

Abstract
Biosimilars are products that contain a similar version of the active substance of an already authorized original biologic medicinal product (reference medicinal product). Their development requires special consideration, as similarity to the reference agent needs to be established through a comprehensive comparability exercise. Given the complex nature of these agents, minor structural differences may emerge, but the process of biosimilarity determination is designed to ascertain that the nature and impact of these differences are not clinically significant. Determination of biosimilarity should follow quality-by-design principles, which provide a deep understanding of the product development process, guided by pre-defined objectives, process control and risk management. Compared with novel biologic development, biosimilar development places greater emphasis on establishing preclinical quality characteristics. Determination of comparability of quality characteristics includes assessment of physicochemical properties, biological activity, immunochemical properties, purity, impurity and quantity, with appropriate in vivo pharmacology studies being conducted thereafter. Head-to-head comparisons are then conducted to determine pharmacokinetic and pharmacodynamic characteristics, and efficacy, safety and tolerability in phase I and phase III clinical studies. Post-approval risk management requirements include implementation of pharmacovigilance systems and risk management through, for example, the conduct of pharmacoepidemiological studies. There are several biosimilars used in the field of rheumatology that are available in the European Union, or in development, that offer the potential to increase affordability/accessibility of biological treatment. The role of these agents in rheumatology will be determined by the confidence placed in them by rheumatologists. These prescribers should expect high-quality data evaluated by an extensive assessment process.
Keywords: biologics, biosimilarity, biosimilar development, comparability, biosimilars, regulatory approval, rheumatology
Rheumatology key messages
A comprehensive process is applied to establish biosimilarity.
Several biosimilar agents are currently licensed for use in rheumatology.
The robust nature of the biosimilarity exercise is an important element when considering these agents.
Introduction
In recent years, we have seen an acceleration in scientific research focused on molecular and cellular biology, which has resulted in the successful development of many biological agents (biologics) that have fundamentally changed clinical practice. Biologics are drugs produced by living cells that mimic the actions of natural endogenous biological moieties [1] or, such as monoclonal antibodies, that are directed against (and affect) particular targets in the human body [2]. Their development has provided clinicians with an alternative therapeutic strategy to that provided by traditional small-chemical-molecule therapeutics. Although biologics have revolutionized the treatment of many chronic conditions [3], they are often expensive in terms of cost per dose [4, 5]. The patent expiry and loss of exclusivity of some of these innovative biologics has opened the door for the introduction of their therapeutically equivalent alternatives, now commonly known as biosimilars.
Generic copies of small-molecule-based therapeutics have been widely used as alternatives to more expensive branded agents. Indeed, a number of generic copies of traditional small-chemical-molecule drugs, such as statins, oral chemotherapeutics and antihistamines, have been approved, based on evidence from relatively small and inexpensive studies demonstrating bioequivalence [5]. However, the manufacturing of biologics is considerably more challenging than that for traditional small-chemical-molecule drugs, as even relatively small changes in an existing manufacturing process may result in changes in efficacy or immunogenicity, making the generic approach scientifically inappropriate for these products. This has led to the development of the concept of biosimilarity and the consequent extensive regulatory approval pathways that are needed for physicochemical, non-clinical and confirmatory clinical comparability [1].
Biosimilars are products that contain a similar version of the active substance of an already authorized original biologic medicinal product (the reference medicinal product) whose data protection has expired [6]. Their development requires special consideration, as similarity to the reference medicinal product needs to be established through extensive comparability exercises in terms of quality characteristics, biological activity, safety and efficacy [6]. This approach needs to be comprehensive so that regulators can review, and clinicians can prescribe, these agents with confidence. The European Medicines Agency (EMA) has taken the lead over the past 10 years in shaping the environment, increasing understanding of comparability and providing realistic expectations of the proof required to confirm biosimilarity [6–8]. In this respect, the EMA has developed guidelines, such as the overarching, quality, non-clinical and class-specific guidelines on different classes of biologics (including mAb) that support a detailed understanding of what is required to thoroughly evaluate biosimilarity [6, 8]; these are being constantly revised and updated to reflect evolving knowledge and cumulative experiences gained with the first generation of biosimilars in Europe [6, 8].
Although the European Union (EU) biosimilar market is relatively new, a preliminary analysis of pricing behaviour indicates that biosimilars in some therapeutic areas are priced below reference biologics, often with discounts of ⩾25% [9]. The prospect of more affordable biosimilar treatment options provides opportunities for health systems to expand access to biologics for more patients, to free up valuable resources for investment in new areas and to relieve the burden on healthcare budgets [10]. In addition, it is anticipated that the development and introduction of a range of biosimilar medicines may generate savings that can be reinvested in healthcare provision, while at the same time driving pharmaceutical innovation that ultimately improves outcomes for patients [10].
In this article, we review the concept of biosimilarity and the regulatory requirements that are needed to establish biosimilarity, as defined by the EMA. We also provide examples of biosimilar agents for use in rheumatology that have been subjected to this assessment and approval process.
The concept of biosimilarity and the rationale for its determination
The scientific concept of biosimilarity is well established, and manufacturers need to ensure that sufficient analyses are performed to demonstrate a high degree of similarity between the reference agents and biosimilars prior to any clinical testing [6, 11]. The EMA has extensive experience in regulating and reviewing comparability exercises [6–8], partly because the manufacturing processes of biologics are often subject to modification, for example, to increase efficiency [12]. Although not expected to be associated with clinical consequences, these changes require an analysis of pre- and post-change products (comparability exercise), with subsequent approval by the EMA [6, 11]. Importantly, these alterations are made with knowledge of the original manufacturing process, which differs from biosimilar development where proprietary manufacturing data are not available [13].
Most biologics are large complex proteins manufactured using a living cell line in a highly controlled setting [14]. Even minor changes in their structure and conformation may have an impact on their pharmaceutical properties, pharmacodynamics (PD), safety or tolerability [11, 14]. For example, conformational changes of mAb and receptor–Fc fusion proteins can affect their binding affinity and biological activity, which in turn may affect their pharmacokinetics (PK) and PD profile, potentially having an impact on their dosing regimen [15]. Furthermore, immunogenicity is an important consideration, and thus should be borne in mind as alterations in manufacturing processes and storage conditions may potentially affect this aspect [11]. Antidrug antibodies could reduce circulating biologic levels, cause a neutralizing antibody effect and/or adverse reactions (e.g. infusion-related reactions) and may lead to a loss of clinical efficacy [16, 17]. Consequently, the integrity of the biosimilar needs to be assured to provide confidence in its use.
The real challenge in biosimilar development is not in determining whether differences exist compared with the reference product, but whether these differences are clinically relevant. Therefore, a comprehensive biosimilarity exercise should cover almost all the structural and functional characteristics of the reference molecule to assess biosimilarity and to ensure that biosimilars have an equivalent risk–benefit balance [6].
Building the evidence for biosimilarity
Biosimilars are characterized by their complex molecular structure and cell culture manufacturing process, which makes them more difficult to characterize, produce and reproduce [18, 19], such that biosimilars and reference biologics cannot be produced identically. Instead, development is undertaken to establish physicochemical, biological and pharmacological critical quality attributes (CQAs) that are intended to guide the clinical profile and establish clinical comparability. CQAs are chemical, physical, biological and microbiological characteristics that can be defined, measured and continually monitored to ensure that the final product outputs remain within acceptable limits of quality. The process of establishing biosimilarity should follow quality by design (QbD) principles. QbD is a systematic approach to establish a deep understanding of the product development process that is guided by predefined objectives and involves implementation of process control, based on sound science and quality risk management [20].
When a manufacturer develops a new chemical or biological product, a well-established, step-wise approach is taken to mitigate the risks of the product development process and patient exposure. Similarly, biosimilar development requires a number of steps, including selection of an appropriate reference biologic, obtaining the reference active pharmaceutical ingredient, identifying the quality target product profile (QTPP) and CQAs of the reference biologic, and developing a manufacturing process to match the attributes of the reference biologic product [18]. Although the concept of a biosimilar is applicable to any biologic, the success of such a development approach will depend on the ability to characterize the reference product and therefore to demonstrate the similar nature of the concerned products [21], using appropriately available comparators [22]. The goal of biosimilar development is to match the QTPP of the reference biologic product and this should form the basis for the development of the biosimilar, with the manufacturing process being appropriately designed [23].
The evidence required to establish biosimilarity is gathered using a step-wise approach, firstly establishing elements related to quality (physicochemical and biological comparability), then pharmacology (non-clinical comparability) and lastly clinical evaluation (clinical comparability) [6, 24]. Quality comparability is established with regard to the molecular structure as well as functionality, and must be demonstrated with comprehensive analytical characterization, relevant receptor-binding studies and bioassays, all of which should be performed with the biosimilar and the reference product in a rigorous comparative manner [24]. The non-clinical and clinical comparability exercise, which is based on a head-to-head comparison between the biosimilar and the reference medicinal product, then provides the confidence that any differences that might be observed at the quality level have no impact on the safety and efficacy of the biosimilar compared with the reference product [24]. Therefore, each step has a critical contribution to the (bio)physical characteristics and should rely on the most advanced state-of-the-art capabilities; no step can refute or overcome significant differences in previous steps, and all three steps must be satisfied to establish biosimilarity. Given the above steps, the development of a biosimilar can be as extensive as that of the original entity but with a different emphasis (Fig. 1) [24]. Indeed, whereas there is a much greater effort placed on establishing clinical efficacy and tolerability for a new biologic agent (i.e. a positive risk–benefit profile), much more emphasis is placed on establishing comparability at the earlier stages of development for a biosimilar. By focusing more closely on establishing analytical similarity, there is increased certainty of a comparable clinical profile and a reduction in the overall need for clinical testing [6].
Fig. 1.
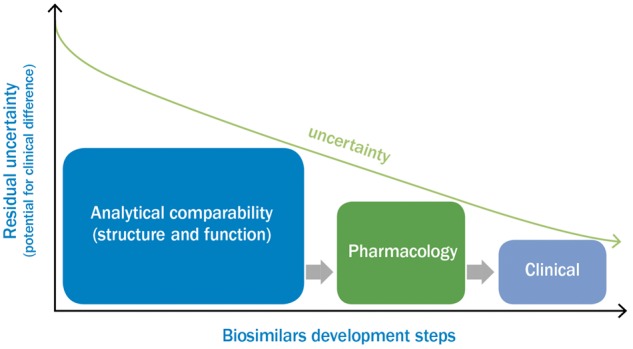
Major elements and emphasis in the development of a biosimilar
Information taken from [24].
Establishing physicochemical and biological comparability
The majority of the work in establishing similarity is performed preclinically, at which point any potential differences between the biosimilar and reference product are more likely to be detected. In this respect, there are two distinct areas on which regulators focus: the comparability of the molecular characteristics and quality attributes of the biosimilar and reference product, and the performance and consistency of the manufacturing process for the biosimilar on its own [23]. This work involves in-depth physicochemical and in vitro biological characterization of the biosimilar and comparison with the original biologic to address structural, functional and immunogenicity concerns [11]. The biosimilarity analytical and quality exercise should involve comprehensive analyses of the proposed biosimilar and the reference agent using sensitive and robust methods to determine not only similarities, but also potential differences, in quality attributes [23]. Furthermore, bioanalytical assays should be appropriate for their intended use and adequately validated [6].
Based on CQAs, key attributes to be evaluated and compared for the biosimilar and reference agent include physicochemical properties, biological activity, immunochemical properties, purity and impurities, quantity and strength (Fig. 2) [23]. The physicochemical comparison comprises the evaluation of physicochemical parameters, and should include a determination of the composition, physical properties, and primary (amino-acid sequence) and higher-order (e.g. local conformation and three-dimensional) structures of the biosimilar [23]. The target amino-acid sequence of the biosimilar, which is expected to be the same as for the reference product, should be confirmed, and the N- and C-terminal amino-acid sequences, free SH groups and disulfide bridges compared. The presence and extent of post-translational modifications (e.g. glycosylation, oxidation, deamidation and truncation) should also be characterized. Finally, if present, carbohydrate structures, such as overall glycan profile and site-specific glycosylation patterns, should be compared [23]. Determination of biological activity is dependent on the nature of the product, but would typically include receptor–ligand binding assays, enzymatic assays, and cell-based and functional assays [23]. This should include comparison of the immunological function of monoclonal antibodies; generally, this would be done by assessing the affinity of the products to the intended target, binding of the Fc to the relevant receptors (e.g. FcγR, C1q, FcRn) and induction of Fab- and Fc-associated effector functions [8]. The purity and impurity profiles of the biosimilar and the reference product should be determined and compared both qualitatively and quantitatively by a combination of analytical procedures. The shelf-life of the reference product and any effect on the quality profile should be accounted for. Process-related impurities (e.g. host cell proteins, host cell DNA, reagents, downstream impurities, etc.) should be determined and the potential risks related to these identified impurities (e.g. immunogenicity) documented [23]. Finally, quantity should be determined and a comparable strength confirmed for the biosimilar and reference product.
Fig. 2.

Key steps in the analytical exercise to establish biosimilarity
Information taken from [23]. FcγR: Fc (gamma) receptor; FcRn: neonatal Fc (fragment crystallizable) receptor; PK: pharmacokinetics.
The role of the manufacturing process
Against this background, the manufacturing process should be tailored to the specific biosimilar and appropriately designed to consistently achieve the key target quality attributes, or QTPP, of the reference biologic product [23]. As the characteristics of a biologic can change over time, as a result of operational variations within a manufacturing process or following storage [25], testing multiple lots of a reference biologic over a period of time is required to build a complete picture of the QTPP and to ensure that the design of a manufacturing process produces a biosimilar that closely reflects the reference biologic product [23]. The formulation of the biosimilar does not need to be identical to that of the reference agent; however, it does need to be appropriate with regard to the originator’s pharmaceutical profile. For example, regardless of the formulation selected, suitability should be determined with regard to the stability, compatibility, integrity, activity and strength of the active substance. If a different formulation/closure system from the reference biologic product is used, its potential impact on the efficacy and safety of the biosimilar also needs to be justified [23].
Establishing non-clinical in vivo biosimilarity
The use of animals in research remains a controversial subject in the wider community. Guidelines recognize this concern and recommend that the use of animals should be minimized or eliminated where possible—for example, by implementing the principles of the 3Rs (Replacement, Reduction and Refinement) for humane animal research. However, non-clinical evaluation using in vivo studies may be necessary to complement information gathered during the analytical phase of biosimilar development [6]. In particular, the need for non-clinical in vivo studies may be prompted by the presence of potentially relevant quality attributes that have not been detected in the reference product, the presence of potentially relevant quantitative differences in quality attributes between the biosimilar and the reference product, and any relevant differences in formulation, for example, use of excipients not widely used for biotechnology-derived proteins [6].
In vivo studies
If an in vivo evaluation is deemed to be necessary, then the focus of the study/studies will depend on the need for additional information [6]. Assessment may include quantitative comparison of the PK and PD profiles of the biosimilar and reference product, including dose–concentration response [6]. For safety studies, a flexible approach should be considered, particularly if non-human primates are the only relevant species [6]. The conduct of toxicity studies in non-relevant species (i.e. to assess non-specific toxicity only, based on impurities) and standard repeated dose-toxicity studies in non-human primates is not recommended [26]. However, if justified, a repeated dose-toxicity study with refined design (e.g. single dose level, one gender) or an in-life evaluation of safety parameters (e.g. clinical signs, body weight and vital functions) may be considered [6]. Quantitative and qualitative differences in product-related variants (e.g. glycosylation), which may give rise to hypersensitivity, should be evaluated clinically [6]. Although immunogenicity studies in animals are not predictive of immunogenicity in humans, blood samples may be taken in animal studies for future evaluations of PK/toxicokinetic data, if required [6]. Studies of safety pharmacology, reproductive toxicology and carcinogenicity are not required [6]. Similarly, studies on local tolerance are generally not required; however, if excipients for which there is little information in relation to the route of administration are introduced, local tolerance may need to be assessed (e.g. as part of other non-clinical in vivo studies) [6].
The confirmatory role of clinical phase I and phase III studies
As discussed previously, the majority of work in establishing biosimilarity focuses on preclinical, and particularly quality, aspects of the biosimilar. This emphasis allows for greater certainty that the clinical profile of the biosimilar and reference biologic are comparable, thereby reducing the need for clinical evaluation. When required, clinical biosimilarity studies are conducted in a step-wise approach, starting with PK, and then (if feasible) PD (phase I) evaluations, followed by clinical efficacy and safety (phase III) studies. However, in some cases (and according to updated EMA guidelines), confirmatory PK and PD studies may suffice to establish clinical biosimilar compatibility [6, 27]. Clinical studies are generally conducted using the manufactured formulation, but if this is not feasible, appropriate supporting data should be generated [6]; for example, comparison of investigational and manufactured formulations that may vary.
Pharmacokinetic and pharmacodynamic phase I studies
Comparative PK studies designed to demonstrate the similar PK profile of the biosimilar and the reference product are an essential element of the biosimilarity exercise [6, 28]. The design of the PK study depends on a number of factors, including clinical context, safety and the PK of the reference agent and should be defined and justified prior to conducting the study [6, 28]. Although a single-dose, cross-over study, with full characterization of the PK profile, including the late elimination phase, is preferred [6], the ordinary cross-over design is not appropriate for therapeutic proteins with a long half-life—for example, therapeutic antibodies and pegylated proteins, or for proteins for which the formation of anti-drug antibodies is likely [28]. In such cases, a parallel-group design may be necessary, with emphasis placed on minimizing the risk for potential imbalance between the groups [28]. If part of the PK information is gathered from healthy volunteers, the validity of extrapolation of that information to the target population needs to be addressed. This is because elimination for some products is largely dependent on target receptor uptake, meaning that differences in receptor density between healthy volunteers and the target population can create some important PK differences—for example, in half-life [28].
PK evaluations for biosimilars include absorption, disposition, dose–time dependency and binding to blood components. In vivo studies should be conducted in healthy volunteers or patients to describe the absorption characteristics of the compound (e.g. extent and rate) unless the IV route is exclusively used [28]. Single-dose studies are generally sufficient to characterize absorption and to compare different administration routes [28]. As soluble receptors may bind to the therapeutic protein, resulting in an altered PK profile through changed clearance or volume, this potential effect should be examined [28]. Likewise, the binding capacity to plasma proteins (albumin, α-acid glycoprotein) should be studied when relevant. When conducting these PK evaluations, it is important to give due consideration to a number of factors, including chemical modification of proteins, inter-subject variability (e.g. demographic factors such as age and weight), immunogenicity, drug–drug interactions and special populations (e.g. patients with renal or hepatic impairment) [28].
It is also necessary to evaluate the PK–PD relationship, and PD markers of clinical relevance can be added to PK studies to cover this requirement [6]. In certain cases, comparative PK/PD studies may be sufficient to demonstrate clinical comparability, provided that the following conditions are met: (1) the selected PD biomarker is an accepted surrogate marker for clinical outcome and comparability has been shown in this respect; (2) a PD marker is chosen that is not an established surrogate for clinical outcome, but is relevant to the PD action of the agent and a dose–response relationship has been established, meaning that a single or multiple dose–response study may be sufficient to waive a clinical efficacy study; (3) the body of evidence from the analytical exercise and phase I PK/PD study may provide sufficiently robust evidence for clinical comparability [6]. If evidence to establish clinical biosimilar comparability is to be derived solely from PK studies, supported by studies with a non-surrogate PD marker/biomarker, a proposal for the size of the equivalence margin(s) with its clinical justification, as well as measures for demonstration of a comparable safety profile, is required [6].
Efficacy studies
The aim of phase III biosimilarity trials is not to establish efficacy per se, but to demonstrate comparable clinical performance [6]. The EMA Committee for Medicinal Products for Human Use has issued disease-specific guidelines on the development of novel agents [29], and where appropriate, product-class-specific guidelines for biosimilars in some areas [6]. In general, comparable clinical efficacy should be established in adequately powered, randomized, parallel-group (preferably double-blind) trials, using an equivalence design [6]. The use of a non-inferiority design is considered to be less acceptable, but may be justified on the basis of a strong scientific rationale and taking into consideration the characteristics of the reference product, for example, safety/tolerability profile and dose range [6]. A non-inferiority trial may only be accepted where the possibility of significant and clinically relevant increases in efficacy can be excluded on scientific and mechanistic grounds [6]. The study population should be representative of those included in the approved indication for the reference agent, and should be sufficiently sensitive to detect potential differences between the biosimilar and the reference product [6]. Occasionally, changes in clinical practice may require a deviation from the most sensitive approved therapeutic indication, for example, in terms of concomitant medication used in a combination treatment, line of therapy or severity of the disease [6]. If a correlation has been demonstrated between the hard clinical endpoints recommended by the guidelines for new active substances and other clinical/PD endpoints that are more sensitive for detecting clinically meaningful differences in previous clinical trials with the reference product, it is not necessary to use the same primary efficacy endpoints as those that were used in the marketing authorization application of the reference product. However, it is advisable to include a selection of common endpoints (e.g. as secondary endpoints) to facilitate comparisons to the clinical trials conducted with the reference product [6].
Safety evaluation
Building knowledge of the safety profile of the biosimilar is a very important aspect of the exercise. As with any new biologic, the safety profile of the biosimilar is captured throughout the clinical programme, during phase I PK/PD studies and in head-to-head phase III comparative studies [6]. As with any safety evaluation, the nature, severity and frequency of adverse events are evaluated, with adverse events between the biosimilar and the reference product being critically compared. In particular, consideration is given to any safety concerns that may arise from differences in the manufacturing process [6]. Furthermore, because of the potential immunogenic nature of biological agents, immunogenicity and infusion-related reactions are closely monitored and comparative studies conducted to assess comparative immunogenicity [6, 30, 31]. The duration of the immunogenicity study should be justified on a case-by-case basis, depending on the duration of the treatment course, clearance of the product from the circulation and the time for emergence of a humoral immune response [6]. Increased immunogenicity compared with the reference product may become an issue for the benefit–risk analysis and would question biosimilarity. However, a lower immunogenicity profile for the biosimilar would not preclude approval as a biosimilar [6].
Post-approval risk management requirements
Use of drugs in the wider clinical community and for longer periods of time may prompt the emergence of rare or concerning adverse reactions [11] that have not been identified during the biosimilar clinical development programme [6]. For this reason, the clinical safety of biosimilars must be monitored closely on an ongoing basis during the post-approval phase to ensure patient safety [6, 24]. To do this, a pharmacovigilance system and risk management plan should be devised, taking into account the identified and possible risks, with immunogenicity being given particular attention [6]. Elements of pharmacovigilance and risk management may include safety monitoring applied to the reference compound or class, participation in new or existing pharmacoepidemiological studies (e.g. for the reference compound) and risk minimization activities applied to the reference compound [6]. Physicians reporting adverse events should provide as much information as possible, noting the specific agent, event and its occurrence linked to source identity, such as brand name and batch number [6, 11]. Such an approach is particularly important when considering the emergence of adverse reactions in relation to switching between reference and biosimilar agents [11].
Overview of biosimilar agents for rheumatology in the EU
DMARDs are the mainstay of rheumatic disease (RD) therapy [32]. The development of conventional synthetic DMARDs (csDMARDs) was followed by the development of TNF inhibitors, the first biological DMARDs (bDMARDs) that were introduced into rheumatology [32]. Today, five different TNF inhibitors (infliximab, adalimumab, etanercept, certolizumab and golimumab) are approved for use in RD. Following TNF inhibitors, new bDMARDs with different modes of action were developed: abatacept, targeting the co-stimulation between T and B cells; rituximab, targeting CD20+ B cells; and tocilizumab, an IL-6 receptor antagonist. Although bDMARDS have been shown to have notable efficacy, they are costly [33]. Consequently, not all patients who are eligible are prescribed these drugs, with preference generally being given to patients with more severe and aggressive disease [34]. Cost containment can however be achieved with bDMARDs. For example, rituximab is a cost-effective alternative to TNF inhibitors during the early course of the disease [35] and bDMARD dose reductions or stoppages of ⩾50% can be achieved [36–38], which would translate into significant cost reductions and wider possible choice. Alternative strategies to delay or mitigate bDMARD implementation include a treat-to-target (or tight control) approach with combination csDMARDs [39–41], and use of combination csDMARDs following inadequate response to csDMARD therapy [42, 43].
As of March 2017, there are four biosimilar medicines that have received approval and are available on the market for patients with RDs in the EU (overviewed in detail in the third article of this supplement, by Schulze-Koops and Skapenko). These comprise two biosimilar versions of infliximab, one of which (CT-P13) is available under two brand names, that is, Inflectra® and Remsima® (manufactured by Celltrion Inc.), and one (SB2) under the brand name Flixabi® (manufactured by Biogen). An etanercept biosimilar (SB4) is available under the brand name Benepali® (manufactured by Biogen). Flixabi®, Inflectra® and Remsima® are approved for use in RA, adult and pediatric Crohn’s disease, adult and pediatric ulcerative colitis, AS, PsA and psoriasis [44–46]. Benepali® is approved for the treatment of adults with RA, PsA, axial spondyloarthritis (AS and non-radiographic axial spondyloarthritis) and plaque psoriasis [47]. Recently, the EMA Committee for Medicinal Products for Human Use has adopted a positive opinion on the type II variation application for the indication extension of Benepali® for the treatment of JIA and pediatric plaque psoriasis in patients weighing >62.5 kg [48]. There are currently some 41 biosimilar medicines for RD in the pipeline for four key reference biologics (adalimumab, etanercept, infliximab and rituximab) [10]; of these, a number (including those already approved) have had data published in peer-reviewed journals or presented at international scientific meetings (Table 1) [49].
Table 1.
Biosimilars for rheumatic diseases for which data have been published in peer-reviewed journals or presented at international scientific meetings
| Reference product | Biosimilars for rheumatic diseases (published or presented data) |
|---|---|
| Adalimumab | ABP 501b |
| BI 695501 | |
| CHS-1420 | |
| GP-2017 | |
| M923 | |
| SB5 | |
| ZRC-3197 (Exemptia®) | |
| PF-06410293 | |
| Etanercept | AVG01 |
| CHS-0214 | |
| GP2015a,b | |
| HD203 | |
| LBEC0101 | |
| SB4 (Benepali®)a | |
| Infliximab | BOW015 |
| CT-P13 (Inflectra®; Remsima®)a,b | |
| PF-06438179 | |
| SB2 (Flixabi®)a | |
| Rituximab | CT-P10 |
| GP2013 | |
| PF-05280586 |
Reproduced from: Ann Rheum Dis. The role of biosimilars in the treatment of rheumatic diseases, Dörner T, Strand V, Cornes P et al. 72:322–8, ©2013 [49]. With permission from BMJ Publishing Group Ltd. aEMA approved. bFDA approved. EMA: European Medicines Agency; FDA: Food and Drug Administration.
Discussion
The pathway for the development and approval of biosimiliars has been well established by the EMA. This development process follows a step-wise approach, with greater emphasis placed on quality assessment and lesser emphasis on clinical assessment than for de novo biologics. This is because proof of biosimilarity largely rests with the molecular and preclinical profile, removing the onus from the clinical pathway, for which only equivalence testing may be required. Rigorous evaluations are followed to establish biosimilarity, and further risk assessment protocols are instigated following authorization. The extensive nature of the biosimilarity approval process emphasizes the quality of the dataset for existing biosimilar agents. This is an important factor when physicians are faced with greater therapeutic choice that can be tailored to patient need; greater physician confidence in prescribing additional agents may well lead to greater patient-tailored care.
More than 80 biologic molecules have been launched globally over the past decade, bringing new treatment options to patients across a large number of therapy areas [10]. Indeed, across the EU, the use of erythropoietins, granulocyte–colony stimulating factors and human growth hormone have all increased following the launch of biosimilar versions. The development of biobetter agents, preferably called next-generation biologics, is also a hot topic under discussion [50–52]. Next generation biologics are versions of existing biologics that have been modified (e.g. through introduction of a different glycosylation profile or through pegylation) to have enhanced properties, such as improved effector function or elimination half-life [53]. The development and possible availability of these agents will provide further choice in this therapeutic area.
To date, four biosimilar agents are available for use in rheumatology, and it is envisaged that they will play an important role in the management of autoimmune RDs by helping to reduce costs, thereby improving patient access to treatment [54, 55]. Moreover, lower costs may also encourage earlier implementation of biologic therapy [56, 57], although further evidence from clinical trials, particularly observational studies, is required. Several other biosimilar agents for use in rheumatology are currently in development, offering further potential to increase affordability of treatment and improve accessibility [10]. The role of these biosimilars in rheumatology will ultimately be determined by the confidence placed in them by rheumatologists, and these prescribers should expect high-quality data, generated in a manner required by leading regulatory authorities such as the EMA and produced by manufacturers with pertinent expertise.
Acknowledgments
The authors would like to acknowledge the editorial support provided by inVentiv Health Medical Communications. Philip Ford and Frances Gambling from inVentiv Health Medical Communications wrote the drafts of the article based on input from all authors, and styled the article per journal requirements. Biogen reviewed and provided feedback on the article to the authors. The authors had full editorial control of the article and provided their final approval of all content. All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this manuscript, take responsibility for the integrity of the work as a whole, and have given final approval of the version to be published.
Funding: This work was supported by Biogen, who provided funding for medical writing and editorial support in the development of this article.
Disclosure statement: P.D. has received speakers’ fee and/or honoraria for lectures or (non-product specific) advisory board meetings from AbbVie, Amgen, Celltrion, Hospira, Pfizer and Roche. M.F.R. is an employee of Biogen International GmbH.
References
- 1. Kumar R, Singh J.. Biosimilar drugs: current status. Int J Appl Basic Med Res 2014;4:63–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Declerck PJ. Biosimilar monoclonal antibodies: a science-based regulatory challenge. Expert Opin Biol Ther 2013;13:153–6. [DOI] [PubMed] [Google Scholar]
- 3. Vincent FB, Morand EF, Murphy K. et al. Antidrug antibodies (ADAb) to tumour necrosis factor (TNF)-specific neutralising agents in chronic inflammatory diseases: a real issue, a clinical perspective. Ann Rheum Dis 2013;72:165–78. [DOI] [PubMed] [Google Scholar]
- 4. Rosman Z, Shoenfeld Y, Zandman-Goddard G.. Biologic therapy for autoimmune diseases: an update. BMC Med 2013;11:88.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Mulcahy AW, Predmore Z, Mattke S. The Cost Savings Potential of Biosimilar Drugs in the United States. 2014. https://www.rand.org/content/dam/rand/pubs/perspectives/PE100/PE127/RAND_PE127.pdf (10 October 2016, date last accessed).
- 6. European Medicines Agency. Guideline on similar biological medicinal products containing biotechnology-derived proteins as active substance: non-clinical and clinical issues. 18 December 2014. http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2015/01/WC500180219.pdf (4 July 2016, date last accessed).
- 7. Weiss M, Bielsky MC, De Smet K. et al. Biosimilars: what clinicians should know. Blood 2012;120:5111–7. [DOI] [PubMed] [Google Scholar]
- 8. European Medicines Agency. Guideline on similar biological medicinal products containing monoclonal antibodies – non-clinical and clinical issues. 30 May 2012. http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2012/06/WC500128686.pdf (10 October 2016, date last accessed).
- 9. Grabowski H, Guha R, Salgado M.. Biosimilar competition: lessons from Europe. Nat Rev Drug Discov 2014;13:99–100. [DOI] [PubMed] [Google Scholar]
- 10. IMS Institute for HealthCare Informatics. Delivering on the potential of biosimilar medicines. The role of functioning competitive markets. March 2016. http://www.imshealth.com/files/web/IMSH%20Institute/Healthcare%20Briefs/Documents/IMS_Institute_Biosimilar_Brief_March_2016.pdf (September 2016, date last accessed).
- 11. Dörner T, Strand V, Cornes P. et al. The changing landscape of biosimilars in rheumatology. Ann Rheum Dis 2016;75:974–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Schiestl M, Stangler T, Torella C. et al. Acceptable changes in quality attributes of glycosylated biopharmaceuticals. Nat Biotechnol 2011;29:310–2. [DOI] [PubMed] [Google Scholar]
- 13. Declerck P, Farouk-Rezk M, Rudd PM.. Biosimilarity versus manufacturing change: two distinct concepts. Pharm Res 2016;33:261–8. [DOI] [PubMed] [Google Scholar]
- 14. Schellekens H. Biosimilar therapeutics—what do we need to consider? NDT Plus 2009;2:i27–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Shealy DJ, Cai A, Staquet K. et al. Characterization of golimumab, a human monoclonal antibody specific for human tumor necrosis factor α. MAbs 2010;2:428–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Baert F, Noman M, Vermeire S. et al. Influence of immunogenicity on the long-term efficacy of infliximab in Crohn's disease. N Engl J Med 2003;348:601–8. [DOI] [PubMed] [Google Scholar]
- 17. Finckh A, Simard JF, Gabay C, Guerne PA; SCQM physicians. Evidence for differential acquired drug resistance to anti-tumour necrosis factor agents in rheumatoid arthritis. Ann Rheum Dis 2006;65:746–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bui LA, Hurst S, Finch GL. et al. Key considerations in the preclinical development of biosimilars. Drug Discov Today 2015;20(Suppl. 1):3–15. [DOI] [PubMed] [Google Scholar]
- 19. Camacho LH, Frost CP, Abella E, Morrow PK, Whittaker S.. Biosimilars 101: considerations for U.S. oncologists in clinical practice. Cancer Med 2014;3:889–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kenett RS, Kenett DA.. Quality by design applications in biosimilar pharmaceutical products. Accred Qual Assur 2008;13:681–90. [Google Scholar]
- 21. European Medicines Agency. Guideline on Similar Biological Medicinal Products. 30 October 2005. http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500003517.pdf (10 October 2016, date last accessed).
- 22. European Medicines Agency. Guideline on Similar Biological Medicinal Products. 22 May 2013. http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2013/05/WC500142978.pdf (10 October 2016, date last accessed).
- 23. European Medicines Agency. Guideline on similar biological medicinal products containing biotechnology-derived proteins as active substances: quality issues (revision 1). 22 May 2014. http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2014/06/WC500167838.pdf (10 October 2016, date last accessed).
- 24. European Commission. What you need to know about biosimilar medicinal products – a consensus information document. ENTR.R.4.DIR 18 December 2014. http://ec.europa.eu/DocsRoom/documents/8242 (10 October 2016, date last accessed).
- 25. Nowicki M. Basic facts about biosimilars. Kidney Blood Press 2007;30:267–72. [DOI] [PubMed] [Google Scholar]
- 26. van Aerts LA, De Smet K, Reichmann G, van der Laan JW, Schneider CK.. Biosimilars entering the clinic without animal studies: a paradigm shift in the European Union. MAbs 2014;6:1155–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. European Medicines Agency. Concept paper on the revision of the guideline on non-clinical and clinical development of similar biological medicinal products containing recombinant granulocyte-colony stimulating factor. 23 July 2015. http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2015/07/WC500190635.pdf (10 October 2016, date last accessed).
- 28. European Medicines Agency. Guideline on the clinical evaluation of the pharmacokinetics of therapeutic proteins. 24 January 2007. http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500003029.pdf (10 October 2016, date last accessed).
- 29. European Medicines Agency. Clinical efficacy and safety guidelines. http://www.ema.europa.eu/ema/index.jsp?curl=pages/regulation/general/general_content_000085.jsp (10 October 2016, date last accessed).
- 30. European Medicines Agency. Guideline on immunogenicity assessment of biotechnology-derived proteins. 13 December 2007. http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500003946.pdf (10 October 2016, date last accessed).
- 31. European Medicines Agency. Guideline on immunogenicity assessment of monoclonal antibodies intended for in vivo clinical use. 24 May 2012. http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2012/06/WC500128688.pdf (10 October 2016, date last accessed).
- 32. Avci AB, Feist E, Burmester GR.. Biologicals in rheumatoid arthritis: current and future. RMD Open 2015;1:e000127.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Putrik P, Ramiro S, Kvien TK. et al. Inequities in access to biologic and synthetic DMARDs across 46 European countries. Ann Rheum Dis 2014;73:198–206. [DOI] [PubMed] [Google Scholar]
- 34. Lapadula G, Ferraccioli GF.. Biosimilars in rheumatology: pharmacological and pharmacoeconomic issues. Clin Exp Rheumatol 2012;30(Suppl. 73):S102–6. [PubMed] [Google Scholar]
- 35. Porter D, van Melckebeke J, Dale J. et al. Tumour necrosis factor inhibition versus rituximab for patients with rheumatoid arthritis who require biological treatment (ORBIT): an open label, randomised controlled, noninferiority, trial. Lancet 2016;388:239–47. [DOI] [PubMed] [Google Scholar]
- 36. Smolen JS, Nash P, Durez P. et al. Maintenance, reduction, or withdrawal of etanercept after treatment with etanercept and methotrexate in patients with moderate rheumatoid arthritis (PRESERVE): a randomised controlled trial. Lancet 2013;381:918–29. [DOI] [PubMed] [Google Scholar]
- 37. van Herwaarden N, van der Maas A, Minten MJ. et al. Disease activity guided dose reduction and withdrawal of adalimumab or etanercept compared with usual care in rheumatoid arthritis: open label, randomised controlled, non-inferiority trial. BMJ 2015;350:h1389.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Fautrel B, Pham T, Alfaiate T. et al. Stepdown strategy of spacing TNF blocker injections for established rheumatoid arthritis in remission: results of the multicentre noninferiority randomised open label controlled trial (STRASS: Spacing of TNFblocker injections in Rheumatoid ArthritiS Study). Ann Rheum Dis 2016;75:59–67. [DOI] [PubMed] [Google Scholar]
- 39. Möttönen T, Hannonen P, Leirisalo-Repo M. et al. Comparison of combination therapy with single-drug therapy in early rheumatoid arthritis: a randomised trial. FIN-RACo Trial Group. Lancet 1999;353:1568–73. [DOI] [PubMed] [Google Scholar]
- 40. Grigor C, Capell H, Stirling A. et al. Effect of a treatment strategy of tight control for rheumatoid arthritis (the TICORA study): a single-blind randomised controlled trial. Lancet 2004;364:263–9. [DOI] [PubMed] [Google Scholar]
- 41. Goekoop-Ruiterman YP, de Vries-Bouwstra JK, Allaart C. et al. Clinical and radiographic outcomes of four different treatment strategies in patients with early rheumatoid arthritis (the BeSt study): a randomized, controlled trial. Arthritis Rheum 2005;52:3381–90. [DOI] [PubMed] [Google Scholar]
- 42. Scott DL, Ibrahim F, Farewell V. et al. Randomised controlled trial of Tumour necrosis factor inhibitors Against Combination Intensive Therapy with conventional disease-modifying antirheumatic drugs in established rheumatoid arthritis: the TACIT trial an associated systematic reviews. Health Technol Assess 2014;18:i-xxiv, 1–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Hazlewood GS, Barnabe C, Tomlinson G, Marshall D, Devoe D, Bombardier C.. Methotrexate monotherapy and methotrexate combination therapy with traditional and biologic disease modifying antirheumatic drugs for rheumatoid arthritis: abridged Cochrane systematic review and network meta-analysis. BMJ 2016;353:i177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Flixabi Summary of Product Characteristics 2016. http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/004020/WC500208356.pdf (10 October 2016, date last accessed).
- 45. Inflectra Summary of Product Characteristics 2016. http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/002778/WC500151489.pdf (10 October 2016, date last accessed).
- 46. Remsima Summary of Product Characteristics 2016. http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/002576/WC500150871.pdf (10 October 2016, date last accessed).
- 47. Benepali Summary of Product Characteristics. http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/004007/WC500200378.pdf (10 October 2016, date last accessed).
- 48. Samsung Bioepis. Benepali gains approval for extra indications. GaBI Online – Generics and Biosimilars Initiative. 13 January 2017. http://www.gabionline.net/Biosimilars/News/Benepali-gains-approval-for-extra-indications (10 October 2016, date last accessed).
- 49. Dörner T, Strand V, Castañeda-Hernández G. et al. The role of biosimilars in the treatment of rheumatic diseases. Ann Rheum Dis 2013;72:322–8. [DOI] [PubMed] [Google Scholar]
- 50. Beck A. Biosimilar, biobetter and next generation therapeutic antibodies. mAbs 2011;3:107–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Elgundi Z, Reslan M, Cruz E, Sifniotis V, Kayser V.. The state-of-play and future of antibody therapeutics. Adv Drug Deliv Rev 2017. (in press) doi: 10.1016/j.addr.2016.11.004. [DOI] [PubMed] [Google Scholar]
- 52. Nickisch K, Bode-Greuel KM.. Biosimilars or biobetters: make your decision wisely. RA J App Res 2016;2:530–5. [Google Scholar]
- 53. Correia IR. Stability of IgG isotypes in serum. mAbs 2010;2:221–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Brodszky V, Baji P, Balogh O, Péntek M.. Budget impact analysis of biosimilar infliximab (CT-P13) for the treatment of rheumatoid arthritis in six Central and Eastern European countries. Eur J Health Econ 2014;15(Suppl. 1):S65–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Jha A, Upton A, Dunlop WC, Akehurst R.. The budget impact of biosimilar infliximab (Remsima®) for the treatment of autoimmune diseases in five European countries. Adv Ther 2015;32:742–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Nikiphorou E, Kautiainen H, Hannonen P. et al. Clinical effectiveness of CT-P13 (infliximab biosimilar) used as a switch from Remicade (infliximab) in patients with established rheumatic disease. Report of clinical experience based on prospective observational data. Expert Opin Biol Ther 2015;15:1677–83. [DOI] [PubMed] [Google Scholar]
- 57. Braun J, Kudrin A.. Switching to biosimilar infliximab (CT-P13): Evidence of clinical safety, effectiveness and impact on public health. Biologicals 2016;44:257–66. [DOI] [PubMed] [Google Scholar]