

Abstract
About 1–6% of the genetic ancestry of modern humans today originates from admixture with archaic humans. It has recently been shown that autosomal genomic regions with a reduced proportion of Neanderthal and Denisovan ancestries (NA and DA) are significantly enriched in genes that are more expressed in testis than in other tissues. To determine whether a cellular segregation pattern would exist, we combined maps of archaic introgression with a cross-analysis of three transcriptomic datasets deciphering the transcriptional landscape of human gonadal cell types. We reveal that the regions deficient in both NA and DA contain a significant enrichment of genes transcribed in meiotic germ cells. The interbreeding of anatomically modern humans with archaic humans may have introduced archaic-derived alleles that contributed to genetic incompatibilities affecting meiosis that were subsequently purged by natural selection.
Keywords: archaic hominin admixture, testis, germ cells, meiosis, genetic incompatibilities
Introduction
Anatomically modern and archaic humans interbred between 40 and 60-thousand years ago during the modern human expansion into Eurasia (Fu et al. 2014; Sankararaman et al. 2014, 2016). About 1–3% of the autosomal genome of non-Africans today originates from admixture with Neanderthals (Green et al. 2010; Sankararaman et al. 2014; Juric et al. 2016), and 3–6% of the autosomal genome of Oceanians originates from admixture with Denisovans (Reich et al. 2010, 2011). However, the proportions of Neanderthal ancestry (NA) and Denisovan ancestry (DA) have been shown to vary across the genomes of present-day non-Africans (Sankararaman et al. 2014, 2016; Vernot and Akey 2014). Two studies notably compared the transcriptomes derived from sixteen adult human tissues (Derrien et al. 2012) with archaic introgression maps that estimate the proportions of NA and DA along the genome (Sankararaman et al. 2014, 2016). The authors observed that the X chromosome has lower levels of NA and DA than autosomes. They also found that autosomal regions of massively reduced NA and DA harbor a disproportionately high number of testis-specific genes—that is, genes that are more highly expressed in the testes than in other tissue types—and hypothesized that natural selection to remove Neanderthal and Denisovan alleles that decreased male fertility in hybrids was driving these patterns (Sankararaman et al. 2014, 2016). The transcriptome analyzed in these studies however was based on the sum of all testicular cellular types. Yet, the complex cellular architecture of the testis (Fawcett 1975) is paralleled by a particular complexity in transcriptomes across the different testicular cell populations (Chalmel et al. 2007, 2012; Gan et al. 2013; Soumillon et al. 2013).
Results
To overcome this limitation, we first interrogated our own published dataset based on human testicular biopsy samples from patients with spermatogenesis arrested at different stages of germ cell development (Chalmel et al. 2012). This subtractive transcriptomic approach allowed us to deduce the gene expression program of the different testicular cell populations. Thirteen expression clusters (termed C1–C13) were identified using the Partitioning Around Medoids (PAM) algorithm (see supplementary table S1, Supplementary Material online). Clusters C1–C7 were associated with genes that show the strongest expression signals in somatic testicular cells whereas those in clusters C8–C13 were consistent with elevated expression levels in germ cells: from the mitotic spermatogonia (C8), through the meiotic spermatocytes (C10), up to the postmeiotic haploid spermatids (C13) that ultimately metamorphose into spermatozoa. We compared these expression clusters to the Neanderthal introgression map (Sankararaman et al. 2014) and found that autosomal genes with low NA were significantly associated with meiotic spermatocytes (C10: Fisher’s exact test P = 1.7e-3 for European individuals, P = 8.5e-5 for east-Asian individuals; hypergeometric test) and, to a lesser extent, with the transient expression clusters between the meiotic and postmeiotic phases (C11: P = 0.026 for Europeans, P = 0.024 for east-Asians; C12: P = 0.236 for Europeans, P = 0.027 for east-Asians; fig. 1A, see supplementary Extended data table S1 and Supplementary information, Supplementary Material online). Note that there was no significant association either with the mitotic phase of spermatogenesis (C8: P = 0.299 for Europeans, P = 0.227 for east-Asians) or with the postmeiotic phase (C13: P = 0.766 for Europeans, P = 0.707 for east-Asians). Despite weaker P-values likely resulting from lower power to infer DA and lower sample size of Oceanian individuals (Sankararaman et al. 2016), we also found the same tendency for DA in Oceanians (C10, P = 0.05; C8, P = 0.886; C13, P ≥ 0.999; fig. 1A, see supplementary Extended data table S1 and supplementary information, Supplementary Material online).
Fig. 1.

Enrichment of meiotic expression genes in regions deficient in Neanderthal and Denisovan ancestries using the Fisher’s exact probability (Gaussian hypergeometric test). (A) Chalmel et al. defined a set of 13 expression clusters (termed C1–13) associated with peak expression in the different testicular cell types (top row) (Chalmel et al. 2012). Clusters C1–C7 correspond to genes expressed in somatic testicular cells including prepubertal somatic cells (C1), steroidogenic Leydig cells (LC, C2–4), and Sertoli cells (SC, C5–7). Clusters C8–C13 are gradually associated with loci preferentially expressed in germ cells: the mitotic spermatogonia (C8 and C9), meiotic spermatocytes (C10), and postmeiotic haploid spermatids (C13). (B and C) Analysis of the transcriptional landscape of testicular cell populations yields three broad expression patterns associated with peak expression in somatic cells (P1), meiotic spermatocytes (P2) and postmeiotic spermatids (P3) (top row). Enrichment of autosomal (A and B) and X-linked (C) genes belonging to each cluster is evaluated for each region deficient in NA and DA by calculating the Fisher’s exact probability (Gaussian hypergeometric test). (A–C) Results are given for the Neanderthal introgression map inferred from the genomes of present-day European and east-Asian (from 1,000 Genomes project) individuals and for the Denisovan introgression map in the genome of present-day Oceanian individuals (Simons Genome Diversity Project, SGDP). The names of expression cluster are indicated on top of each column. Associated P-values as well as numbers of genes as observed and expected by chance are given within rectangles that are color-coded with red and blue indicating over- and under-representation, respectively, according to the scale bar on bottom. Numbers in bold indicate significant over-representation.
A possible concern with this analysis is that the germ cell transcriptomes derived from infertile patients could in principle be atypical. We therefore produced a new transcriptomic dataset based on enriched populations of five testicular cell types from fertile men (see supplementary information, Supplementary Material online). This allowed us to define three broad groups of gene transcripts with elevated expression signals in testicular somatic cells (P1), meiotic spermatocytes (P2), and early haploid spermatids (P3; see supplementary table S1, fig. S1 and supplementary information, Supplementary Material online). Our data reveal again that only genes with a meiotic expression pattern (P2) were significantly enriched in autosomal regions of low NA (Fisher’s exact test P = 3.7e-8 for European and P = 1.1e-9 for East-Asian populations) and DA (P = 0.01 for Oceanians populations; fig. 1B and see supplementary Extended data table S1, Supplementary Material online). We also found that meiotic genes (P2) in reduced DA regions (inferred in Oceanian populations) overlap significantly with those in reduced NA regions (hypergeometric test, P = 9.7e-6 for European and P = 1.8e-4 for East-Asian populations; see supplementary fig. S6, Supplementary Material online). In addition, the overlap in meiotic genes (P2) significantly exceeds what might be expected based on the overall overlap of reduced archaic introgressions (hypergeometric test, P = 4.6e-12 and P = 5.3e-14 for the overlap between DA regions and NA regions inferred from European and East-Asian populations, respectively), suggesting that alleles in common genes tended to be purged in two groups of anatomically modern hominids.
As it has been observed that the entire X chromosome has lower NA/DA levels than autosomes (Sankararaman et al. 2014, 2016), we investigated whether X-linked genes would display a significant reduction of NA/DA, independent of their expression. Using the Fisher’s exact test (see supplementary information, Supplementary Material online), our data confirm that X-linked genes are all significantly associated with regions of reduced ancestry, independent of their somatic (P1, Fisher’s exact test, P < 4.3e-10), meiotic (P2, P < 3.6e-2), or postmeiotic (P3, P < 7.7e-4) expression pattern (fig. 1C). When we performed gene-level analyses of the X chromosome (performing a logistic regression of the NA status in European populations of a gene against the expression pattern) none of the expression patterns are associated with NA status (logistic regression one-sided P-value P = 0.76, 0.07, and 0.62, respectively for P1, P2, and P3 states). The only association that we find to be statistically significant for chromosome X is between pattern C9 and NA (logistic regression one-sided P-value P = 6e-4). We conjecture that this lack of power is a result of the smaller number of genes on chromosome X combined with the overall lower NA on the X (see supplementary table S2, Supplementary Material online). Finally, we further wanted to investigate whether meiotic genes (P2) of low archaic ancestry would be uniformly organized all along the human chromosomes and observed that they do not cluster in the genome physically (see supplementary fig. S2, Supplementary Material online).
Because of a massive chromatin remodeling leading to progressive genomic silencing in haploid spermatids, several genes critical for the spermiogenic phase are expressed earlier on in meiotic spermatocytes (Iguchi et al. 2006; Gan et al. 2013). Thus, it is conceivable that low NA/DA genes transcribed during the meiotic phase may be translated later on during spermiogenesis and not be acting during meiosis. To investigate whether regions of reduced ancestry would be significantly associated with genes involved in the meiotic process, we performed a Gene Ontology enrichment analysis (see supplementary figs. S3 and S4, supplementary information, Supplementary Material online). We observed that meiosis-transcribed genes (P2 pattern) fulfill a range of biological functions necessary for spermatogenesis, including meiotic machinery and spermatid differentiation up to spermatozoa (see supplementary fig. S3, Supplementary Material online). Notably, we found that meiosis-related terms such as “meiotic nuclear division” were significantly associated with the P2 meiotic pattern (P = 2.3e-8; see supplementary fig. S3, Supplementary Material online) and, unlike terms related to post-meiosis, still remained highly significant when restricted to autosomal “low NA” genes (P = 6.3e-3; see supplementary fig. S4, Supplementary Material online).
As molecular factors are shared by male and female meiotic germ cells, we further explored whether similar phenomena would also be observed with autosomal loci expressed in oocytes. Whereas meiosis occurs after puberty in males, by contrast, meiotic prophase I in the ovary initiates in primary oocytes during embryonic development, is then arrested in the diplotene stage, and resumes after puberty, at the time of ovulation (Gondos et al. 1986). We thus interrogated a transcriptomic dataset in which the transcriptional landmarks of human prenatal germline development was established (Gkountela et al. 2015). In the latter study, modules 1, 16, 24, and 2 were found to be highly representative of human primitive embryonic stem cells (hESCs), primordial germ cells (PGCs), male germ cells between 74 and 98 days of development termed “male advanced germ cells” (mAGCs), and female meiotic germ cells between 67 and 93 days termed “female advanced germ cells” (fAGCs), respectively (see supplementary table S1, Supplementary Material online). Our results show that the female meiotic germline module (module 2) overlaps significantly with male meiotic genes (P2 pattern, hypergeometric test, P = 1.5e-91; see supplementary Extended data table S1, Supplementary Material online). Consistently, our data also reveal that regions of low NA/DA are significantly associated with female meiotic genes (module 2; NA: P = 1.6e-3 for Europeans and P = 6.1e-3 for east-Asians; DA: P = 3.8e-3 for Oceanians) just as they are for male meiotic genes (fig. 2 and see supplementary Extended data table S1, Supplementary Material online).
Fig. 2.
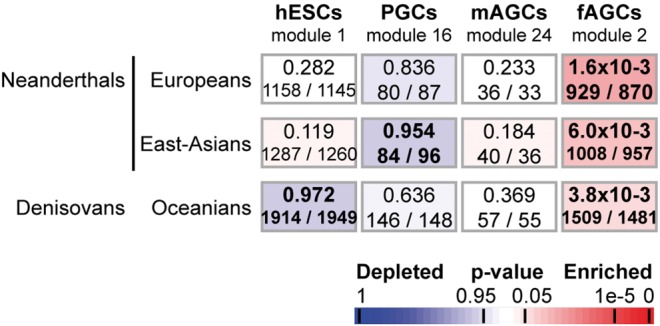
Enrichment of genes expressed in human prenatal germline cells in regions deficient in ancient hominin ancestries. Gkountela et al. defined a set of 39 modules of co-expressed transcripts including four highly representative of human embryonic stem cells (hESCs, module 1), primordial germ cells (module 16, PGCs), male advanced germ cells (module 24, mAGCs) and female advanced germ cells (fAGCs, module 2) (Gkountela et al. 2015). As in figure 1, P-values corresponding to enrichment of autosomal genes belonging to each of those four modules are calculated for each genomic region of reduced archaic ancestry using the Fisher’s exact probability (Gaussian hypergeometric test).
Knowing that genes involved in reproduction, notably testis-expressed genes, are subject to intense adaptive selection (Wyckoff et al. 2000; Swanson and Vacquier 2002; Khaitovich et al. 2005), we were also concerned that meiotic genes might be more subject to linked natural selection to remove deleterious variation than most other genes in the genome—a process that profoundly shaped the distribution of archaic ancestry in modern humans (Juric et al.2016; Harris and Nielsen 2016). To also address this important issue, we measured the heterozygosity in each gene in the genome in sub-Saharan Africans without archaic admixture, which should be sensitive to linked selection. We then repeated our enrichment statistics within a logistic regression framework after controlling for the heterozygosity within a gene in sub-Saharan Africans as a surrogate for linked selection (see supplementary information, Supplementary Material online). The deficiency in archaic ancestry associated with meiosis (P2 expression pattern) actually became stronger after this control (logistic regression one-sided P-values of 3.3e-9 for NA in 1,000 Genomes Europeans, 5.0e-11 for NA in 1,000 Genomes East Asians, 9.7e-8 for NA for mainland Eurasians in the Simons Genome Diversity Project (SGDP) and 9.9e-4 for DA inferred from Oceanians individuals in SGDP; see supplementary table S3, Supplementary Material online). We also replicated this result after controlling for a B-statistic which measures the strength of background selection within a gene (see supplementary table S3 and supplementary information, Supplementary Material online).
Genes that control reproduction have been shown to evolve rapidly likely due to the effects of positive selection (Wyckoff et al. 2000; Swanson and Vacquier 2002). To test if positive selection could explain the enrichment of archaic ancestry in reproductive genes, we repeated the above analyses after removing: 1) Genes that show evidence for recent selection (as identified from composite of multiple signals as well as long-range haplotype tests; Grossman et al. 2013) in the CEU population (Utah residents with northern and western European ancestry); 2) In each of CEU, CHB + JPT (Han Chinese from Beijing, China, and Japanese from Tokyo, Japan) and YRI (Yoruba from Ibadan, Nigeria) populations; and 3) Positively-selected genes on the human lineage identified by human–chimp comparisons (Bakewell et al. 2007). A logistic regression of NA in 1,000 Genomes Europeans on expression stage revealed a consistent and statistically significant association of NA with meiotic expression pattern P2 (logistic regression P-values of 7.8e-11, 4.3e-11 and 4.9e-11 in analysis 1, 2, and 3, respectively; see supplementary table S4, Supplementary Material online). Neither the somatic expression patterns P1 nor the post-meiotic expression pattern P3 showed a significant association across any of the analyses. We also repeated each of the above three analyses by including local heterozygosity in sub-Saharan Africans and obtained consistent results (logistic regression one-sided P-values of 4.9e-9, 2.2e-9, and 3.4e-9 for P2 across the three analyses; see supplementary table S4, Supplementary Material online). Thus, the deficiency of NA ancestry in genes expressed in meiotic spermatocytes persists after removing genes subject to strong positive selection (the reduction in significance of P-values in this analysis suggests some contribution from positive selection but could also be a result of the smaller number of genes in the analysis).
Finally, to assess the robustness of our results to the binary classification of each gene (as depleted in archaic ancestry or not), we repeated our analyses by instead performing a linear regression of the average of the marginal probability of NA inferred in 1,000 Genomes European populations (Sankararaman et al. 2014) and obtained concordant results (Linear regression one-sided P-value = 3.1e-13 for P2; 0.99 for P1; and 0.49 for P3; see supplementary tables S5 and S6, Supplementary Material online).
Discussion
Recently, it has been shown that testis-specific loci are enriched in autosomal genomic regions with a reduced frequency in archaic hominin alleles (Sankararaman et al. 2014, 2016). Only very few of those genes predominantly expressed in the testis (10%) are transcribed during the mitotic phase of spermatogenesis or in the somatic cell populations (Chalmel et al. 2012). Indeed, the vast majority (90%) of testis-specific genes are exclusively produced during the meiotic phase (∼30%), when genetic recombination occurs thanks to exchange of genetic material between homologous chromosomes, and during the postmeiotic phase (∼60%), when haploid spermatids metamorphose into spermatozoa (Jegou 1993; Wassarman et al. 2001; Chalmel et al. 2012). It was thus hypothesized that, among all genes expressed in the testis, low NA or DA regions would be significantly associated with autosomal genes showing peak expression in haploid spermatids and, to a lesser extent, in meiotic spermatocytes. The current study reveals that a significant association was only detected with the meiotic phase but not with the somatic cell populations, the mitotic phase, nor even with the postmeiotic phase. It is known that genes showing peak expression in spermatocytes are not strictly committed to the meiotic process as a number of them encodes proteins involved in the later steps of spermatogenesis (see supplementary fig. S3, Supplementary Material online; Iguchi et al. 2006; Gan et al. 2013). Our functional analysis indicates that meiosis-transcribed genes present in low NA/DA autosomal regions are predominantly associated to the meiotic machinery itself but not to spermatid differentiation (see supplementary fig. S4, Supplementary Material online). In agreement with this, we demonstrate that the enrichment of meiotic genes in regions with reduced archaic ancestry also applies to oogenesis when meiosis occurs, suggesting that natural selection most probably worked to remove deleterious archaic-derived alleles from genes involved in meiosis as such. Notably, among the 3,950 meiotic genes present in low NA/DA autosomal regions (see supplementary Extended data table S1, Supplementary Material online), 249 and 236 are related to gamete defects and infertility, respectively, including 89 directly associated to abnormal meiosis such as RAD51C and DMC1 (Matzuk and Lamb 2008). Although it has already been shown that the hybrid infertility may also occur in the homogametic sex (Barbash and Ashburner 2003) including in the early stages of speciation in rodents (Suzuki and Nachman 2015), it is worth noting that our findings still remains compatible with “Haldane’s Rule” (Coyne 1985) as the gene sets expressed in male and female meiotic germ cell types are correlated.
Our findings also suggest that meiosis involves a high number of speciation genes (Mihola et al. 2009; Nosil and Schluter 2011). The reduced hybrid fertility is plausibly due to “Dobzhansky–Muller incompatibilities”, that is, combinations of alleles on meiotic genes that have never previously been exposed to each other. These incompatibilities can result in perturbation of meiosis in germ cells through abnormal chromosomal behavior interfering with gamete production and quality, as well as with embryonic development (Martin 2006; Handyside 2012). These observations are also consistent with the suggestion that the reproductive fitness of archaic–modern human hybrids was probably marginal, as in hybrids of genetically divergent lineages in other taxa (Rhymer and Simberloff 1996; Corbett-Detig et al. 2013; Banes et al. 2016). Recently, it has been suggested that the reduced fitness of Neanderthal-modern human hybrids is a result of the increased load of deleterious alleles in Neanderthals due to their reduced effective population size since speciation (Harris and Nielsen 2016; Juric et al. 2016).
An investigation on recent changes in the human genome identified that chromosomal rearrangements between modern humans and Neanderthals (although not Denisovans) also occurred preferentially in the vicinity of testis-specific genes, consistent with the hypothesis of hybrid infertility (Rogers 2015). In addition, a comparison of the Neanderthal and modern human Y chromosomes has shown that all three genes with functional missense mutations that differentiate Neanderthals from modern humans are male-specific minor histocompatibility genes, providing some additional evidence of reproductive isolation between the two groups (Mendez et al. 2016). We note that hybridization between modern and archaic humans was not only deleterious; it also had positive consequences for certain classes of genes, probably helping modern humans to expand into environments to which the archaic humans were already adapted due to hundreds of thousands of years of local evolution (Racimo et al. 2015). Understanding the relative importance of positive and negative selection on archaic alleles segregating in non-African people today—that is, in successful populations that emerged from the Neanderthal-modern hybridization events—is an important direction for future research.
Materials and Methods
Detailed descriptions of materials and methods are provided in supplementary information, Supplementary Material online.
Identification of Genes Depleted in the Archaic Ancestries
To assess whether a gene is depleted for each of the archaic ancestries, we used previously published maps of NA and DA (Sankararaman et al. 2014, 2016). As defined these studies, we declared a gene as depleted in NA/DA if all SNPs across all individuals were assigned a marginal probability of NA/DA ≤ 10% (Sankararaman et al. 2014). We derived sets of genes depleted in archaic ancestry from four maps: maps of NA inferred from Europeans and East Asians in the 1,000 Genomes Project (Sankararaman et al. 2014), a map of NA inferred from mainland Eurasians in the SGDP data (Sankararaman et al. 2016) and a map of DA in Oceanians also inferred from the SGDP data (Sankararaman et al. 2016). In total, these studies identified 8,475 genes (7,874 autosomal loci) of European individuals and 9,300 genes (8,738 autosomal loci) of East-Asian individuals associated with low NA regions as well as 14,475 genes (13,751 autosomal loci) of Oceanians associated with low DA regions (see supplementary table S1 and Extended data table S1, Supplementary Material online). To assess the robustness of our results to the binary classification of each gene, we also repeated our analyses using the marginal probability of archaic ancestry at each gene (a number between 0 and 1; see supplementary tables S5 and S6, Supplementary Material online). Specifically, for each gene we computed the average of the marginal probability of NA inferred in 1,000 Genomes European populations (Sankararaman et al. 2014). Note that we also used the number of NA/DA single nucleotide polymorphisms (NA/DA SNPs) per gene as defined in both studies (Sankararaman et al. 2014, 2016).
Testing the Relationship between Archaic Ancestry and Testicular Cell Populations
We wanted to test if archaic ancestry is reduced in genes that are highly expressed in a given testicular cell population using the Fisher’s exact probability (Gaussian hypergeometric test).
To account for potential differences in the strength of linked selection across these sets of genes, we set up two distinct statistical tests (heterozygosity, B-statistics, c.f. see supplementary information, supplementary tables S2 and S3, Supplementary Material online) for each cell population where the response is whether a gene is depleted in archaic ancestry and the covariates consist of 1) whether the gene is expressed in the chosen cell population and 2) a statistic that measures the strength of linked selection near the gene. We restricted the statistical analysis to genes that are found to be differentially expressed in the dataset analyzed. We applied these tests in a number of different settings: 1) Archaic introgression maps: NA in 1,000 genomes Europeans, NA in 1,000 genomes East Asians, NA in SGDP Europeans, DA in SGDP Oceanians; 2) Genes on the autosomes and chromosome X; 3) Autosomal genes after removing genes listed in positive selection scans. Reported is the one-sided P-value associated with the cell population of the test under the null hypothesis that archaic ancestry is not depleted in genes that are expressed in that cell population (see supplementary table S4, Supplementary Material online).
Finally, we also employed linear regression to test the relationship between the average marginal probability of NA in 1,000 Genomes Europeans and whether a gene is expressed in a chosen cell population (see supplementary tables S5 and S6, Supplementary Material online).
Supplementary Material
Supplementary data are available at Molecular Biology and Evolution online.
Author Contributions
F.C., B.J., and D.R. conceived the study, supervised research and wrote the manuscript with help from all co-authors. F.C. and S.S. performed the analyses. A.D.R. prepared the testicular samples and contributed to the analysis.
Supplementary Material
Acknowledgments
This work was supported by Rennes Métropole (“Défis scientifiques émergents 2011” to F.C., “Défis scientifiques émergents 2013” to A.D.R.), the National Institutes of Health (GM100233 to D.R., 4R00GM111744 to S.S.), the National Science Foundation (HO BCS-1032255 to D.R.). B.J. and F.C. were supported by the Institut national de la santé et de la recherche médicale (Inserm), the Université de Rennes 1, the Ecole des hautes études en santé publique (EHESP—School of Public Health), the Université Sorbonne Paris Cité (USPC). D.R. is an investigator of the Howard Hughes Medical Institute. We thank E. Lecluze and S. Condemi for stimulating discussions and critical comments. We are grateful for helpful advice from all members of the SEQanswers forums. We thank S. Salzberg and C. Trapnell for continuous support with the “Tuxedo” suite; members of the UCSC genome browser. Sequencing was performed by the GenomEast platform, member of the France Genomique program. Raw data (RNA-seq) have been deposited in the Gene Expression Omnibus under accession number GSE74896. RNA-seq data are also conveniently accessible through the ReproGenomics Viewer at http://rgv.genouest.org.
References
- Bakewell MA, Shi P, Zhang J.. 2007. More genes underwent positive selection in chimpanzee evolution than in human evolution. Proc Natl Acad Sci U S A. 104:7489–7494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banes GL, Galdikas BM, Vigilant L.. 2016. Reintroduction of confiscated and displaced mammals risks outbreeding and introgression in natural populations, as evidenced by orang-utans of divergent subspecies. Sci Rep. 6:22026.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbash DA, Ashburner M.. 2003. A novel system of fertility rescue in Drosophila hybrids reveals a link between hybrid lethality and female sterility. Genetics 163:217–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalmel F, Lardenois A, Evrard B, Mathieu R, Feig C, Demougin P, Gattiker A, Schulze W, Jegou B, Kirchhoff C, et al. 2012. Global human tissue profiling and protein network analysis reveals distinct levels of transcriptional germline-specificity and identifies target genes for male infertility. Hum Reprod. 27:3233–3248. [DOI] [PubMed] [Google Scholar]
- Chalmel F, Rolland AD, Niederhauser-Wiederkehr C, Chung SS, Demougin P, Gattiker A, Moore J, Patard JJ, Wolgemuth DJ, Jegou B, et al. 2007. The conserved transcriptome in human and rodent male gametogenesis. Proc Natl Acad Sci U S A. 104:8346–8351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corbett-Detig RB, Zhou J, Clark AG, Hartl DL, Ayroles JF.. 2013. Genetic incompatibilities are widespread within species. Nature 504:135–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coyne JA. 1985. The genetic basis of Haldane's rule. Nature 314:736–738. [DOI] [PubMed] [Google Scholar]
- Derrien T, Johnson R, Bussotti G, Tanzer A, Djebali S, Tilgner H, Guernec G, Martin D, Merkel A, Knowles DG, et al. 2012. The GENCODE v7 catalog of human long noncoding RNAs: analysis of their gene structure, evolution, and expression. Genome Res. 22:1775–1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fawcett DW. 1975. Ultrastructure and function of the Sertoli cell In: Handbook of Physiology, Section 7, vol. V, Male Reproductive System, pp 21–55. D. W. Hamilton & R. O. Greep, editors. Washington, D.C: American Physiological Society. [Google Scholar]
- Fu Q, Li H, Moorjani P, Jay F, Slepchenko SM, Bondarev AA, Johnson PL, Aximu-Petri A, Prufer K, de Filippo C, et al. 2014. Genome sequence of a 45,000-year-old modern human from western Siberia. Nature 514:445–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gan H, Cai T, Lin X, Wu Y, Wang X, Yang F, Han C.. 2013. Integrative proteomic and transcriptomic analyses reveal multiple post-transcriptional regulatory mechanisms of mouse spermatogenesis. Mol Cell Proteomics. 12:1144–1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gan H, Wen L, Liao S, Lin X, Ma T, Liu J, Song CX, Wang M, He C, Han C, et al. 2013. Dynamics of 5-hydroxymethylcytosine during mouse spermatogenesis. Nat Commun. 4:1995.. [DOI] [PubMed] [Google Scholar]
- Gkountela S, Zhang KX, Shafiq TA, Liao WW, Hargan-Calvopina J, Chen PY, Clark AT.. 2015. DNA demethylation dynamics in the human prenatal germline. Cell 161:1425–1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gondos B, Westergaard L, Byskov AG.. 1986. Initiation of oogenesis in the human fetal ovary: ultrastructural and squash preparation study. Am J Obstet Gynecol. 155:189–195. [DOI] [PubMed] [Google Scholar]
- Green RE, Krause J, Briggs AW, Maricic T, Stenzel U, Kircher M, Patterson N, Li H, Zhai W, Fritz MH, et al. 2010. A draft sequence of the Neandertal genome. Science 328:710–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grossman SR, Andersen KG, Shlyakhter I, Tabrizi S, Winnicki S, Yen A, Park DJ, Griesemer D, Karlsson EK, Wong SH, et al. 2013. Identifying recent adaptations in large-scale genomic data. Cell 152:703–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Handyside AH. 2012. Molecular origin of female meiotic aneuploidies. Biochim Biophys Acta. 1822:1913–1920. [DOI] [PubMed] [Google Scholar]
- Harris K, Nielsen R.. 2016. The genetic cost of Neanderthal introgression. Genetics 203:881–891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iguchi N, Tobias JW, Hecht NB.. 2006. Expression profiling reveals meiotic male germ cell mRNAs that are translationally up- and down-regulated. Proc Natl Acad Sci U S A. 103:7712–7717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jegou B. 1993. The Sertoli-germ cell communication network in mammals. Int Rev Cytol. 147:25–96. [PubMed] [Google Scholar]
- Juric I, Aeschbacher S, Coop G.. 2016. The strength of selection against Neanderthal introgression. PLoS Genet. 12:e1006340.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khaitovich P, Hellmann I, Enard W, Nowick K, Leinweber M, Franz H, Weiss G, Lachmann M, Paabo S.. 2005. Parallel patterns of evolution in the genomes and transcriptomes of humans and chimpanzees. Science 309:1850–1854. [DOI] [PubMed] [Google Scholar]
- Martin RH. 2006. Meiotic chromosome abnormalities in human spermatogenesis. Reprod Toxicol. 22:142–147. [DOI] [PubMed] [Google Scholar]
- Matzuk MM, Lamb DJ.. 2008. The biology of infertility: research advances and clinical challenges. Nat Med. 14:1197–1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendez FL, Poznik GD, Castellano S, Bustamante CD.. 2016. The divergence of Neandertal and modern human Y chromosomes. Am J Hum Genet. 98:728–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mihola O, Trachtulec Z, Vlcek C, Schimenti JC, Forejt J.. 2009. A mouse speciation gene encodes a meiotic histone H3 methyltransferase. Science 323:373–375. [DOI] [PubMed] [Google Scholar]
- Nosil P, Schluter D.. 2011. The genes underlying the process of speciation. Trends Ecol Evol. 26:160–167. [DOI] [PubMed] [Google Scholar]
- Racimo F, Sankararaman S, Nielsen R, Huerta-Sanchez E.. 2015. Evidence for archaic adaptive introgression in humans. Nat Rev Genet. 16:359–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reich D, Green RE, Kircher M, Krause J, Patterson N, Durand EY, Viola B, Briggs AW, Stenzel U, Johnson PL, et al. 2010. Genetic history of an archaic hominin group from Denisova Cave in Siberia. Nature 468:1053–1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reich D, Patterson N, Kircher M, Delfin F, Nandineni MR, Pugach I, Ko AM, Ko YC, Jinam TA, Phipps ME, et al. 2011. Denisova admixture and the first modern human dispersals into Southeast Asia and Oceania. Am J Hum Genet. 89:516–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhymer JM, Simberloff D.. 1996. Extinction by hybridization and introgression. Ann Rev Ecol Syst. 27:83–109. [Google Scholar]
- Rogers RL. 2015. Chromosomal rearrangements as barriers to genetic homogenization between archaic and modern humans. Mol Biol Evol. 32:3064–3078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sankararaman S, Mallick S, Dannemann M, Prufer K, Kelso J, Paabo S, Patterson N, Reich D.. 2014. The genomic landscape of Neanderthal ancestry in present-day humans. Nature 507:354–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sankararaman S, Mallick S, Patterson N, Reich D.. 2016. The combined landscape of Denisovan and Neanderthal ancestry in present-day humans. Curr Biol. 269:1241–1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soumillon M, Necsulea A, Weier M, Brawand D, Zhang X, Gu H, Barthes P, Kokkinaki M, Nef S, Gnirke A, et al. 2013. Cellular source and mechanisms of high transcriptome complexity in the mammalian testis. Cell Rep. 3:2179–2190. [DOI] [PubMed] [Google Scholar]
- Suzuki TA, Nachman MW.. 2015. Speciation and reduced hybrid female fertility in house mice. Evolution 69:2468–2481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanson WJ, Vacquier VD.. 2002. The rapid evolution of reproductive proteins. Nat Rev Genet. 3:137–144. [DOI] [PubMed] [Google Scholar]
- Vernot B, Akey JM.. 2014. Resurrecting surviving Neandertal lineages from modern human genomes. Science 343:1017–1021. [DOI] [PubMed] [Google Scholar]
- Wassarman PM, Jovine L, Litscher ES.. 2001. A profile of fertilization in mammals. Nat Cell Biol. 3:E59–E64. [DOI] [PubMed] [Google Scholar]
- Wyckoff GJ, Wang W, Wu CI.. 2000. Rapid evolution of male reproductive genes in the descent of man. Nature 403:304–309. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.