

Abstract
Developing drugs that target KRAS, the most frequently mutated oncogene in cancer, has not been successful despite much concerted efforts dedicated towards it in the last thirty years. Considering the key role this driver oncogene plays, the pharmacological drugging of KRAS remains a key challenge for cancer research. In this review, we highlight the emerging experimental strategies for blocking KRAS function and signaling and its direct targeting. We also report on the results in this field of research produced by our group.
Introduction
Colorectal cancer (CRC) is one of the most common cancers worldwide with 1.4 million new cases and 694.000 deaths reported in 2012 and where it is the fourth most deadly cancer after lung, liver and stomach [1]. Interestingly, the last decade has seen a decrease in the number of new CRC cases particularly in Western countries due to a variety of reasons including the implementation of colonoscopies as screening tests, the use of chemopreventive agents such as non-steroidal anti-inflammatory drugs (NSAIDs) and the clustering of diets, nutritional supplements and regular exercise in a healthy lifestyle [2–4].
At the same time, the outcome of CRC patients, in particular that of metastatic CRC (mCRC) patients, has markedly improved given that the median overall survival more than doubled in the last twenty years reaching approximately 30 months. This amelioration in the treatment of mCRC has been obtained by combining different cytotoxic drugs, in particular the protracted infusion of 5-fluorouracil modulated by leucovorin in combination with irinotecan (FOLFIRI) or with oxaliplatin (FOLFOX), capecitabine and oxaliplatin combination (XELOX) or 5-fluorouracil, leucovorin, irinotecan and oxaliplatin (FOLFOXIRI) [5, 6] and subsequently, by the introduction of targeted-therapy [7].
As for other types of cancer, genomic instability plays a major role in CRC and where three different groups have been identified as the pathogenic mechanisms, including mainly microsatellite instability (MSI), CpG island methylator phenotype (CIMP) and chromosomal instability (CIN), which represents up to 80–85% of the causes of all CRC cases [8]. Within these types, affected pathways have been reported to be involved in cell proliferation and survival such as WNT, MAPK/PI3K, TGF-β, TP53 and mutations in different genes including c-MYC, BRAF, PIK3CA, PTEN, SMAD2 and SMAD4 and finally RAS.
The three human RAS genes (KRAS, NRAS and HRAS) are the most frequently mutated oncogenes in human cancer appearing in 90% of pancreatic, 35% of lung and in 45% of colon cancers. These high occurrences make RAS one of the most important targets in oncology for drug development [9]. In particular, KRAS is the isoform prevalently mutated in pancreas, lung and colon cancer, while NRAS is the predominant isoform mutated in cutaneous melanomas and acute myelogenous leukemia and HRAS is the predominant isoform mutated in the bladder [9]. The reasons for this high prevalence of RAS mutation in cancers and for the preferential mutation in some kind of cancers still remains to be elucidated. The three human RAS genes that encode four small guanosine triphosphatase (GTPases) are KRAS4A, KRAS4B, HRAS and NRAS. RAS is the component of the mitogen activated protein kinase (MAPK) signaling pathway, which is activated by a ligand binding to a receptor tyrosine kinase (RTK) such as the epidermal growth factor receptor (EGFR). RAS exists in the non-active (GDP, guanosine diphosphatase) or active-state (GTP) and the transition between these two states is responsible for signal transduction events occurring from the cell surface receptor to the inside of the cell which is crucial for cell growth and differentiation [9]. In physiological conditions, this switch is catalysed by two guanine exchange factors known as SOS1/2, that promote the activation of RAS proteins by stimulating GDP for GTP exchange and GTPase-activating proteins (GAPs), which in turn accelerate RAS-mediated GTP hydrolysis. The GTP-bound form of RAS is the activated state and, together with the insensitivity to cytosolic GAP [9], represents the biochemical key defect of mutant RAS proteins, which results in persistent accumulation of the active, GTP-bound protein and activation of multiple downstream effectors (Fig. 1).
Fig. 1.
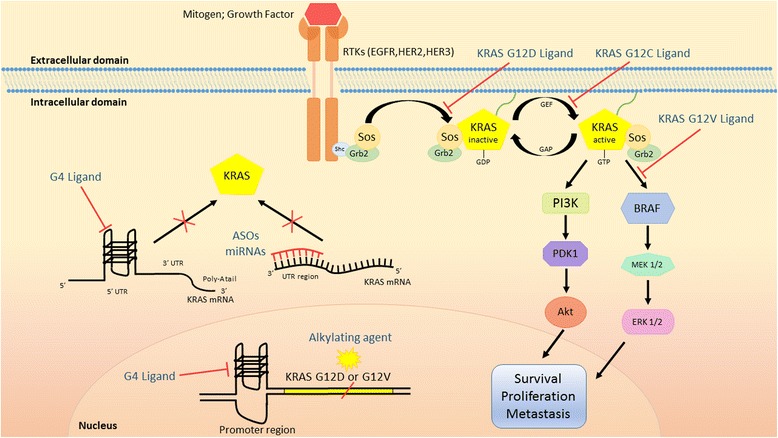
Direct targeting of KRAS. ASOs: Antisense Oligonucleotides; GAP: GTP-ase Activating Proteins; GEF: Guanine Nucleotide Exchange Factor; G4: G-quadruplex; RTKs: Receptor Tyrosine Kinases
Mutated KRAS is a major driver for malignant transformation in pancreatic tumors and in lung adenocarcinoma, as G12C mutations are detected in early lesions, retained in all metastases and are a hallmark in the exposure to tobacco smoke, respectively [10]. Regarding CRC, although KRAS mutations occur as an early event in about 50% of cases, they are probably not the primary initiating events being but the loss of APC or mutations in β-catenin in mismatch repair deficient tumors. The degree to which these tumors depend on KRAS is still underinvestigation [11] but these high occurrences make RAS one of the most important drug targets for cancer, including CRC.
Emerging biomarkers of resistance to anti-EGFR treatment
Most human epithelial cancer are characterized by the functional activation of growth factors and receptors of the epidermal growth factor receptor (EGFR) family. EGFR is a receptor tyrosine kinase which, by triggering a series of intracellular signals, controls proliferation of cancer cells, differentiation, angiogenesis and metastasis dissemination. In particular, 60–80% of patients with CRC overexpress EGFR, this has been associated with the progression of the malignancy and with poor prognosis. A clinical benefit for mCRC patients has been observed after the approval of humanized monoclonal antibodies cetuximab and panitumumab, directed against the extracellular domain of the EGFR, which block ligand binding and lead to inhibition of the downstream RAS-RAF-MEK-ERK signaling pathway. Subsequently, it was observed that EGFR status did not predict the efficacy of treatment [7]. In contrast, it has been found that resistance to anti-EGFR therapy is mediated by mutations in KRAS gene which result in constitutive activation of the RAS-RAF-MEK-ERK pathway. This means that RAS testing is mandatory before treatment with anti-EGFR therapy and and RAS analysis should include at least KRAS exons 2, 3 and 4 (codons 12, 13, 59, 61, 117 and 146) and NRAS exons 2, 3 and 4 (codons 12, 13, 59, 61 and 117) [7]. In addition, the presence of BRAF V600E mutation, which is found in 8–12% of patients with mCRC, is almost exclusively non-overlapping with RAS mutations and should be assessed before the initiation of therapy with anti-EGFR. In fact, there are accumulating data showing that BRAF mutations are predictive of resistance to anti-EGFR therapy [12].
Despite wild-type RAS, mCRC patients initially respond to cetuximab or panitumumab, treatment is effective in less than 10% of patients and the majority of them will exhibit disease progression [13, 14], thus suggesting the onset of acquired resistance to anti-EGFR therapy. In the last-five years many biomarkers and pathways involved in anti-EGFR resistance have been identified and are continuously on the rise, mainly due to the use of novel technologies and advanced approaches.
From this perspective, the integration of the classical tumour biopsy with liquid biopsy and the use of next-generation sequencing (NGS) covering hundred of genes, represents a very potent approach that permits to dissect molecular tumour heterogeneity and to detect emerging mutations responsible for the failure of anti-EGFR therapy that are not captured by biopsy in single lesions. In particular, the analysis of plasma DNA permitted to Bardelli’s group [15] to reveal a novel mechanism of acquired resistance to anti-EGFR therapy in patient which initially responded and subsequently progressed. In fact, they observed the presence of KRAS mutated alleles in blood samples obtained at progression accompanied by EGFR ectodomain mutations and KRAS or MET gene amplification. Interestingly, the KRAS mutant alleles, which emerge at the time of disease progression, decline at the suspension of anti-EGFR therapy, thus indicating the dynamic mode of the tumor cell population that was confirmed in experimental models. Accordingly to these observations, the authors planned to re-challenge patients which progressed after anti-EGFR therapy but showed a subsequent decline of KRAS mutant clones in circulating tumor DNA, thus leading authors to postulate that clonal evolution of tumor cells that survive treatment with anti-EGFR antibodies, continues beyond the clinical progression. The lack of response to anti-EGFR therapy has been associated with alterations in different genes. In particular, one patient with KRAS and NRAS wild-type who initially responded to chemotherapy plus cetuximab but that successively progressed, evidenced a K57 T MEK1 mutation in the NGS analysis of the primary tumour and liver metastasis [16]. This mutation was not observed prior to cetuximab treatment, suggesting MEK1 as a novel mechanism of acquired resistance to cetuximab. Subsequent in vitro experiments, showing that the combination with the MEK inhibitor trametinib restored sensitivity to anti-EGFR antibodies, the patient was treated with trametinib and panitumumab that elicited a reduction in size of the liver metastasis previously analyzed, however other metastatic lesions had in the meantime progressed. The analysis of circulating (ct) DNA from peripheral blood confirmed the presence of MAP2K1 p.K57 T variants prior to starting therapy with trametinib and panitumumab and the decline of the levels in the course of treatment, thus indicating the specific targeting of MEK1 mutant clones. Surprisingly, the ctDNA analysis revealed a KRAS p.Q61H mutation which lay undetected in the liver metastasis prior to therapy with trametinib and panitumumab. Moreover, the levels of mutated DNA markedly increased during treatment, thus suggesting the pre-existence of resistant KRAS-mutant clone responsible for the failure of cetuximab-based therapy.
Very recently, Pietrantonio et al. [17] analyzed tumor biopsies and ctDNA of 22 RAS-BRAF-wild-type, HER2/MET-negative mCRC patients, which benefited from anti-EGFR treatment and then developed resistance. Interestingly, this is the first study in which acquired resistance was studied in the tissue and the liquid biopsy of the same patient. They observed that the occurrence of anti-EGFR resistance was associated with RAS mutation and HER2/MET amplification and in particular an intralesion and interlesion heterogeneity of the resistance mechanism, thus highlighting the complexity of clonal evolution of resistance to EGFR-blockade. To this purpose, this high genetic heterogeneity of mCRC permits the authors to point out the differences with mutated-EGFR adenocarcinoma resistant to first generation TKIs, but sensitive to third generation of TKIs targeting T790-mutated EGFR [18].
All these results highlight the key role of RAS mutations and molecular heterogeneity of the resistance to anti-EGFR therapy and reinforce the need for more effective strategies to improve the outcome of mCRC patients.
Approaches in targeting KRAS function and signaling
The knowledge of the biochemical defects in mutant RAS proteins was the starting point in developing the first RAS inhibitor but attempts to discover small molecules able to antagonize RAS-GTP binding failed due to the picomolar affinity of RAS for GTP and the millimolar cellular concentrations of GTP [9, 19]. These unfortunate early efforts in discovering anti-RAS drugs and the lack of well-defined hydrophobic pockets on the surface of RAS proteins, contributed to the general perception that the direct targeting of RAS may not be actionable [9]. For these reasons, indirect approaches to inhibit mutant RAS have been investigated and most advances have been represented by targeting the association of RAS with the plasma membrane, which is necessary for trafficking the protein from the cytoplasm to the inner face of the cell and for activating the effector pathway. Since the lipid modification of RAS proteins by a farnesyl isoprenoid is a key step for this association in this context, farnesyl transferase inhibitors (FTI) were developed and in particular two compounds (lonafarnib and tipifarnib) were tested in solid tumors with KRAS mutation in clinical trials, including CRC. Unfortunately, FTIs resulted ineffective as anti-cancer drugs in advanced phase III clinical trials, as H-RAS, N-RAS and K-RAS were shown to be refractory to inactivation by FTIs and can still associate with membranes as well as continue to function [20].
A recent approach towards disrupting the association of RAS with the plasma membrane is represented by the targeting of the prenyl-binding protein PDEδ which is crucial for the plasma membrane localization of prenylated RAS. Understanding the role of PDEδ in the trafficking and signaling of RAS led to the development of two small-molecules deltarasin and deltazinone, which occupy the farnesyl–binding pocket of PDEδ and inhibit the localization of Ras to plasma membrane. These compounds, used at low micromolar levels, were able to inhibit in vitro growth and tumorigenic ability of mutant KRAS tumor cell lines [21, 22].
The inhibition of RAF-MEK-ERK mitogen-activated kinase (MAPK) signaling pathways represents a further approach for targeting RAS activity. In this context, being RAF kinases the first to be activated in the MAPK pathway, the drug discovery led to the approval of two RAF-inhibitors in melanoma, vemurafenib and dafravenib. Paradoxically, when these compounds were evaluated in KRAS-mutant tumors, ERK was activated instead of being inhibited as expected. This effect was explained by the fact that vemurafenib and dabrafenib are selective for BRAF only, while, in KRAS-mutant tumors, BRAF inhibitors drive BRAF-CRAF binding which finally activated ERK [19, 23]. Based on these lines of evidence, pan-inhibitors are being developed such as LY3009120 and PLX8394, which inhibit BRAF and CRAF isoforms with similar affinity and do not activate the MAPK pathway when a RAS mutation is present. In particular, these compounds showed activity against in vitro cultured cells and in xenografts from tumors carrying KRAS, NRAS, or BRAF mutation [24–27].
Direct targeting of KRAS
Despite several challenges and many failed attempts in targeting KRAS, researchers remain determined to search for new and effective approaches, and where the most promising of them will be discussed in the last sections of this review and highlighted in Fig. 1.
a. Targeting of mutated sites. Several studies have been performed to identify molecules able to bind the mutated sites of KRAS or inhibit the synthesis at the DNA level of the mutated protein and consequently blocking KRAS biological activity. In particular, groups from academic institutions or private companies are currently working on developing small molecules that are able to bind KRAS G12D mutant, compete with the KRAS-SOS interaction and prevent the formation of active KRAS-GTP [28, 29]. A different approach was represented by the use of Kobe0065-family compounds which inhibit the interaction between GTP-bound RAS and RAF and showed antitumor activity against CRC xenografts KRAS G12 V mutant [30]. As reported by Wilson and Tolias [31], these are promising compounds that could serve as a basis for the optimization of clinically active compounds.
A further strategy for developing small molecules like RAS-inhibitors, is the targeting of the cysteine in the mutated KRASG12C, thus impacting only the KRAS-mutated form. In particular, Ostrem et al. [32] have identified compounds that irreversibly bind to KRASG12C, favour the nucleotide GDP state over GTP, impair the binding to Raf and finally decrease viability and increase apoptosis of G12C-containing cancer cell lines. Following these results, Patricelli et al. [33] after testing several candidates for their activity against purified recombinant KRASG12C, as well as for their ability to engage KRASG12C in cells, identified ARS-853 as the most potent compound. The in vitro experiments demonstrated that the treatment of KRASG12C-mutated cells with ARS-853 inhibited the downstream signaling through both MAPK (including pMEK, pERK, and pRSK) and PI3K signaling (pAKT) pathways finally leading to a reduction in cell proliferation. In general, these studies provide convincing evidence that the targeting of inactive GDP-bound form could be a promising approach for generating novel and effective anti-RAS compounds. Obviously, in vivo experiments need to be performed before demonstrating that these compounds might be useful in the clinical setting. Moreover, given that G12C mutation is very rare in CRC while the most abundant is the G12D, the use of these compounds will be used only for a limited number of patients. Nevertheless, these results reinforce the idea of drugging a protein that has been deemed “undruggable” for longer.
Very recently Ross et al. [34] proposed a strategy against KRAS based on the use of the AZD4785, a 16-mer antisense oligonucleotides (ASO) complementary to the 3′ untranslated region (3′UTR) of KRAS mRNA sequences in a distinctly different region from the mutation codon sites, thus targeting both wild-type or mutant-KRAS. In particular, they used 2′-4′ constrained ethyl (cEt)-modified residues ASOs which showed a high stability and did not present the off-target toxicity effects which was the main issue of the first-generation phosphorotioate ASO [35]. The experiments performed in vitro on a panel of tumor cell lines of different histotypes including CRC, showed that despite AZD4785 induces a potent down-regulation of both wild-type and mutant KRAS protein, AZD4785 treatment reduces the proliferation only in mutant KRAS, while wild-type cells are not affected. Importantly, a selective inhibition of downstream MAPK and PI3K pathway signaling pathway was observed in mutant KRAS cells treated with AZD4785 and the inhibition of KRAS was not limited by the feedback reactivation of MAPK pathway, which could be a critical factor for the clinical use of these compounds. The experiments performed in vivo on xenografts and on PDXs demonstrate the high therapeutic efficacy of AZD4785 delivered systematically in mice. Finally, treatment with AZD4785 was well tolerated as only minimal changes in some clinical chemistry parameters and clinical pathology profiles were observed in mice and monkeys, thus confirming the potential use of this direct anti-KRAS strategy in the clinical setting [34].
To inhibit the mutated form of KRAS, Hiraoka et al. [36] proposed the use of a synthetic alkylating agent called KR12 (pyrrole-imidazole polyamide indole-seco-CBI conjugate). This small molecule was designed to recognize and alkylate adenine residues on the template strand at codon 12 (GTT and GAT), exon 2 of mutant KRAS. The authors demonstrated that KR12 produced a selective alkylation-mediated DNA strand cleavage at the G12D/G12 V mutations and this biochemical effect is associated with a greater reduction in the proliferation of CRC cells with G12D/G12 V mutation than that observed in G12C mutant cell, since KR12 does not recognize the latter mutation. Finally, the immunoblot analysis confirmed that the biological effects observed after KR12 treatment were related to the inhibition of KRAS, as a significant reduction in KRAS protein levels and in its GST-Raf-bound, active GTP-bound form, was observed. Interestingly, the in vivo experiments performed in immunosuppressed mice implanted with HT29 (KRAS WT), LS180 (heterozygous 12D/WT) or SW480 (homozygous 12 V) CRC cells, demonstrated that SW480 line was the most sensitive to the KR12 treatment since a marked inhibition of tumor growth was observed, while only a slight or no antitumor effect was observed on LS180 or HT29 models, respectively. Importantly, treatment was safe as mice remained healthy during as well as after the end of treatment, thus suggesting that KR12 could be a potential candidate for applying it in humans.
b. Targeting of G4 structures. G-quadruplex (G4) structures, formed in guanine-rich nucleic acid sequences, are abundant in regulatory regions of the genome, particularly in the promoters of a wide range of oncogenes, tumor suppressor genes and somatic copy number alterations related to cancer development [37, 38]. In particular, three G4 motifs were reported in the human KRAS promoter, called G4-proximal, G4-middle and G4-distal based on their proximity to the transcription start site [39]. Further studies have demonstrated that the most distal regions do not form an inducible nor stable G4 structure, whereas two more of the proximal regions do, particularly the G4-middle structure which seems to be the most critical for transcriptional silencing and for the development of targeted therapeutics [40]. Furthermore, in the KRAS promoter i-motifs have been identified on the complementary C-rich region which act as transcription activators. These observations led to the discovery of a benzophenanthridine alkaloid following a combined effect on the Mid-region i-motif and on the stabilization of G4, which down-regulated KRAS and cytotoxic activity against tumor cells [41]. Other than DNA, G4 structures were found in RNA sequences, including the 5′ untranslated region (UTRs) of KRAS mRNA [42]. Given that these sequences are important for post-transcriptional regulation of gene expression, such RNA motifs represent a potential target for G4-ligand small molecules.
A promising contribution to this field of research comes from Paulo’s group [43] which identified natural alkaloids Indoloquinolines as potential G4-ligand compounds for targeting of KRAS in CRC. In fact, they demonstrated the high selectivity of indolo[3,2-b]quinolines with a 7-carboxylate group and three alkylamine side chains (IQ3A) against G4 structures compared to duplex DNA and the ability on decreasing KRAS expression in HCT116 CRC lines. Successively, the same group showed that IQ3A compounds were able to inhibit KRAS mRNA and protein expression by the interaction with G4 structures present in the KRAS gene promoter and as a result, a decrease in cell proliferation and apoptosis induction was reported [44]. Interestingly, the IQ3A compounds were not selective for KRAS mutant cells as they are active both in wild-type and mutant CRC cells. It is important to underline that these compounds need to be evaluated in more complex experimental models than established cell lines, such xenografts or PDXs models of CRC, to establish the potential use against KRAS mutant CRC human tumors.
For several years our group focused on the study of novel G4 ligands as potential antineoplastic drugs and their mechanism of action [45–47]. In particular, we synthesized a new molecule, namely EMICORON, which has one piperidinyl group bound to the perylene bay area, sufficient to guarantee a good selectivity and an extended aromatic core able to increase the stacking interactions with the ending tetrad of the G4 [48]. Interestingly, we demonstrated that in addition to the ability of EMICORON to target G4 structures at telomeres, this compound is able to bind and stabilize the G-rich sequences located within the promoters of c-MYC, BCL-2 and VEGFR-2. Importantly, following EMICORON treatment the expression of genes was affected and this led to the inhibition of proliferation, induction of apoptosis and impairment of angiogenesis both in vitro and in vivo [49].
Moreover, this compound has shown promising toxicological characteristics, as we observed that treated mice were free of adverse effects, and showed a marked antitumor efficacy against different CRC preclinical models [50], including KRAS-mutated patient-derived xenografts (PDXs). These models, preserving the genomic integrity and tumor heterogeneity of the human tumor of origin, can better mimic the response of humans to new anticancer therapeutics [51].
To better detail the role of EMICORON in targeting KRAS-mutated CRC, experiments were performed in vitro on HCT-116 CRC cells, which were sensitive to EMICORON treatment in an in vivo model of dissemination metastases [50]. To this end, we showed that after treatment with EMICORON both KRAS mRNA and protein expression (Fig. 2a and b) were down-regulated in these cells, thus suggesting the direct targeting by this compound of G4 structures present in this gene. Interestingly, in our subsequent experiments in vivo performed in a panel of PDXs bearing KRAS mutations, we confirmed that this compound is very active against this tumor population (Fig. 3) for which no clinically effective therapy currently exists. In fact, we observed that EMICORON has a marked antitumoral activity against all the three different PDXs evaluated, as a reduced tumor mass was observed in treated mice compared to the untreated ones. Moreover, since most patients with advanced mutated KRAS mCRC and treated with systemic chemotherapy unfortunately showed disease progression, we investigated the ability of EMICORON to improve the efficacy of chemotherapy. In particular, EMICORON was added to the standard regimen FOLFIRI and the therapeutic efficacy of FOLFIRI alone was compared to the FOLFIRI plus EMICORON on CRC PDXs. As shown in Table I, after treatment with FOLFIRI one mouse exhibited tumor regression and three mice showed stabilization of the disease, while one mouse progressed. Interestingly, it is evident that the combination with EMICORON consistently improved the antitumor efficacy of FOLFIRI as regression in three mice treated and stabilization in the remaining two mice was observed. While the molecular mechanisms of EMICORON activity against KRAS-mutated tumors need to be fully elucidated, our data on HCT-116 cells showing the effect of EMICORON in down-regulating both KRAS mRNA and protein expression accompanied by the strong in vivo antitumor efficacy against a panel of KRAS-mutated CRC PDXs, suggest the potential of EMICORON for the clinical application of EMICORON in this mCRC clinical setting.
Fig. 2.
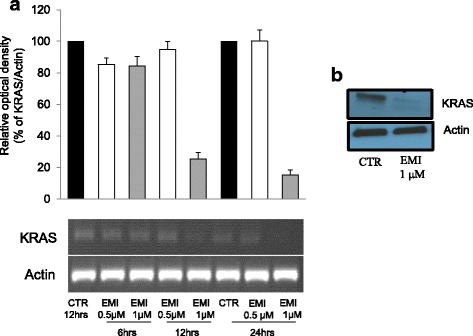
EMICORON downregulates KRAS expression. a HCT116 colon cancer cells were treated with EMICORON at doses of 0.5 and 1 μM for 6, 12, 24 h. The DNA extracted was amplified by PCR. The histogram shows the relative optical density of KRAS. Histograms show the mean values ± SD. A representative picture of PCR products is shown. Densitometry was performed with ImageJ software version 1.40. b KRAS protein levels were detected by Western blotting. The total cell extracts of HCT116 cells treated with EMICORON 1 μM for 24 h, were prepared and immunoblotting was conducted by antibodies against KRAS (Santa Cruz) or Actin (Santa Cruz)
Fig. 3.
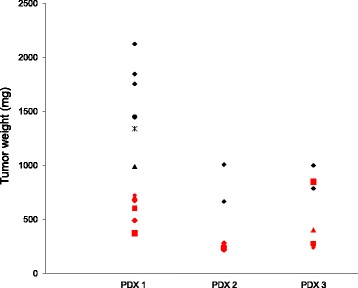
EMICORON has antitumoral activity against CRC KRAS-mutated PDXs. PDXs were obtained by the implant in mice of tumor fragments from three CRC patients at IRCCS Regina Elena National Cancer Institute (Rome, Italy). Briefly, surgical specimens, not required for histopathologic analysis, were placed in medium supplemented with antibiotics, diced into 15–20 mm3 pieces, coated in Matrigel and implanted in NOD.SCID mice by a small incision and subcutaneous pocket made in one side of the lower back [50]. After mass formation in mice (Passage 0), tumors were passed in four mice (Passage 1) and afterwards expanded in further mice for the drug experimentation (Passage 2). When tumors reached a mass of 250–300 mm3, EMICORON was administered per os at 15 mg/kg for seven days. Tumor growth was followed by caliper measurements. The tumor weight in untreated or EMICORON-treated mice was reported at the nadir of the effect. PDX 1 experiment included six untreated and six EMICORON-treated mice; PDX 2 experiment included two untreated and four EMICORON-treated mice; PDX 3 experiment included two untreated and four EMICORON-treated mice. The use of human specimens was approved by the Ethics Committee of the IRCCS Regina Elena National Cancer Institute (N. 823/2016) and animal procedures were in compliance with the national and international directives (D.L. March 4, 2014, no. 26; directive 2010/63/EU of the European Parliament and of the council) and were approved by the Ethics Committee of the IRCCS Regina Elena National Cancer Institute (N. 823/2016) and by the Italian Ministry of Health (N. 183/2017-PR)
Table 1.
EMICORON increase therapeutic efficacy of FOLFIRI regimen against CRC PDXs
| Treatmenta | Regressionb | Stable diseaseb | Progressionb |
|---|---|---|---|
| EMICORON | 0/5 | 1/5 | 4/5 |
| FOLFIRI | 1/5 | 3/5 | 1/5 |
| FOLFIRI plus EMICORON | 3/5 | 2/5 | 0/5 |
aTumor fragments derived from passage 1, were coated in Matrigel and implanted s.c. in mice (15–20 mm3/mouse) as described in the legend of fig. 2. When the tumor mass reached about 250 to 300 mm3, treatment started by giving mice FOLFIRI regimen consisting in Irinotecan (CPT-11) i.p. at 15 mg/kg for five consecutive days, followed by 5-Fluorouracil (5-FU) i.p. at 19 mg/kg plus Leucovorin at 20 mg/kg for five consecutive days. EMICORON was administered per os at 15 mg/kg for the next seven days. Five mice were included in each experimental group
bRegression or stable disease were considered when was observed the reduction or the maintenance, respectively, of the same tumor weight for at least two weeks after the starting of treatment
Moreover, this compound could be an attractive therapeutic also for KRAS wild type mCRC tumors, given that the majority of these patients acquired resistance to anti-EGFR monoclonal antibodies.
Targeting KRAS by microRNAs
We devoted a separate chapter to the emerging role of microRNAs (miRNAs) in the regulation of KRAS for two main reasons: 1) the increasing interest in these molecules for their role in crucial biological aspect of cancer, including CRC; 2) the mechanism of action which includes both the binding to regulatory regions of KRAS or to genes involved in KRAS-driven pathways. MiRNAs are small (~ 18–25 nucleotide) non-coding RNAs which silence gene expression post-transcriptionally by binding to specific regions in the 3′UTR of mRNA [52]. Many studies have demonstrated that miRNAs play a key role in many crucial biological processes including cancer mechanisms such as cell proliferation, metastasis, apoptosis and angiogenesis [53].
The first set of evidence demonstrating the involvement of miRNA as tumor suppressor in human cancer was obtained in 2002 by Croce’s group from studies in B-cell chronic lymphocytic leukemia cells [54]. Nowadays, an increasing number of groups are identifying a number of miRNAs as potential biomarkers for human cancer diagnosis, prognosis and therapeutic targets. Aberrant expression of miRNAs has been reported for most types of cancers, including CRC [55], and in particular miRNAs regulate critical pathways involved in the CRC pathogenesis such as the p53, PI3K, RAS, MAPK, EMT transcription factors, and Wnt/β-catenin pathways [56].
The miRNA-mediated regulation of RAS protein was firstly identified in 2005 by Johnson’s group which described the well studied Let-7 family of miRNAs [57]. The human Let-7 family contains 13 members that regulate cell proliferation and differentiation by directly targeting various oncogenes and where the Let-7 expression is deregulated in many human cancers [58, 59]. In particular, Let-7b miRNA have been shown to repress KRAS expression and inhibit mutant KRAS-dependent cell growth in vitro and tumor growth in vivo in lung and pancreatic cancer cell models [60, 61].
The role of some miRNA can vary between the different cancer histotypes. In particular, miR-96 can act as either oncomirs or tumor suppressors depending on the cell context. For instance, its expression has increased in lung, prostate, bladder, colorectal and breast cancer [62]. By contrast, the expression of miR-96 is strongly down-regulated in pancreatic cancer. Yu et al. [63] demonstrated that miR-96 can affect cell proliferation, migration and apoptosis in pancreatic cancer cells by binding the 3’UTR region of KRASG12C mRNA, inhibit its protein expression and consequently decrease Akt signaling. Other miRNAs have been shown to behave like KRAS tumor suppressors, in particular miRNA-181a in oral squamous cell carcinoma [64], and finally miRNA-30b in CRC [65]. Moreover, it has been reported that reduced expression of miR-143 contributes to CRC development through the derepression of KRAS expression [66]. In fact, these authors observed an inverse correlation between KRAS protein and miR-143 and the treatment with miR-143 mimic reduced KRAS expression while miR-143 inhibitor increased KRAS protein level. Moreover, after treatment with miR-143 inhibitor CRC cells exhibited an increased cell proliferation, whereas miR-143 overexpression reduced the proliferation. These biological effects have been correlated to the direct recognition by miR-143 of the 3′-untranslated region of KRAS transcripts followed by the inhibition of constitutive phosphorylation of ERK1/2. More recently, by using a synthetic lethal strategy, Zhou et al. [67] identified miR-1298 and its targets, FAK and LAMB3, as potential suppressors of KRAS-dependent cell growth in both CRC and NSCLC cells. In fact, they observed that the miR-1298 mimic was lethal in all the KRAS mutant cells analyzed, both in vitro and in vivo. Moreover, miR-1298 functions by the binding and degradation of target FAK (focal adhesion kinase) and LAMB3 (laminin β3 subunit) which are transcriptionally regulated by mutant KRAS.
The role of miRNAs in the regulation of KRAS in the CRC context summarized here suggests that miRNAs could be a further potential strategy to explore effective targeting of KRAS-driven tumors.
Conclusions
Even though it has been more than 30 years following the discovery of the role of KRAS in transforming cells and in driving cancer growth and development, there are no drugs in this current moment targeting the activating mutations of KRAS or selectively down-regulating KRAS mRNA and proteins, neither are there ones inhibiting downstream effector pathways in clinical trials. This represents a major problem in the treatment of three of the deadliest cancers, including pancreas, non-small cell lung and CRC, as KRAS mutations are associated with unfavourable prognosis in these tumor histotypes. In this review, we presented the most recent and promising advances in the development of new compounds able to target KRAS. We covered the many challenges emerging in this field of research, as most of these compounds are in the very early stages of preclinical experimentation or for other, mechanisms of action need to be elucidated. In summary, the road to discovering the first approved KRAS inhibitor is still long and winding, considering the genetic heterogeneity of KRAS-mutant cancers and the cell context. Nevertheless, these results represent fascinating efforts which could overcome the perception that KRAS is basically undruggable and that can help to develop effective drugs for targeting traditionally difficult pathways in the clinical setting.
Acknowledgements
We are grateful to Dr. Adele Petricca for secretarial assistance and to Dr. Tania Merlino for the English revision.
Funding
This work was supported by grants from the Italian Association for Cancer Research AIRC IG 16910 (AB) and AIRC IG 18637 (CL).
Competing interests
The authors declare that they have no competing interests.
Authors’contribution
MP and LP carried out the experiments presented in Figs 2, 3 and in Table 1. LP prepared the Fig. 1. AB and CL designed experiments and wrote the manuscript. CC discussed data and participated inthe writing of the manuscript. All authors read and approved the final manuscript.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Annamaria Biroccio, Email: annamaria.biroccio@ifo.gov.it.
Carlo Leonetti, Email: carlo.leonetti@ifo.gov.it.
References
- 1.Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M, et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015;136:E359–E386. doi: 10.1002/ijc.29210. [DOI] [PubMed] [Google Scholar]
- 2.Gurpinar E, Grizzle WE, Piazza GA. NSAIDs inhibit tumorigenesis, but how? Clin Cancer Res. 2014;20:1104–1113. doi: 10.1158/1078-0432.CCR-13-1573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Meyskens FL Jr, McLaren CE, Pelot D, Fujikawa-Brooks S, Carpenter PM, Hawk E, et al. Difluoromethylornithine plus sulindac for the prevention of sporadic colorectal adenomas: a randomized placebo-controlled, double-blind trial. Cancer Prev Res (Phila) 2008;1:32–38. [DOI] [PMC free article] [PubMed]
- 4.Gravitz L. Prevention: tending the gut. Nature. 2015;521:S6–S8. doi: 10.1038/521S6a. [DOI] [PubMed] [Google Scholar]
- 5.Cassidy J, Clarke S, Díaz-Rubio E, Scheithauer W, Figer A, Wong R, et al. Randomized phase III study of capecitabine plus oxaliplatin compared with fluorouracil/folinic acid plus oxaliplatin as first-line therapy for metastatic colorectal cancer. J Clin Oncol. 2008;26(12):2006. doi: 10.1200/JCO.2007.14.9898. [DOI] [PubMed] [Google Scholar]
- 6.Falcone A, Ricci S, Brunetti I, Pfanner E, Allegrini G, Barbara C, et al. Phase III trial of infusional fluorouracil, leucovorin, oxaliplatin, and irinotecan (FOLFOXIRI) compared with infusional fluorouracil, leucovorin, and irinotecan (FOLFIRI) as first-line treatment for metastatic colorectal cancer: the Gruppo Oncologico Nord Ovest. J Clin Oncol. 2007;25:1670–1676. doi: 10.1200/JCO.2006.09.0928. [DOI] [PubMed] [Google Scholar]
- 7.Van Cutsem E, Cervantes A, Adam R, Sobrero A, Van Krieken JH, Aderka D, et al. ESMO consensus guidelines for the management of patients with metastatic colorectal cancer. Ann Oncol. 2016;27:1386–1422. doi: 10.1093/annonc/mdw235. [DOI] [PubMed] [Google Scholar]
- 8.Grady WM, Carethers JM. Genomic and epigenetic instability in colorectal cancer pathogenesis. Gastroenterology. 2008;135:1079–1099. doi: 10.1053/j.gastro.2008.07.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cox AD, Fesik SW, Kimmelman AC, Luo J, Der CJ. Drugging the undruggable RAS: mission possible? Nat Rev Drug Discov. 2014;13:828–851. doi: 10.1038/nrd4389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Prior IA, Lewis PD, Mattos C. A comprehensive survey of Ras mutations in cancer. Cancer Res. 2012;72:2457–2467. doi: 10.1158/0008-5472.CAN-11-2612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McCormick F. KRAS as a therapeutic target. Clin Cancer Res. 2015;21:1797–1801. doi: 10.1158/1078-0432.CCR-14-2662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tol J, Nagtegaal ID, Punt CJ. BRAF mutation in metastatic colorectal cancer. N Engl J Med. 2009;361:98–99. doi: 10.1056/NEJMc0904160. [DOI] [PubMed] [Google Scholar]
- 13.Cunningham D, Humblet Y, Siena S, Khayat D, Bleiberg H, Santoro A, et al. Cetuximab monotherapy and cetuximab plus irinotecan in irinotecan-refractory metastatic colorectal cancer. N Engl J Med. 2004;351:337–345. doi: 10.1056/NEJMoa033025. [DOI] [PubMed] [Google Scholar]
- 14.Van Cutsem E, Peeters M, Siena S, Humblet Y, Hendlisz A, Neyns B, et al. Open-label phase III trial of panitumumab plus best supportive care compared with best supportive care alone in patients with chemotherapy-refractory metastatic colorectal cancer. J Clin Oncol. 2007;25:1658–1664. doi: 10.1200/JCO.2006.08.1620. [DOI] [PubMed] [Google Scholar]
- 15.Siravegna G, Mussolin B, Buscarino M, Corti G, Cassingena A, Crisafulli G, et al. Clonal evolution and resistance to EGFR blockade in the blood of colorectal cancer patients. Nat Med. 2015;21:795–801. doi: 10.1038/nm.3870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Russo M, Siravegna G, Blaszkowsky LS, Corti G, Crisafulli G, Ahronian LG, et al. Tumor heterogeneity and lesion-specific response to targeted therapy in colorectal cancer. Cancer Discov. 2016;6:147–153. doi: 10.1158/2159-8290.CD-15-1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pietrantonio F, Vernieri C, Siravegna G, Mennitto A, Berenato R, Perrone F, et al. Heterogeneity of acquired resistance to anti-EGFR monoclonal antibodies in patients with metastatic colorectal cancer. Clin Cancer Res. 2017;23:2414–2422. doi: 10.1158/1078-0432.CCR-16-1863. [DOI] [PubMed] [Google Scholar]
- 18.Cross DA, Ashton SE, Ghiorghiu S, Eberlein C, Nebhan CA, Spitzler PJ, et al. AZD9291, an irreversible EGFR TKI, overcomes T790M-mediated resistance to EGFR inhibitors in lung cancer. Cancer Discov. 2014;4:1046–1061. doi: 10.1158/2159-8290.CD-14-0337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Papke B, Der CJ, Drugging RAS. Know the enemy. Science. 2017;355:1158–1163. doi: 10.1126/science.aam7622. [DOI] [PubMed] [Google Scholar]
- 20.Karnoub AE, Weinberg RA. Ras oncogenes: split personalities. Nat Rev Mol Cell Biol. 2008;9:517–531. doi: 10.1038/nrm2438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Papke B, Murarka S, Vogel HA, Martín-Gago P, Kovacevic M, Truxius DC, et al. Identification of pyrazolopyridazinones as PDEδ inhibitors. Nat Commun. 2016;7:11360. doi: 10.1038/ncomms11360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.G Z, Schultz-Fademrecht C, Küchler P, Murarka S, Ismail S, Triola G, et al. Structure guided design and kinetic analysis of highly potent benzimidazole inhibitors targeting the PDEδprenyl binding site. J Med Chem. 2014;57:5435–5448. doi: 10.1021/jm500632s. [DOI] [PubMed] [Google Scholar]
- 23.Heidorn SJ1, Milagre C, Whittaker S, Nourry A, Niculescu-Duvas I, Dhomen N, et al. Kinase-dead BRAF and oncogenic RAS cooperate to drive tumor progression through CRAF. Cell 2010;140:209–221. [DOI] [PMC free article] [PubMed]
- 24.Peng SB, Henry JR, Kaufman MD, Lu WP, Smith BD, Vogeti S, et al. Inhibition of RAF isoforms and active dimers by LY3009120 leads to anti-tumor activities in RAS or BRAF mutant cancers. Cancer Cell. 2015;28:384–398. doi: 10.1016/j.ccell.2015.08.002. [DOI] [PubMed] [Google Scholar]
- 25.Vakana E, Pratt S, Blosser W, Dowless M, Simpson N, Yuan XJ, et al. LY3009120, a panRAF inhibitor, has significant anti-tumor activity in BRAF and KRAS mutant preclinical models of colorectal cancer. Oncotarget. 2017;8:9251–9266. doi: 10.18632/oncotarget.14002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang C, Spevak W, Zhang Y, Burton EA, Ma Y, Habets G, et al. RAF inhibitors that evade paradoxical MAPK pathway activation. Nature. 2015;526:583–586. doi: 10.1038/nature14982. [DOI] [PubMed] [Google Scholar]
- 27.Tutuka CSA, Andrews MC, Mariadason JM, Ioannidis P, Hudson C, Cebon J, et al. PLX8394, a new generation BRAF inhibitor, selectively inhibits BRAF in colonic adenocarcinoma cells and prevents paradoxical MAPK pathway activation. Mol Cancer. 2017;16:112. doi: 10.1186/s12943-017-0684-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Maurer T, Garrenton LS, Oh A, Pitts K, Anderson DJ, Skelton NJ, et al. Small-molecule ligands bind to a distinct pocket in Ras and inhibit SOS-mediated nucleotide exchange activity. Proc Natl Acad Sci U S A. 2012;109:5299–5304. doi: 10.1073/pnas.1116510109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sun Q, Burke JP, Phan J, Burns MC, Olejniczak ET, Waterson AG, et al. Discovery of small molecules that bind to K-Ras and inhibit Sos-mediated activation. Angew Chem Int Ed Engl. 2012;51:6140–6143. doi: 10.1002/anie.201201358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shima F, Yoshikawa Y, Ye M, Araki M, Matsumoto S, Liao J, et al. In silico discovery of small-molecule Ras inhibitors that display antitumor activity by blocking the Ras-effector interaction. Proc Natl Acad Sci U S A. 2013;110:8182–8187. doi: 10.1073/pnas.1217730110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wilson CY, Tolias P. Recent advances in cancer drug discovery targeting RAS. Drug Discov Today. 2016;21:1915–1919. doi: 10.1016/j.drudis.2016.08.002. [DOI] [PubMed] [Google Scholar]
- 32.Ostrem JM, Peters U, Sos ML, Wells JA, Shokat KM. K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature. 2013;503:548–551. doi: 10.1038/nature12796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Patricelli MP, Janes MR, Li LS, Hansen R, Peters U, Kessler LV, et al. Selective inhibition of oncogenic KRAS output with small molecules targeting the inactive state. Cancer Discov. 2016;6:316–329. doi: 10.1158/2159-8290.CD-15-1105. [DOI] [PubMed] [Google Scholar]
- 34.Ross SJ, Revenko AS, Hanson LL, Ellston R, Staniszewska A, Whalley N, et al. Targeting KRAS-dependent tumors with AZD4785, a high-affinity therapeutic antisense oligonucleotide inhibitor of KRAS. Sci Transl Med. 2017;9(394):eaal5253. [DOI] [PubMed]
- 35.Jones D. The long march of antisense. Nat Rev Drug Discov. 2011;10:401–402. doi: 10.1038/nrd3474. [DOI] [PubMed] [Google Scholar]
- 36.Hiraoka K, Inoue T, Taylor RD, Watanabe T, Koshikawa N, Yoda H, et al. Inhibition of KRAS codon 12 mutants using a novel DNA-alkylating pyrrole-imidazole polyamide conjugate. Nat Commun. 2015;6:6706. doi: 10.1038/ncomms7706. [DOI] [PubMed] [Google Scholar]
- 37.Balasubramanian S, Hurley LH, Neidle S. Targeting G-quadruplexes in gene promoters: a novel anticancer strategy? Nat Rev Drug Discov. 2011;10:261–275. doi: 10.1038/nrd3428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chambers VS, Marsico G, Boutell JM, Di Antonio M, Smith GP, Balasubramanian S. High-throughput sequencing of DNA G-quadruplex structures in the human genome. Nat Biotechnol. 2015;33:877–881. doi: 10.1038/nbt.3295. [DOI] [PubMed] [Google Scholar]
- 39.Cogoi S, Xodo LE. G4 DNA in ras genes and its potential in cancer therapy. Biochim Biophys Acta. 2016;1859:663–674. doi: 10.1016/j.bbagrm.2016.02.002. [DOI] [PubMed] [Google Scholar]
- 40.Morgan RK, Batra H, Gaerig VC, Hockings J, Brooks TA. Identification and characterization of a new G-quadruplex forming region within the kRAS promoter as a transcriptional regulator. Biochim Biophys Acta. 2016;1859:235–245. doi: 10.1016/j.bbagrm.2015.11.004. [DOI] [PubMed] [Google Scholar]
- 41.Kaiser CE, Van Ert NA, Agrawal P, Chawla R, Yang D, Hurley LH. Insight into the complexity of the i-motif and G-Quadruplex DNA structures formed in the KRAS promoter and subsequent drug-induced gene repression. J Am Chem Soc. 2017;139:8522–8536. doi: 10.1021/jacs.7b02046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bugaut A, Balasubramanian S. 5'-UTR RNA G-quadruplexes: translation regulation and targeting. Nucleic Acids Res. 2012;40:4727–4741. doi: 10.1093/nar/gks068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lavrado J, Borralho PM, Ohnmacht SA, Castro RE, Rodrigues CM, Moreira R, et al. Synthesis, G-quadruplexstabilisation, docking studies, and effect on cancer cells of indolo[3,2-b]quinolines with one, two, or three basic side chains. Chem Med Chem 2013;8:1648–1661. [DOI] [PubMed]
- 44.Brito H, Martins AC, Lavrado J, Mendes E, Francisco AP, Santos SA, et al. Targeting KRAS oncogene in colon cancer cells with 7-carboxylate Indolo[3,2-b]quinoline tri-Alkylamine derivatives. PLoS One. 2015;10:e0126891. doi: 10.1371/journal.pone.0126891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Leonetti C, Scarsella M, Riggio G, Rizzo A, Salvati E, D’Incalci M, et al. G-quadruplex ligand RHPS4 potentiates the antitumor activity of camptothecins in preclinical models of solid tumors. Clin Cancer Res. 2008;14:7284–7291. doi: 10.1158/1078-0432.CCR-08-0941. [DOI] [PubMed] [Google Scholar]
- 46.Salvati E, Scarsella M, Porru M, Rizzo A, Iachettini S, Tentori L, et al. PARP1 is activated at telomeres upon G4 stabilization: possible target for telomere-based therapy. Oncogene. 2010;29:6280–6293. doi: 10.1038/onc.2010.344. [DOI] [PubMed] [Google Scholar]
- 47.Biroccio A, Porru M, Rizzo A, Salvati E, D’Angelo C, Orlandi A, et al. DNA damage persistence as determinant of tumor sensitivity to the combination of topo I inhibitors and telomere-targeting agents. Clin Cancer Res. 2011;17:2227–2236. doi: 10.1158/1078-0432.CCR-10-3033. [DOI] [PubMed] [Google Scholar]
- 48.Franceschin M, Rizzo A, Casagrande V, Salvati E, Alvino A, Altieri A, et al. Aromatic core extension in the series of N-cyclic bay-substituted perylene G-quadruplex ligands: increased telomere damage, antitumor activity, and strong selectivity for neoplastic over healthy cells. ChemMedChem. 2012;7:2144–2154. doi: 10.1002/cmdc.201200348. [DOI] [PubMed] [Google Scholar]
- 49.Porru M, Zizza P, Franceschin M, Leonetti C, Biroccio A. EMICORON: A multi-targeting G4 ligand with a promising preclinical profile. Biochim Biophys Acta. 2017;1861(5 Pt B):1362–1370. doi: 10.1016/j.bbagen.2016.11.010. [DOI] [PubMed] [Google Scholar]
- 50.Porru M, Artuso S, Salvati E, Bianco A, Franceschin M, Diodoro MG, et al. Targeting G-quadruplex DNA structures by EMICORON has a strong antitumor efficacy against advanced models of human colon cancer. Mol Cancer Ther. 2015;14:2541–2551. doi: 10.1158/1535-7163.MCT-15-0253. [DOI] [PubMed] [Google Scholar]
- 51.Pompili L, Porru M, Caruso C, Biroccio A, Leonetti C. Patient-derived xenografts: a relevant preclinical model for drug development. J Exp Clin Cancer Res. 2016;35:189. doi: 10.1186/s13046-016-0462-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ha M, Kim VM. Regulation of microRNA biogenesis. Nat Rev Mol Cell Biol. 2014;15:509–524. doi: 10.1038/nrm3838. [DOI] [PubMed] [Google Scholar]
- 53.Peng Y, Croce CM. The role of MicroRNAs in human cancer. Signal Transduct Target Ther. 2016;1:15004. doi: 10.1038/sigtrans.2015.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Calin GA, Dumitru CD, Shimizu M, Bichi R, Zupo S, Noch E, et al. Frequent deletions and down-regulation of micro- RNA genes miR15 and miR16 at 13q14 in chronic lymphocytic leukemia. Proc Natl Acad Sci U S A. 2002;99:15524–15529. doi: 10.1073/pnas.242606799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhang GJ, Li JS, Zhou H, Xiao HX, Li Y, Zhou T. MicroRNA-106b promotes colorectal cancer cell migration and invasion by directly targeting DLC1. J Exp Clin Cancer Res. 2015;30:34–73. doi: 10.1186/s13046-015-0189-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chi Y, Zhou D. MicroRNAs in colorectal carcinoma from pathogenesis to therapy. J Exp Clin Cancer Res. 2016;10:35–43. doi: 10.1186/s13046-016-0320-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Johnson SM, Grosshans H, Shingara J, Byrom M, Jarvis R, Cheng A, et al. RAS is regulated by the Let-7 MicroRNA family. Cell. 2005;120:635–647. doi: 10.1016/j.cell.2005.01.014. [DOI] [PubMed] [Google Scholar]
- 58.Boyerinas B, Park SM, Hau A, Murmann AE, Peter ME. The role of let-7 in cell differentiation and cancer. Endocr Relat Cancer. 2010;17:F19–F36. doi: 10.1677/ERC-09-0184. [DOI] [PubMed] [Google Scholar]
- 59.Kim M, Slack FJ. MicroRNA-mediated regulation of KRAS in cancer. J Hematol Oncol. 2014;7:84. doi: 10.1186/s13045-014-0084-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Johnson CD, Esquela-Kerscher A, Stefani G, Byrom M, Kelnar K, Ovcharenko D, et al. The let-7 MicroRNA represses cell proliferation pathways in human cells. Cancer Res. 2007;67(16):7713–7722. doi: 10.1158/0008-5472.CAN-07-1083. [DOI] [PubMed] [Google Scholar]
- 61.Rachagani S, Macha MA, Menning MS, Dey P, Pai P, Smith LM, et al. Changes in microRNA (miRNA) expression during pancreatic cancer development and progression in a genetically engineered KrasG12D; Pdx1-Cre mouse (KC) model. Oncotarget. 2015;6:40295–40309. doi: 10.18632/oncotarget.5641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hong Y, Liang H, Uzair-Ur-Rehman WY, Zhang W, Zhou Y, et al. miR-96 promotes cell proliferation, migration and invasion by targeting PTPN9 in breast cancer. Sci Rep. 2016;6:37421. doi: 10.1038/srep37421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yu S, Lu Z, Liu C, Meng Y, Ma Y, Zhao W, et al. miRNA-96 suppresses KRAS and functions as a tumor suppressor gene in pancreatic cancer. Cancer Res. 2010;70:6015–6025. doi: 10.1158/0008-5472.CAN-09-4531. [DOI] [PubMed] [Google Scholar]
- 64.Shin KH, Bae SD, Hong HS, Kim RH, Kang MK, Park NH. miR-181a shows tumor suppressive effect against oral squamous cell carcinoma cells by downregulating K-ras. Biochem Biophys Res Commun. 2011;404:896–902. doi: 10.1016/j.bbrc.2010.12.055. [DOI] [PubMed] [Google Scholar]
- 65.Liao WT, Ye YP, Zhang NJ, Li TT, Wang SY, Cui YM, et al. MicroRNA-30b functions as a tumour suppressor in human colorectal cancer by targeting KRAS, PIK3CD and BCL2. J Pathol. 2014;232:415–427. doi: 10.1002/path.4309. [DOI] [PubMed] [Google Scholar]
- 66.Chen X, Guo X, Zhang H, Xiang Y, Chen J, Yin Y, et al. Role of miR-143 targeting KRAS in colorectal tumorigenesis. Oncogene. 2009;28:1385–1392. doi: 10.1038/onc.2008.474. [DOI] [PubMed] [Google Scholar]
- 67.Zhou Y, Dang J, Chang KY, Yau E, Aza-Blanc P, Moscat J, et al. miR-1298 inhibits mutant KRAS-driven tumor growth by repressing FAK and LAMB3. Cancer Res. 2016;76:5777–5787. doi: 10.1158/0008-5472.CAN-15-2936. [DOI] [PMC free article] [PubMed] [Google Scholar]