, the Maremma region (western Tuscany) confirmed to be a local hotspot of hybridization, as previously described for the wolf (Caniglia et al. 2013) and other mammals, such as the wildcat (Mattucci et al. 2013) and the roe deer (Mucci et al. 2012). However, mapping the individual sampling locations cannot properly take into account the movements and dispersals of individuals through the generations following the hybridization event.
The prevalent dog contribution to admixture appears to come from German Shepherds, which interestingly represented the most common breed in Italy in the last decades (Ente Nazionale della Cinofilia Italiana, http://www.enci.it, last accessed March 1, 2017) and is also consistent with the presumably higher probability of successful mating of wolves with wolf-sized dogs compared with other breeds.
Ancestry mapping enabled us to identify several regions with significant excess of dog or wolf alleles and with steeper or milder genetic clines in the hybrids, which are discussed into details in supplementary text S1, Supplementary Material online.
The phenotype–genotype association tests on the black coat, used as a control trait, confirmed the ability of our data set to correctly identify the genomic region hosting the causative mutation for such phenotype, although the exclusion of dogs from the data set due to missing phenotypic information might have reduced the power of the test. Interestingly, both the local genome ancestry analysis and the haplotype networks revealed the dog derivation of this region in all the analyzed cases, confirming previous hypotheses derived from North American wolves (Anderson et al. 2009). However, the extension of dog-derived haplotype blocks and their rooting within the network suggest the occurrence of at least two separate events. The most ancient one included seven individuals sampled in the Northern Apennines, only one detected as a hybrid by Admixture and the other six completely assigned to the wolf cluster (qw = 1.00), indicating a genetic legacy not detectable anymore at the genome-wide level. Conversely, the most recent event traced back 3–8 generations and the corresponding haplotypes were mostly detected in hybrids from the Maremma region (0.841 < qw < 0.978), where a high frequency of black-coated canids was recently documented (Caniglia et al. 2013).
Also for the white claws we identified a single highly associated peak on chr20, though relatively large (c. 20 Mb) and not explaining all the cases. This region hosted several genes related to pigmentation, such as MITF (microphthalmia-associated transcription factor), EOGT (EGF domain-specific O-linked N-acetylglucosamine transferase), PRICKLE2 (prickle homolog 2), WNT5A (wingless-type MMTV integration site family member 5A), and GNAI2 (guanine nucleotide binding protein, alpha inhibiting activity). However, a series of successive highly significant SNPs located between 36.92 and 37.65 Mb was found just 70 kb upstream of WTN5A. This gene belongs to the WNT family, whose members encode for secreted glycoproteins with signaling functions (Bachmann et al. 2005) and was described to be targeted by a miRNA (mir-202) highly expressed in the epidermal cells of white-colored alpacas (Tian et al. 2012). However, finding the causal mutations for white-coloring alleles in the claws of hybrid wolves needs further investigations and the role of adjacent genes could also concur to the expression of such trait. In particular, MITF (which is located right at the beginning of the peak) is regulated by the same miRNA of WNT5A (Tian et al. 2012) and is responsible for the major white spotting coloration in Boxer dogs (Karlsson et al. 2007), whereas PRICLKE2 is involved in the planar cell polarity signaling pathway, which controls the differentiation of follicles and thus affects the patterning of the skin and of its keratin annexes (Chen and Chuong 2012). Again, the reconstructed haplotype network and the local ancestry analysis revealed that in all the explained cases the region hosting the significantly associated SNPs was clearly dog-derived, confirming previous suggestions based on morphological bases (Ciucci et al. 2003). Coherently, all the cases explained by the associated variants were detected as possible hybrids (0.781 < qw < 0.993) and their estimated time of admixture traced back 4–9 generations.
A more complex pattern emerged from the analyses of the genetic variants associated with the spur, showing a number of peaks on several chromosomes, none of them able to explain all the cases per se. However, this is not surprising since a number of different genes are known to play a role in the development of polydactyly, both in humans (Biesecker 2011) and in dogs, where it occurs more commonly in large breeds, such as Saint Bernard and Bernese (Galis et al. 2001). The regions hosting the significantly associated SNPs included several of such genes. In particular PITX1 (paired-like homeodomain 1) on chr11 and BMP7 (bone morphogenetic protein 7) on chr24 are known to be implicated in the development of polydactyly in a number of species (Galis et al. 2001; Marcil 2003; Klopocki et al. 2012), similar to LMBR1 (limb development membrane protein 1) on chr16, that Park et al. (2008) demonstrated to be responsible for such trait in a Korean dog breed. However, genes included in other significantly associated regions, such as FGF14 (fibroblast growth factor 14) on chr22 and WNT8B (wingless-type MMTV integration site family member 8A) on chr28 could also play a significant role in the complex development of canine polydactyly (Towle and Breur 2004). Again, although the Network results are not conclusive, from PCAdmix the main associated region appears to be dog-derived in the analyzed individuals, as confirmed by their low assignment values (0.781 < qw < 0.862) and compatible with admixture events occurred from 5 to 9 generations in the past.
Therefore, the three atypical phenotypic traits we focused on seem to have genetic bases and their presence in wild-living canids is likely a signal of introgression from the dog to the wolf gene pool, although several caveats should be considered. First, not all the cases of white claws are explained by the associated genomic variants; therefore their presence cannot be considered an absolute signal of hybridization, but it could be due to environmental factors (e.g., nutrition, diseases, carcass degradation, or loss of the external keratin shell). Second, several individuals carrying a black coat could not be identified as hybrids even by genome-wide assignment procedures carried out with thousands of markers. This indicates that the hybridization event originating such gene flow occurred several generations in the past and most of the dog-derived alleles in such individuals got lost, resulting in almost pure wolf genomes.
Therefore, any classification of hybrids based on sole phenotypic traits for management purposes (e.g., for hybrid removal) would be highly hazardous. Rather, we think that the likely dog ancestry of these traits should be used as a phenotypic marker for a simpler identification of potential hybrids, but a final assessment of their status should be based on careful genetic investigations. To this purpose, the reduced panels of AIMs we identified turned out to be highly concordant with the 25 k SNP data set and appeared to perform better than previously used microsatellites in the identification of hybrids, although such a direct comparison should be treated with caution given the different programs applied to analyze these two types of markers. Therefore, a small panel of highly informative SNPs could be applied to more extensive monitoring plans in areas of supposed or documented hybridization through microfluidic or quantitative PCR techniques (vonHoldt et al. 2013). Such approaches would allow the cost-effective analysis of dozens of samples and markers at a time (e.g., 48 samples × 48 SNPs), even starting from noninvasively collected materials such as faeces (Kraus et al. 2015; Norman and Spong 2015). However, any future management practice for the removal of hybrids should be undertaken considering the serious possibility to incur in both type I (removing nonadmixed wolves erroneously identified as hybrids) and type II errors (not removing hybrid individuals falsely identified as wolves; Allendorf et al. 2001; Randi et al. 2014). Therefore, these problems should be carefully evaluated especially in a population that is undergoing a recent reexpansion but is still threatened by strong poaching and accidental killings, which can weaken the pack structure and further promote hybridization (Rutledge et al. 2012). Moreover, hybrids originated several generations in the past can act as good ecological surrogates of nonadmixed wild-living wolves (Caniglia et al. 2013; Bassi et al. 2017), as documented also for wolf-coyote crosses (Benson et al. 2012), and should not be necessarily removed (VonHoldt et al. 2016a, 2016b; Wayne and Shaffer 2016).
Consequently, management actions should be primarily aimed at reducing the high number of free-ranging dogs within the current wolf distribution, and particularly in their current expansion range (the Alps), as indicated by the latest action plan for the conservation of the wolf in Italy (Boitani et al., in prep.). Secondary, attention should be focused on recent hybrids or hybrid packs, in order to prevent the diffusion of large dog-derived genomic regions and a lower acceptance by local communities (M. Apollonio, personal communication).
In conclusion, the application of multiple genome-wide ancestry reconstruction methods allows to clarify the patterns and dynamics of admixture even in highly introgressed populations, map the genetic bases of phenotypical indicators of hybridization, identify optimal ancestry-informative markers and support appropriate management practices.
Materials and Methods
Sample Selection, DNA Extraction, and SNP Genotyping
We genotyped DNA extracted from blood or muscular tissue samples from 118 wolves, 31 village dogs, and 72 putative wolf x dog hybrids. Wolves were sampled from the whole species’ distribution in Italy from 1992 to 2015. Putative hybrids were previously identified based on genetic evidences (Randi and Lucchini 2002; Caniglia et al. 2013; Randi et al. 2014) and/or on atypical phenotypes such as black coat color, white claws, or spur on the hind legs (Ciucci et al. 2003). Wolves and hybrids were sampled from carcasses and individuals live-trapped for scientific purposes or rescuing operations, under the Italian Ministry of the Environment’s permit. Dog blood samples were collected in local shelters by veterinary personnel, always respecting animal welfare procedures. No animal was hurt nor sacrificed for the purposes of this study. Genomic DNA was extracted using a DNeasy Tissue Kit (Qiagen Inc., Hilden, Germany) according to the manufacturer’s instructions. We quantified double-stranded DNA concentrations by the PicoGreen assay on a Quant-iT fluorometer (TermoFisher Scientific, Vantaa, Finland) and visually evaluated DNA quality by 1% agarose gel electrophoresis, selecting only samples with a minimum of 50 ng DNA (average DNA concentration: 60.63 ± 3.22 ng/µl) and showing no signs of degradation. We used the CanineHD BeadChip microarray (Illumina, Inc., San Diego, California, USA) to genotype the DNA samples at c. 170 k SNPs, following the Infinium HD Ultra Assay protocol and calling genotypes with GenomeStudio (http://www.illumina.com/documents/products/datasheets/datasheet_genomestudio_software.pdf, last accessed March 1, 2017). We then added 456 dogs from 30 breeds that were genotyped with the same microarray in the LUPA project and that were publicly available (Lequarré et al. 2011; Vaysse et al. 2011). Although the use of the CanineHD BeadChip, mostly developed from known dog variants, could introduce a limited ascertainment bias against wolves (VonHoldt et al. 2011), our haplotype-based analyses, although being based on dog recombination maps, are designed to minimize such an effect when detecting dog genomic segments that have introgressed into the wolf population.
Data Filtering
All genotypes were imported into the SNP&Variant Suite 8.0.1 (SVS, Golden Helix Inc., Bozeman, MT) and were checked for the possible presence of pairs of individuals with an identity-by-descent score higher than 0.5 (closely related individuals). Genotypes were filtered to ensure high call rates (> 95%) per sample and per locus (hereafter: quality-pruned data set), after discarding SNPs mapping on chromosomes X and Y. We used Plink 1.07 (Purcell et al. 2007) to further filter out loci in linkage disequilibrium (LD) at threshold r2 = 0.1 (LD-pruned data set), using the –dog option in order to manage the correct number of chromosomes. We then developed an ad hoc Unix pipeline (available upon request) to integrate and automate the subsequent analysis steps.
Assignment and Admixture Tests
We carried out an exploratory principal component analysis (PCA; Novembre and Stephens 2008) for the first five components in SVS to visualize the distribution of samples in the genetic space, using the additive genetic model (Price et al. 2006) on the quality-pruned data set. We then used Admixture 1.23 (Alexander et al. 2009) on the 25 k LD-pruned data set to reassign each sample to its population of origin, assuming K values from 1 to 40. The most likely number of clusters was identified based on the lowest cross-validation error (Alexander et al. 2009) and the results were plotted in R 3.0.2 (www.r-project.org, last accessed March 1, 2017). Introgression fractions assessed with Admixture (K = 2) were then used to select the reference dogs, reference wolves and admixed individuals for all the subsequent analyses (see results). Individual assignment values of hybrids were verified running again the software using the “supervised approach”, which allows to fix the reference populations (Alexander and Lange 2011), and confirmed by PCAdmix (Brisbin et al. 2012; see below), which is more appropriate in evaluating the actual admixture proportions (Falush et al. 2016). We used the f3 test in ThreePop (TreeMix package 1.12; Pickrell and Pritchard 2012) to formally assess the occurrence of admixture in the Italian wolf population, using blocks of 500 SNPs each and considering Z-score values < −3 to be indicative of admixture (Pickrell and Pritchard 2012).
Estimating the Timing of Admixture
We reconstructed chromosomal haplotypes from the quality-pruned data set in Shapeit 2.837 (Delaneau et al. 2012) with standard parameters and based on dog recombination maps derived from Muñoz-Fuentes et al. (2015), which were the best proxy in absence of any wolf-specific map. All coordinates were referred to the canFam2 dog genome, namely the genomic build that was used to originally design the CanineHD BeadChip. We then estimated the average timing of the admixture events using Alder 1.03 (Loh et al. 2013), which exploits information derived from the haplotype structure and the extent of LD decay among neighboring loci. The putative hybrids were grouped into generation cohorts based on their sampling dates. When in a given cohort the number of samples was <4 we pooled adjacent generations, then the software was run independently on each cohort or pool of cohorts. When the admixture was significant (at P values < 0.01), we retained the estimated number of generations since the admixture event. The average date of admixture for each cohort was then calculated as Y = sY – (nG * g), where Y is the inferred year of admixture, sY is the average sampling year of the cohort, and nG is the number of generations from the admixture event calculated by the software, assuming a generation time g = 3 years (Skoglund et al. 2011).
Moreover, we estimated individual admixture times with the PCA-based admixture deconvolution approach implemented in PCAdmix 1.0 (Brisbin et al. 2012), which assigns the most likely ancestry of haplotype blocks identified in hybrid individuals to their parental populations. PCAdmix was run with blocks of 20 consecutive, nonoverlapping SNPs. The average genome-wide proportion of blocks assigned to each reference population was calculated for each sample and compared with the assignment proportions estimated by Admixture. The number of generations since the admixture for each individual was then estimated based on the number of switches from dog to wolf ancestry blocks (or viceversa) using the formula developed by Johnson et al. (2011), modified according to the dog genome length, conditional on the proportion of admixture estimated from PCAdmix. The number of generations from admixture was converted into years since sampling using the same value of 3 years per generation. Summary plots across all samples were then compared with those obtained from Alder. Finally, results for single admixed individuals were visualized in a map to identify possible geographic patterns of admixture through time.
Deviations from Admixture Neutrality
According to the neutral theory, the proportions of local ancestry in the hybrid samples should stochastically fluctuate around the average genome-wide proportion of dog and wolf ancestral components. However, selective pressures acting on each locus or linkage group can result in some genetic regions being introgressed more frequently than others in the admixed genotypes. We exploited recently developed Bayesian statistical approaches (Alexander et al. 2009; Brisbin et al. 2012; Gompert and Buerkle 2012) to identify genomic regions that are significantly more or less frequent than expected by chance in the hybrid individuals identified from Admixture.
First, haplotype blocks identified by PCAdmix were ranked according to their relative proportion of “dog” or “wolf” assignment in the hybrids. Blocks falling into the top and bottom 1% of the genome-wide distribution were labeled as admixture outliers and adjacent top-ranking blocks were joined into single segments. These regions were then compared with those identified in the hybrids with the Bayesian Genomic Cline analysis in BGC (Gompert and Buerkle 2012), which retrieves SNPs with excess of ancestry in one of the parental populations (the reference wolves and the reference dogs identified from Admixture) compared with random expectations (α parameter), as well as those with excess of or resistance to introgression (ẞ parameter). The software was run after filtering for LD and removing all sites with missing genotypes for 10,000 iterations, sampled every fifth one, using the ICARrho model based upon dog recombination maps derived from Muñoz-Fuentes et al. (2015), in absence of any wolf-specific recombination map. Results were gathered with the estpost utility (Gompert and Buerkle 2012) and the adequate number of burn-in iterations were discarded after visual inspection of the log likelihood values. SNPs were retained as significant when the 95% credible intervals of their median α or ẞ values did not include zero and their median value was included in the top or bottom 1% of the genome-wide distribution (Trier et al. 2014).
Ancestry-Informative Marker Selection
Starting from the LD-pruned data set, we selected three panels of the 192, 96, and 48 most ancestry-informative markers (AIMs), based on their values of FST between dogs and nonadmixed wolves (with Admixtureqw ≥ 0.999, see Results) calculated in SVS. These three panels of SNPs were used to rerun the PCA and Admixture analyses and to verify their power to identify wolves, dogs and their hybrids. Finally, their performance was compared with that of 39 microsatellite markers previously used to investigate wolf x dog admixture in Italian and European wolves (Randi et al. 2014), based on their Structureqw at K = 2 (run with admixture model, independent allele frequencies, 40 k burn-ins and 400 k MCMC for four iterations).
Genotype–Phenotype Association Testing and Identification of Trait Ancestry
All individuals showing the spur (SP) and white claws (WC) were ranked as “cases” in an exploratory genotype–phenotype association study, using as “controls” all the wild-type wolves and putative hybrids for which phenotypic information was available. Dogs had to be excluded since no phenotypic information was available for such traits.
We performed the association test in SVS (Gorlova et al. 2011) using the “basic allele model”, in absence of any hypotheses on the dominance of these traits. We then applied a Bonferroni correction to identify significantly associated SNPs (at nominal P < 0.01) and we plotted the values of the –log10 Chi-Squared P for all chromosomes using the GenomeBrowse tool in SVS.
To evaluate whether the mutations associated with the atypical phenotypes had wolf or dog ancestry, the significantly associated SNPs were concatenated and their haplotypes were reconstructed for each chromosomal region with at least ten SNPs in Phase (Stephens and Donnelly 2003) via DnaSP (Librado and Rozas 2009), including the village dog samples for comparison. We then used these haplotypes to build median-joining networks in Network 5.0 (Bandelt et al. 1999). Finally, we used PCAdmix to further evaluate the ancestry of these atypical traits by verifying the assignment of the haplotype blocks that included the significant SNPs to the wolf or dog clusters.
The black coat (BC) phenotype, whose genetic basis and likely dog origin are well known (Candille et al. 2007; Anderson et al. 2009), was used for validation in all these analyses, with known black individuals used as cases and wild-type ones as controls.
Gene Search, Gene Ontology Enrichment, and Functional Networks
Coordinates of the outlier blocks from PCAdmix and the outlier SNPs from BGC, as well as the significant SNPs from the association tests, were converted into the canFam3.1 assembly using the liftover tool (https://genome.ucsc.edu/cgi-bin/hgLiftOver) in order to exploit the more complete gene annotation available for such build. Information for the genes mapping within 50 kb of distance from the significant SNPs (which is approximately half of the distance between two nearby SNPs of the 25 k data set) was retrieved in Ensembl Biomart (http://www.ensembl.org/biomart/martview/, last accessed March 1, 2017) from the Ensembl Genes 84 annotation database. The genes obtained from each analysis were then independently checked for possible enrichment towards specific Gene Ontology (GO) and Human Phenotypes (HP) categories in G-profiler (Reimand et al. 2016), only retaining categories significant at P < 0.05 after Benjamini–Hockberg correction. Possible functional relationships between the identified genes were searched in string v.10 (Szklarczyk et al. 2015), including all the available types of evidences.
Supplementary Material
Supplementary data are available at Molecular Biology and Evolution online.
Supplementary Material
Acknowledgments
We warmly thank all the people who contributed to the collection of samples, in particular D. Bigi and M. Delogu (University of Bologna), W. Reggioni, L. Molinari and M. Canestrini (Wolf Apennine Center), and E. Berti (CRAS Monte Adone). We specially thank P. Milanesi (ISPRA) for providing the geographical maps of the hybrids, L. Montana (University of Sherbrooke) for the assistance in blood sample extraction and D. Lawson (University of Bristol) for a preliminary exploration of data. We also thank M. Scandura and M. Apollonio (University of Sassari) for the useful discussions. We are indebted with the Associate Editor and two anonymous referees for their constructive and useful comments that deeply improved the manuscript.
References
- Alexander DH, Lange K.. 2011. Enhancements to the Admixture algorithm for individual ancestry estimation. BMC Bioinformatics 12:246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander DH, Novembre J, Lange K.. 2009. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 19:1655–1664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allendorf FW, Leary RF, Spruell P, Wenburg JK.. 2001. The problems with hybrids : setting conservation guidelines. Trends Ecol Evol. 16:613–622. [Google Scholar]
- Anderson TM, VonHoldt BM, Candille SI, Musiani M, Greco C, Stahler DR, Smith DW, Padhukasahasram B, Randi E, Leonard JA, et al. 2009. Molecular and evolutionary history of melanism in North American gray wolves. Science 323:1339–1343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold ML, Martin NH.. 2010. Hybrid fitness across time and habitats. Trends Ecol Evol. 25:530–536. [DOI] [PubMed] [Google Scholar]
- Bachmann IM, Straume O, Puntervoll HE, Kalvenes MB, Akslen LA.. 2005. Importance of P-cadherin, β-catenin, and Wnt5a/Frizzled for progression of melanocytic tumors and prognosis in cutaneous melanoma. Clin Cancer Res. 11:8606–8614. [DOI] [PubMed] [Google Scholar]
- Bailey RI, Eroukhmanoff F, Sætre PG.. 2013. Hybridization and genome evolution II: mechanisms of species divergence and their effects on evolution in hybrids. Curr Zool. 59:675–685. [Google Scholar]
- Bandelt HJ, Forster P, Röhl A.. 1999. Median-joining networks for inferring intraspecific phylogenies. Mol Biol Evol. 16:37–48. [DOI] [PubMed] [Google Scholar]
- Bassi E, Canu A, Firmo I, Mattioli L, Scandura M, Apollonio M.. 2017. Trophic overlap between wolves and free-ranging wolf × dog hybrids in the Apennine Mountains, Italy. Global Ecol Conserv. 9:39–49. [Google Scholar]
- Benson JF, Patterson BR, Wheeldon TJ.. 2012. Spatial genetic and morphologic structure of wolves and coyotes in relation to environmental heterogeneity in a Canis hybrid zone. Mol Ecol. 21:5934–5954. [DOI] [PubMed] [Google Scholar]
- Biesecker LG. 2011. Polydactyly: how many disorders and how many genes? 2010 update. Dev Dyn. 240:931–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennan AC, Woodward G, Seehausen O, Munoz-Fuentes V, Moritz C, Guelmami A, Abbott RJ, Edelaar P.. 2015. Hybridization due to changing species distributions: adding problems or solutions to conservation of biodiversity during global change?. Evol Ecol Res. 16:475–491. [Google Scholar]
- Brisbin A, Bryc K, Byrnes J, Zakharia F, Omberg L, Degenhardt J, Reynolds A, Ostrer H, Mezey JG, Bustamante CD.. 2012. PCAdmix: principal components-based assignment of ancestry along each chromosome in individuals with admixed ancestry from two or more populations. Hum Biol. 84:343–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Candille SI, Kaelin CB, Cattanach BM, Yu B, Thompson D. a, Nix M. a, Kerns J. a, Schmutz SM, Millhauser GL, Barsh GS.. 2007. A b-defensin mutation causes black coat color in domestic dogs. Science 318:1418–1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caniglia R, Fabbri E, Greco C, Galaverni M, Manghi L, Boitani L, Sforzi A, Randi E.. 2013. Black coats in an admixed wolf × dog pack is melanism an indicator of hybridization in wolves?. Eur J Wildl Res. 59:543–555. [Google Scholar]
- Caniglia R, Fabbri E, Greco C, Galaverni M, Randi E.. 2010. Forensic DNA against wildlife poaching: identification of a serial wolf killing in Italy. Forensic Sci Int Genet. 4:334–338. [DOI] [PubMed] [Google Scholar]
- Chen J, Chuong CM.. 2012. Patterning skin by planar cell polarity: the multi-talented hair designer. Exp Dermatol. 21:81–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciucci P, Lucchini V, Boitani L, Randi E.. 2003. Dewclaws in wolves as evidence of admixed ancestry with dogs. Can J Zool. 81:2077–2081. [Google Scholar]
- Coulson T, MacNulty DR, Stahler DR, vonHoldt B, Wayne RK, Smith DW.. 2011. Modeling effects of environmental change on wolf population dynamics, trait evolution, and life history. Science 334:1275–1278. [DOI] [PubMed] [Google Scholar]
- Currat M, Excoffier L.. 2011. Strong reproductive isolation between humans and Neanderthals inferred from observed patterns of introgression. Proc Natl Acad Sci U S A. 108:15129–15134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Currat M, Ruedi M, Petit RJ, Excoffier L.. 2008. The hidden side of invasions: massive introgression by local genes. Evolution 62:1908–1920. [DOI] [PubMed] [Google Scholar]
- Delaneau O, Marchini J, Zagury J-F.. 2012. A linear complexity phasing method for thousands of genomes. Nat Methods 9:179–181. [DOI] [PubMed] [Google Scholar]
- Deng H, Tan T, Yuan L.. 2015. Advances in the molecular genetics of non-syndromic polydactyly. Expert Rev Mol Med. 17:e18. [DOI] [PubMed] [Google Scholar]
- Fabbri E, Miquel C, Lucchini V, Santini A, Caniglia R, Duchamp C, Weber JM, Lequette B, Marucco F, Boitani L, et al. 2007. From the Apennines to the Alps: colonization genetics of the naturally expanding Italian wolf (Canis lupus) population. Mol Ecol. 16:1661–1671. [DOI] [PubMed] [Google Scholar]
- Falush D, van Dorp L, Lawson D.. 2016. A tutorial on how (not) to over-interpret Structure/Admixture bar plots. BioRxiv 066431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fredrickson RJ, Hedrick PW.. 2006. Dynamics of hybridization and introgression in red wolves and coyotes. Conserv Biol. 20:1272–1283. [DOI] [PubMed] [Google Scholar]
- Freedman AH, Gronau I, Schweizer RM, Ortega-Del Vecchyo D, Han E, Silva PM, Galaverni M, Fan Z, Marx P, Lorente-Galdos B, et al. 2014. Genome sequencing highlights the dynamic early history of dogs. PLoS Genet. 10:e1004016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fulgione D, Rippa D, Buglione M, Trapanese M, Petrelli S, Maselli V.. 2016. Unexpected but welcome. Artificially selected traits may increase fitness in wild boar. Evol Appl. 9:769–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galaverni M, Caniglia R, Fabbri E, Milanesi P, Randi E.. 2016. One, no one, or one hundred thousand: how many wolves are there currently in Italy?. Mammal Res. 61:13–24. [Google Scholar]
- Galis F, van Alphen JJM, Metz JAJ.. 2001. Why five fingers? Evolutionary constraints on digit numbers. Trends Ecol Evol. 16:637–646. [Google Scholar]
- Galov A, Fabbri E, Caniglia R, Arbanasić H, Lapalombella S, Florijančić T, Bošković I, Galaverni M, Randi E.. 2015. First evidence of hybridization between golden jackal (Canis aureus) and domestic dog (Canis familiaris) as revealed by genetic markers. R Soc Open Sci. 2:150450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gese E, Knowlton F, Adams J, Beck K.. 2015. Managing hybridization of a recovering endangered species: the red wolf Canis rufus as a case study. Curr Zool. 61:191–205. [Google Scholar]
- Gompert Z, Buerkle CA.. 2012. bgc: software for Bayesian estimation of genomic clines. Mol Ecol Resour. 12:1168–1176. [DOI] [PubMed] [Google Scholar]
- Gompert Z, Buerkle CA.. 2016. What, if anything, are hybrids: enduring truths and challenges associated with population structure and gene flow. Evol Appl. 9:909–923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorlova O, Martin J-E, Rueda B, Koeleman BPC, Ying J, Teruel M, Diaz-Gallo L-M, Broen JC, Vonk MC, Simeon CP, et al. 2011. Identification of novel genetic markers associated with clinical phenotypes of systemic sclerosis through a genome-wide association strategy. PLoS Genet. 7:e1002178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grossen C, Keller L, Biebach I, Zhang W, Tosser-Klopp G, Ajmone P, Amills M, Boitard S, Chen W, Cheng S, et al. 2014. Introgression from domestic goat generated variation at the major histocompatibility complex of Alpine ibex. PLoS Genet. 10:d459–d467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedrick PW, Smith DW, Stahler DR.. 2016. Negative-assortative mating for color in wolves. Evolution 70:757–766. [DOI] [PubMed] [Google Scholar]
- Hovick SM, Whitney KD.. 2014. Hybridisation is associated with increased fecundity and size in invasive taxa: Meta-analytic support for the hybridisation-invasion hypothesis. Ecol Lett. 17:1464–1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imbert C, Caniglia R, Fabbri E, Milanesi P, Randi E, Serafini M, Torretta E, Meriggi A.. 2016. Why do wolves eat livestock?. Biol Conserv. 195:156–168. [Google Scholar]
- Johnson NA, Coram MA, Shriver MD, Romieu I, Barsh GS, London SJ, Tang H.. 2011. Ancestral components of admixed genomes in a Mexican cohort. PLoS Genet. 7:e1002410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlsson EK, Baranowska I, Wade CM, Salmon Hillbertz NHC, Zody MC, Anderson N, Biagi TM, Patterson N, Pielberg GR, Kulbokas EJ, et al. 2007. Efficient mapping of mendelian traits in dogs through genome-wide association. Nat Genet. 39:1321–1328. [DOI] [PubMed] [Google Scholar]
- Klopocki E, Kähler C, Foulds N, Shah H, Joseph B, Vogel H, Lüttgen S, Bald R, Besoke R, Held K, et al. 2012. Deletions in PITX1 cause a spectrum of lower-limb malformations including mirror-image polydactyly. Eur J Hum Genet. 20:705–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koblmuller S, Nord M, Wayne RK, Leonard JA.. 2009. Origin and status of the Great Lakes wolf. Mol Ecol. 18:2313–2326. [DOI] [PubMed] [Google Scholar]
- Kraus RHS, VonHoldt B, Cocchiararo B, Harms V, Bayerl H, Kuhn R, Forster DW, Fickel J, Roos C, Nowak C.. 2015. A single-nucleotide polymorphism-based approach for rapid and cost-effective genetic wolf monitoring in Europe based on noninvasively collected samples. Mol Ecol Resour. 15:295–305. [DOI] [PubMed] [Google Scholar]
- Laikre L, Schwartz MK, Waples RS, Ryman N.. 2010. Compromising genetic diversity in the wild: Unmonitored large-scale release of plants and animals. Trends Ecol Evol. 25:520–529. [DOI] [PubMed] [Google Scholar]
- Larsen PA, Marchán-Rivadeneira MR, Baker RJ.. 2010. Natural hybridization generates mammalian lineage with species characteristics. Proc Natl Acad Sci U S A. 107:11447–11452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavrenchenko LA, Bulatova NS.. 2016. The role of hybrid zones in speciation: a case study on chromosome races of the house mouse Mus domesticus and common shrew Sorex araneus. Biol Bull Rev. 6:232–244. [PubMed] [Google Scholar]
- Lequarré A-S, Andersson L, André C, Fredholm M, Hitte C, Leeb T, Lohi H, Lindblad-Toh K, Georges M.. 2011. LUPA: a European initiative taking advantage of the canine genome architecture for unravelling complex disorders in both human and dogs. Vet J. 189:155–159. [DOI] [PubMed] [Google Scholar]
- Librado P, Rozas J.. 2009. DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25:1451–1452. [DOI] [PubMed] [Google Scholar]
- Lindblad-Toh K, Wade CM, Mikkelsen TS, Karlsson EK, Jaffe DB, Kamal M, Clamp M, Chang JL, Kulbokas EJ, Zody MC, et al. 2005. Genome sequence, comparative analysis and haplotype structure of the domestic dog. Nature 438:803–819. [DOI] [PubMed] [Google Scholar]
- Loh PR, Lipson M, Patterson N, Moorjani P, Pickrell JK, Reich D, Berger B.. 2013. Inferring admixture histories of human populations using linkage disequilibrium. Genetics 193:1233–1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucchini V, Fabbri E, Marucco F, Ricci S, Boitani L, Randi E.. 2002. Noninvasive molecular tracking of colonizing wolf (Canis lupus) packs in the western Italian Alps. Mol Ecol. 11:857–868. [DOI] [PubMed] [Google Scholar]
- Lucchini V, Galov A, Randi E.. 2004. Evidence of genetic distinction and long-term population decline in wolves (Canis lupus) in the Italian Apennines. Mol Ecol. 13:523–536. [DOI] [PubMed] [Google Scholar]
- Mallet J. 2008. Hybridization, ecological races and the nature of species: empirical evidence for the ease of speciation. Philos Trans R Soc B Biol Sci. 363:2971–2986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcil A. 2003. Pitx1 and Pitx2 are required for development of hindlimb buds. Development 130:45–55. [DOI] [PubMed] [Google Scholar]
- Mattucci F, Oliveira R, Bizzarri L, Vercillo F, Anile S, Ragni B, Lapini L, Sforzi A, Alves PC, Lyons LA, et al. 2013. Genetic structure of wildcat (Felis silvestris) populations in Italy. Ecol Evol. 3:2443–2458. [Google Scholar]
- Monzón J, Kays R, Dykhuizen DE.. 2014. Assessment of coyote-wolf-dog admixture using ancestry-informative diagnostic SNPs. Mol Ecol. 23:182–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moorjani P, Patterson N, Hirschhorn JN, Keinan A, Hao L, Atzmon G, Burns E, Ostrer H, Price AL, Reich D.. 2011. The history of African gene flow into Southern Europeans, Levantines, and Jews. PLoS Genet. 7:e1001373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moura AE, Tsingarska E, Dąbrowski MJ, Czarnomska SD, Jędrzejewska B, Pilot M.. 2014. Unregulated hunting and genetic recovery from a severe population decline: the cautionary case of Bulgarian wolves. Conserv Genet. 15:405–417. [Google Scholar]
- Mucci N, Mattucci F, Randi E.. 2012. Conservation of threatened local gene pools: landscape genetics of the Italian roe deer (Capreolus c. italicus) populations. Evol Ecol Res. 14:897–920. [Google Scholar]
- Muhlfeld CC, Kovach RP, Jones LA, Al-Chokhachy R, Boyer MC, Leary RF, Lowe WH, Luikart G, Allendorf FW.. 2014. Invasive hybridization in a threatened species is accelerated by climate change. Nat Clim Change 4:620–624. [Google Scholar]
- Muñoz-Fuentes V, Marcet-Ortega M, Alkorta-Aranburu G, Linde Forsberg C, Morrell JM, Manzano-Piedras E, Söderberg A, Daniel K, Villalba A, Toth A, et al. 2015. Strong artificial selection in domestic mammals did not result in an increased recombination rate. Mol Biol Evol. 32:510–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norman AJ, Spong G.. 2015. Single nucleotide polymorphism-based dispersal estimates using noninvasive sampling. Ecol Evol. 5:3056–3065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novembre J, Stephens M.. 2008. Interpreting principal component analyses of spatial population genetic variation. Nat Genet. 40:646–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parham JF, Papenfuss TJ, Dijk PP, van Wilson BS, Marte C, Schettino LR, Brian Simison W.. 2013. Genetic introgression and hybridization in Antillean freshwater turtles (Trachemys) revealed by coalescent analyses of mitochondrial and cloned nuclear markers. Mol Phylogenet Evol. 67:176–187. [DOI] [PubMed] [Google Scholar]
- Park K, Kang J, Subedi KP, Ha J-H, Park C.. 2008. Canine polydactyl mutations with heterogeneous origin in the conserved intronic sequence of LMBR1. Genetics 179:2163–2172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson N, Moorjani P, Luo Y, Mallick S, Rohland N, Zhan Y, Genschoreck T, Webster T, Reich D.. 2012. Ancient admixture in human history. Genetics 192:1065–1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson N, Petersen DC, Ross RE. v d, Sudoyo H, Glashoff RH, Marzuki S, Reich D, Hayes VM.. 2010. Genetic structure of a unique admixed population: implications for medical research. Hum Mol Genet. 19:411–419. [DOI] [PubMed] [Google Scholar]
- Pickrell JK, Pritchard JK.. 2012. Inference of population splits and mixtures from genome-wide allele frequency data. PLoS Genet. 8:e1002967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pilot M, Greco C, Vonholdt BM, Jędrzejewska B, Randi E, Jędrzejewski W, Sidorovich VE, Ostrander E. a, Wayne RK.. 2014. Genome-wide signatures of population bottlenecks and diversifying selection in European wolves. Heredity 112:428–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price AL, Patterson NJ, Plenge RM, Weinblatt ME, Shadick NA, Reich D.. 2006. Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet. 38:904–909. [DOI] [PubMed] [Google Scholar]
- Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MAR, Bender D, Maller J, Sklar P, de Bakker PIW, Daly MJ, et al. 2007. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 81:559–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Randi E. 2008. Detecting hybridization between wild species and their domesticated relatives. Mol Ecol. 17:285–293. [DOI] [PubMed] [Google Scholar]
- Randi E, Hulva P, Fabbri E, Galaverni M, Galov A, Kusak J, Bigi D, Bolfíková BČ, Smetanová M, Caniglia R.. 2014. Multilocus detection of wolf x dog hybridization in italy, and guidelines for marker selection. PLoS One 9:e86409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Randi E, Lucchini V.. 2002. Detecting rare introgression of domestic dog genes into wild wolf (Canis lupus) populations by Bayesian admixture analyses of microsatellite variation. Conserv Genet. 3:31–45. [Google Scholar]
- Reimand J, Arak T, Adler P, Kolberg L, Reisberg S, Peterson H, Vilo J.. 2016. g:Profiler—a web server for functional interpretation of gene lists (2016 update). Nucleic Acids Res. 44:W83–W89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhymer JM, Simberloff D.. 1996. Extinction By Hybridization and Introgression. Annu Rev Ecol Syst. 27:83–109. [Google Scholar]
- Rieseberg LH, Whitton J, Gardner K, Arnold ML, Barton NH, Hewitt GM, Barton NH, Hewitt GM, Bishop YMM, Fienberg SE, et al. 1999. Hybrid zones and the genetic architecture of a barrier to gene flow between two sunflower species. Genetics 152:367–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rius M, Darling JA.. 2014. How important is intraspecific genetic admixture to the success of colonising populations?. Trends Ecol Evol. 29:233–242. [DOI] [PubMed] [Google Scholar]
- Rosenberg NA, Huang L, Jewett EM, Szpiech ZA, Jankovic I, Boehnke M.. 2010. Genome-wide association studies in diverse populations. Nat Rev Genet. 11:356–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutledge LY, Devillard S, Boone JQ, Hohenlohe PA, White BN, Drive EB, Canada KJ, Biome LD.. 2015. RAD sequencing and genomic simulations resolve hybrid origins within North American Canis. Biol Lett. 11:20150303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutledge LY, Patterson BR, Mills KJ, Loveless KM, Murray DL, White BN.. 2010. Protection from harvesting restores the natural social structure of eastern wolf packs. Biol Conserv. 143:332–339. [Google Scholar]
- Rutledge LY, White BN, Row JR, Patterson BR.. 2012. Intense harvesting of eastern wolves facilitated hybridization with coyotes. Ecol Evol. 2:19–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saetre G-P. 2013. Hybridization is important in evolution, but is speciation?. J Evol Biol. 26:256–258. [DOI] [PubMed] [Google Scholar]
- Skoglund P, Gotherstrom A, Jakobsson M.. 2011. Estimation of population divergence times from non-overlapping genomic sequences: examples from dogs and wolves. Mol Biol Evol. 28:1505–1517. [DOI] [PubMed] [Google Scholar]
- Stephens M, Donnelly P.. 2003. A comparison of bayesian methods for haplotype reconstruction from population genotype data. Am J Hum Genet. 73:1162–1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stern DL. 2013. The genetic causes of convergent evolution. Nat Rev Genet. 14:751–764. [DOI] [PubMed] [Google Scholar]
- Stronen AV, Paquet PC.. 2013. Perspectives on the conservation of wild hybrids. Biol Conserv. 167:390–395. [Google Scholar]
- Szklarczyk D, Franceschini A, Wyder S, Forslund K, Heller D, Huerta-Cepas J, Simonovic M, Roth A, Santos A, Tsafou KP, et al. 2015. STRING v10: protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 43:D447–D452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian X, Jiang J, Fan R, Wang H, Meng X, He X, He J, Li H, Geng J, Yu X, et al. 2012. Identification and characterization of microRNAs in white and brown alpaca skin. BMC Genomics 13:555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todesco M, Pascual MA, Owens GL, Ostevik KL, Moyers BT, Hübner S, Heredia SM, Hahn MA, Caseys C, Bock DG, et al. 2016. Hybridization and extinction. Evol Appl. 9:892–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Towle HAM, Breur GJ.. 2004. Dysostoses of the canine and feline appendicular skeleton. J Am Vet Med Assoc. 225:1685–1692. [DOI] [PubMed] [Google Scholar]
- Trier CN, Hermansen JS, Sætre GP, Bailey RI.. 2014. Evidence for Mito-Nuclear and Sex-Linked Reproductive Barriers between the Hybrid Italian Sparrow and Its Parent Species. PLoS Genet. 10:e1004075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Twyford AD, Ennos RA.. 2012. Next-generation hybridization and introgression. Heredity 108:179–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vähä J-P, Primmer CR.. 2006. Efficiency of model-based Bayesian methods for detecting hybrid individuals under different hybridization scenarios and with different numbers of loci. Mol Ecol. 15:63–72. [DOI] [PubMed] [Google Scholar]
- Vaysse A, Ratnakumar A, Derrien T, Axelsson E, Rosengren Pielberg G, Sigurdsson S, Fall T, Seppälä EH, Hansen MST, Lawley CT, et al. 2011. Identification of genomic regions associated with phenotypic variation between dog breeds using selection mapping. PLoS Genet. 7:e1002316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VonHoldt BM, Cahill JA, Fan Z, Gronau I, Robinson J, Pollinger JP, Shapiro B, Wall J, Wayne RK.. 2016a. Whole-genome sequence analysis shows that two endemic species of North American wolf are admixtures of the coyote and gray wolf. Sci Adv. 2:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VonHoldt BM, Kays R, Pollinger JP, Wayne RK.. 2016b. Admixture mapping identifies introgressed genomic regions in North American canids. Mol Ecol. 25:2443–2453. [DOI] [PubMed] [Google Scholar]
- VonHoldt BM, Pollinger JP, Earl DA, Knowles JC, Boyko AR, Parker H, Geffen E, Pilot M, Jedrzejewski W, Jedrzejewska B, et al. 2011. A genome-wide perspective on the evolutionary history of enigmatic wolf-like canids. Genome Res. 21:1294–1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- vonHoldt BM, Pollinger JP, Earl DA, Parker HG, Ostrander EA, Wayne RK.. 2013. Identification of recent hybridization between gray wolves and domesticated dogs by SNP genotyping. Mamm Genome 24:80–88. [DOI] [PubMed] [Google Scholar]
- Vonholdt BM, Pollinger JP, Lohmueller KE, Han E, Parker HG, Quignon P, Degenhardt JD, Boyko AR, Earl DA, Auton A, et al. 2010. Genome-wide SNP and haplotype analyses reveal a rich history underlying dog domestication. Nature 464:898–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wayne RK, Shaffer HB.. 2016. Hybridization and endangered species protection in the molecular era. Mol Ecol. 81:778–793. [DOI] [PubMed] [Google Scholar]
- Zimen E, Boitani L.. 1975. Number and distribution of wolf in Italy. Zeitschrift Fur Saugetierkunde 40:102–112. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.