

Abstract
We previously showed that the mitochondrial fatty acid oxidation enzyme long-chain acyl-CoA dehydrogenase (LCAD) is expressed in alveolar type II pneumocytes and that LCAD-/- mice have altered breathing mechanics and surfactant defects. Here, we hypothesized that LCAD-/-mice would be susceptible to influenza infection. Indeed, LCAD-/- mice demonstrated increased mortality following infection with 2009 pandemic influenza (A/CA/07/09). However, the mortality was not due to increased lung injury, as inflammatory cell counts, viral titers, and histology scores all showed non-significant trends toward milder injury in LCAD-/- mice. To confirm this, LCAD-/- were infected with a second, mouse-adapted H1N1 virus (A/PR/8/34), to which they responded with significantly less lung injury. While both strains become increasingly hypoglycemic over the first week post-infection, LCAD-/- mice lose body weight more rapidly than wild-type mice. Surprisingly, while acutely fasted LCAD-/- mice develop hepatic steatosis, influenza-infected LCAD-/- mice do not. They do, however, become more hypothermic than wild-type mice and demonstrate increased blood lactate values. We conclude that LCAD-/- mice succumb to influenza from bioenergetic starvation, likely due to increased reliance upon glucose for energy.
Keywords: fatty acid oxidation, long-chain acyl-CoA dehydrogenase, lung injury, influenza
1. Introduction
Respiratory viral infections such as influenza have long been known to serve as triggers for metabolic decompensation and mortality in patients with genetic fatty acid oxidation (FAO) disorders [1]. The mechanisms behind the metabolic decompensation are not well understood. We previously showed that the alveolar type II pneumocyte (ATII), a key mitochondria-rich cell type in the lung, catalyzes FAO at high rates [2]. Both mouse and human ATII cells abundantly express the FAO enzyme long-chain acyl-CoA dehydrogenase (LCAD). LCAD-/- mice have increased lung epithelial permeability, altered breathing mechanics, and dysfunctional pulmonary surfactant [2]. Based on this, we hypothesized that LCAD-/- mice would show enhanced sensitivity to lung injury during a respiratory infection. To test this, LCAD-/- mice were infected with two different strains of influenza and evaluated for lung pathology as well as indicators of energy metabolism.
2. Methods
2.1. Animals and influenza treatments
All protocols were approved by the University of Pittsburgh Institutional Animal Care and Use Committee. LCAD+/- mice (B6.129S6-Acadltm1Uab) were purchased from the Mutant Mouse Regional Resource Center (University of Missouri, Columbia, MO) on a C57Bl/6 strain background. Due to infertility on the C57Bl/6 background, the LCAD-/- mice used here were maintained on a mixed C57Bl/6 and 129S6 background. Age and gender-matched wild-type B6/129S6 mice served as controls for all experiments. For influenza infection, the mouse-adapted A/PR/8/34 H1N1 virus (PR8) was propagated in chicken eggs as described [3]. The 2009 pandemic virus A/CA07/09 H1N1 (CA07) was propagated in MDCK cells [4]. Female mice age 6-8 weeks were given 80 plaque forming units (pfu) in 50 μl of sterile PBS by oropharyngeal aspiration. Body weights were tracked daily. Blood glucose and lactate were measured in unrestrained animals by nicking the tip of the tail and collecting droplets of blood into assay strips for handheld analyzers. A digital rodent rectal temperature probe (Physitemps Instruments, Clifton, NJ) was used to monitor core body temperature.
2.2. Bronchoalveolar lavage fluid (BALF) collection and analysis
Mice were anesthetized, tracheotomized, and bronchoalveolar lavage performed using 1-ml of 0.9% NaCl. The amount of saline recovered was measured and recorded. The fluid was centrifuged at 300 × g for 10 min to pellet the cells, which were immediately resuspended in PBS, centrifuged onto glass slides, and stained for differential counting. The cell-free BALF supernatants were snap frozen in liquid nitrogen and stored at -80°C for later viral titer assays performed by standard MDCK plaque assay [4].
2.3. Lung histology
The lungs were inflated with 10% neutral-buffered formalin at a pressure of 25 cm of H2O for 10 min, then placed in fresh 10% neutral-buffered formalin for 24 h before processing for H&E staining. A pathologist blind to the genotypes scored lung injury on a severity scale of 1 (mild) to 5 (severe).
2.4. Liver triglycerides
Snap-frozen liver tissue was assayed for triglyceride content exactly as described [5].
2.5. Western blotting
Western blotting was carried out as described. Primary anti-surfactant protein-A (SP-A) antibody (Proteintech, Inc) was used at a dilution of 1:300.
3. Results & Discussion
3.1 Increased mortality in LCAD-/- mice
Based on our previous work demonstrating altered breathing mechanics, surfactant defects, and increased epithelial permeability [2], we hypothesized that LCAD-/- mice would be more sensitive to influenza infection. Indeed, 8-week old LCAD-/- female mice infected with the CA07 virus displayed significantly reduced survival compared to wild-type control mice (Fig 1A). LCAD-/- mice began to succumb by Day 5 post-infection compared to Day 8 for the wild-type controls. Weight loss is considered a good marker of disease severity in influenza-infected mice [6]; here, we observed significantly exacerbated weight loss in the LCAD-/- mice by Day 4 post-infection (Fig 1B).
Figure 1. Increased mortality in LCAD-/- mice following CA07 infection.
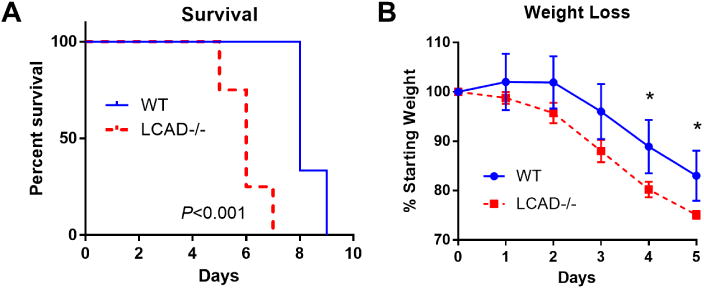
LCAD-/- female mice (N=6) and matched wild-type controls (N=6) were infected with the 2009 pandemic CA07 influenza virus. A) LCAD-/- mice exhibited poorer survival and B) greater weight loss than control mice. *P<0.05.
3.2 Increased mortality in LCAD-/- mice is not due to lung injury
Given the baseline lung defects previously established in LCAD-/- mice we hypothesized that the shortened survival time following infection was caused by enhanced lung injury. To test this, we repeated the CA07 infection but with tissue harvest on Day 5, just prior to the onset of mortality. The mice were subjected to bronchoalveolar lavage and lung tissue was processed for histopathology. Contrary to our hypothesis, the LCAD-/- mice did not show signs of enhanced lung injury following CA07 infection. There was a nonsignificant trend toward reduced inflammatory cells in bronchoalveolar lavage fluid (BALF) from LCAD-/- mice (Figs 2A), and the distribution of these cell counts across neutrophils, monocytes, and lymphocytes were not different between genotypes (Fig 2B). Equal titers of influenza virus in BALF indicated that viral replication and infectivity were not altered in LCAD-/- lungs (Fig 2C). In keeping with the lack of difference in these parameters, histological examination of lung tissue revealed no significant difference in severity of injury as scored by a pathologist blind to the genotypes (Fig 2D).
Figure 2. Increased mortality in CA07-infected LCAD-/- mice is not due to lung injury.
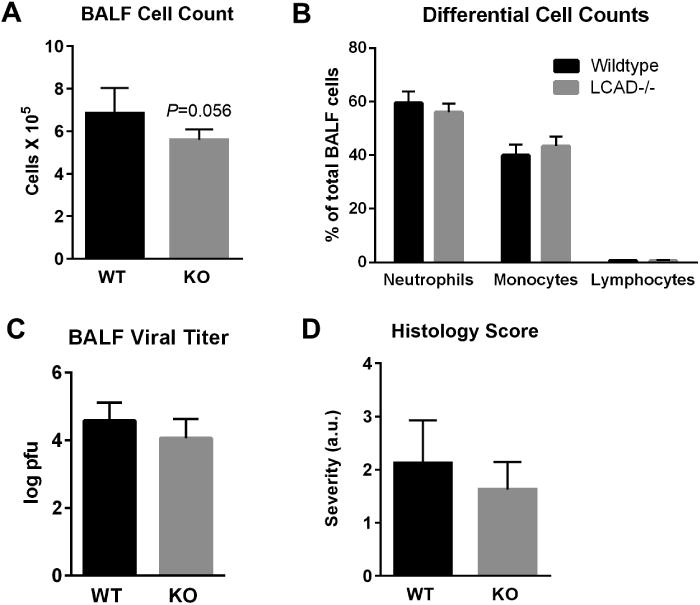
LCAD-/- female mice (N=6) and matched wild-type controls (N=6) were subjected to bronchoalveolar lavage and tissue harvest on Day 5 following infection with CA07. A) Total cell count in bronchoalveolar lavage fluid (BALF) trended toward being lower in LCAD-/- mice, but B) the distribution of these cells (% of total) across different cell types was not different. C) BALF viral titer by plaque assay, and D) lung injury severity scores as judged by a pathologist blind to the genotypes.
Seasonal H1N1 strains such as CA07 have been shown to cause less severe lung injury in mice than the more commonly used mouse-adapted PR8 strain [6,7]. We therefore infected a cohort of LCAD-/- and wild-type control mice with PR8. Compared to wild-type, LCAD-/- mice displayed significantly less lung tissue injury (Figs 3A-C). The protection against injury in LCAD-/- lungs was limited to the parenchyma, as histology scores were not significantly different between genotypes in the perivascular or peribronchial regions of the lung.
Figure 3. LCAD-/- mice infected with PR8 show less lung injury than wild-type mice.
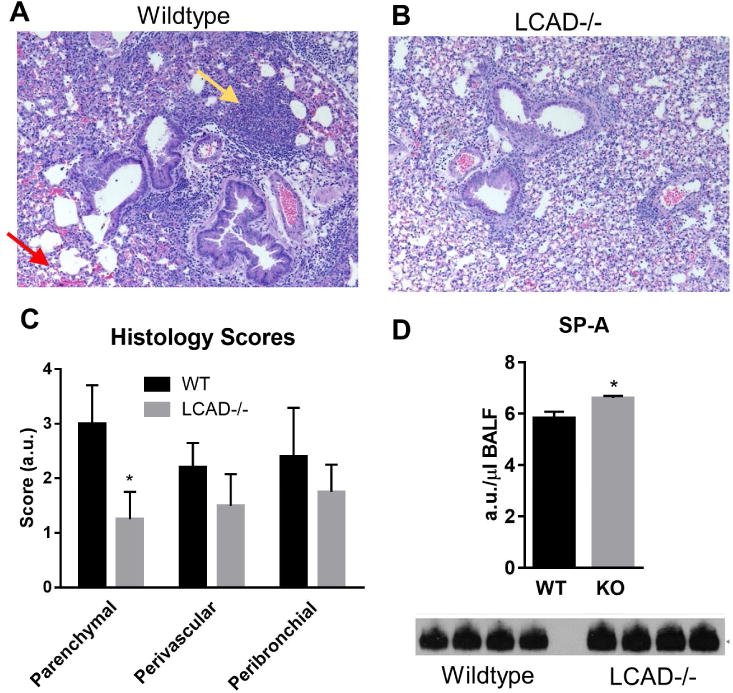
LCAD-/- female mice (N=5) and matched wild-type controls (N=5) were infected with mouse-adapted PR8 influenza. Lung histology showed more inflammatory cell invasion (yellow arrow) and less red blood cell leakage across the epithelial barrier (red arrow) in wild-type (A) compared to LCAD-/- mice (B). Severity scores generated by a pathologist blind to the genotypes (C) indicated significantly less parenchymal tissue damage in LCAD-/- lungs. Reduced injury in LCAD-/- lung may be related to increased BALF content of the collectin surfactant protein-A (SP-A) as measured by western-blotting of equal volumes of BALF (D). *P<0.01.
The protection of LCAD-/- lung parenchyma from PR8-induced injury was an unexpected finding. Our previous work implicated surfactant defects as the cause of the altered breathing mechanics in LCAD-/- mice [2]. LCAD-/- had significantly less total surfactant as well as relative changes in composition in terms of acyl chain lengths and degree of unsaturation [2]. Surfactant is known to be the first line of defense against pathogens, but this defensive property is mostly ascribed to the surfactant proteins A (SP-A) and D (SP-D), which are innate immune system collectins that opsonize pathogens and mark them for phagocytosis by alveolar macrophages [8]. The lipid component of pulmonary surfactant has actually been shown to promote viral infection of the alveolar epithelium, particularly dipalmitoylphosphatidylcholine (DPPC), a highly abundant surfactant phospholipid with two saturated acyl chains [8]. DPPC undergoes a constant cycle of cellular uptake and recycling. Viral particles can bind to DPPC and thereby obtain entry into epithelial cells [9]. In contrast to DPPC, phosphatidylcholine species with unsaturated acyl chains retard entry of viral particles into cells [9]. Our previous characterization of the surfactant profile in LCAD-/- lung indicated a paucity of DPPC with increased abundance of unsaturated phosphatidylcholine species [2]. This may be the mechanism that allows either normal (CA07) or suppressed (PR8) lung injury in the face of a viral infection. Here, we additionally show that the key surfactant collectin SP-A is increased in BALF from LCAD-/- mice under basal conditions (Fig 3D). Based on our previous and current observations we postulate that a combination of low DPPC and high SP-A helps protect LCAD-/- lungs against infection, particularly from the mouse-adapted PR8 virus.
3.3 Metabolic effects of influenza infection
The data discussed above suggested that extra-pulmonary factors must contribute to the increased mortality in LCAD-/- mice. LCAD-/- mice are known to be sensitive to fasting and develop fasting-induced hepatic steatosis, hypoglycemia, and hypothermia [10,11]. Given that influenza has been reported to severely reduce food intake in mice [12], we predicted that CA07 would drive severe hypoglycemia, fatty liver, and hypothermia in LCAD-/- mice. Surprisingly, while CA07 did indeed induce hypoglycemia, there was no difference in blood glucose values between genotypes (Fig 4A). Likewise, while LCAD-/- livers from uninfected mice had significantly more triglycerides at baseline and especially after an overnight fast (Fig 4B), the levels of triglycerides on Day 5 post-infection with CA07 were the same as wild-type. Core body temperatures were significantly lower in LCAD-/- mice after an acute 16-hr fast as well as on Day 5 post-infection with CA07 (Fig 4C).
Figure 4. Metabolic effects of CA07 infection.
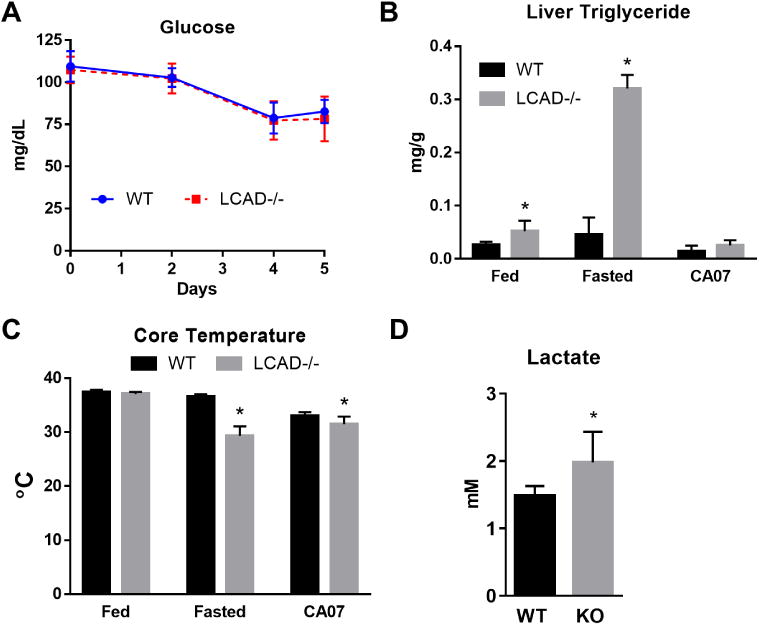
A) Both LCAD-/- (N=6) and wild-type (N=6) mice show progressive hypoglycemia over five days following infection with CA07. B) LCAD-/-mice have hepatic steatosis basally (fed) and after an acute 16-hr fast (fasted), but not on Day 5 post CA07 infection. C) Compared to wild-type, LCAD-/- mice display significantly greater hypothermia after an acute 16-hr fast (fasted) or on Day 5 post-CA07 infection. D) LCAD-/- mice have significantly higher blood lactate values on Day 5 post-CA07 infection. *p<0.05.
Our metabolic data indicate that the effects of influenza are different than those of an acute, overnight fast. The course of influenza is that of a slow starvation rather than the rapid and massive mobilization of adipose-derived lipids seen in mice subjected to overnight fasting. The latter overwhelms the liver of humans and animals with FAO deficiencies resulting in hepatic steatosis and hypoketotic hypoglycemia [1,13]. LCAD-/- mice retain a partial ∼40% capacity for hepatic FAO due to very long-chain acyl-CoA dehydrogenase (VLCAD) which has overlapping substrate specificity [14]. We speculate that during influenza the presence of VLCAD in LCAD-/- mice is sufficient to maintain liver function, thereby explaining the lack of lipid deposition and the similar blood glucose values between LCAD-/- and wild-type mice during the course of infection. The cause of mortality appears to be wasting, as the enhanced weight loss was the most prominent difference we observed between genotypes of mice (Fig 1B). LCAD-/- mice have an increased reliance upon glucose for energy in peripheral tissues and thus a higher respiratory exchange ratio (RER)[15,16]. In the current study this is supported by the observation of higher blood lactate values in LCAD-/- mice after infection with CA07 (Fig 4D). We propose that the decreased metabolic efficiency in LCAD-/- mice leads to faster depletion of energy stores and muscle wasting in order to fuel gluconeogenesis, ultimately leading to more severe morbidity and death. Future studies are needed to determine the inter-relationships between inborn errors of metabolism, weight loss, and mortality from influenza.
Supplementary Material
Highlights.
LCAD-/- mice show increased mortality after influenza infection.
LCAD-/- mice exhibit equal or less lung injury depending upon the influenza strain.
LCAD-/- mice lose more body weight and become more hypothermic during infection.
Acknowledgments
This work was supported by National Institutes of Health grants DK090242 (E.S.G.), HL113655 (J.W.), and HL107380 (J.F.A.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Vishwanath VA. Fatty Acid Beta-Oxidation Disorders: A Brief Review. Ann Neurosci. 2016;23:51–55. doi: 10.1159/000443556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Goetzman ES, Alcorn JF, Bharathi SS, Uppala R, McHugh KJ, Kosmider B, Chen R, Zuo YY, Beck ME, McKinney RW, Skilling H, Suhrie KR, Karunanidhi A, Yeasted R, Otsubo C, Ellis B, Tyurina YY, Kagan VE, Mallampalli RK, Vockley J. Long-chain acyl-CoA dehydrogenase deficiency as a cause of pulmonary surfactant dysfunction. J Biol Chem. 2014;289:10668–10679. doi: 10.1074/jbc.M113.540260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Braciale TJ. Immunologic recognition of influenza virus-infected cells. I. Generation of a virus-strain specific and a cross-reactive subpopulation of cytotoxic T cells in the response to type A influenza viruses of different subtypes. Cell Immunol. 1977;33:423–436. doi: 10.1016/0008-8749(77)90170-8. [DOI] [PubMed] [Google Scholar]
- 4.Zhang H, Luo J, Alcorn JF, Chen K, Fan S, Pilewski J, Liu A, Chen W, Kolls JK, Wang J. AIM2 Inflammasome Is Critical for Influenza-Induced Lung Injury and Mortality. J Immunol. 2017;198:4383–4393. doi: 10.4049/jimmunol.1600714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jouihan H. Measurement of Liver Triglyceride Content. Bio-Protocls. 2012;2:e223. [Google Scholar]
- 6.Bouvier NM, Lowen AC. Animal Models for Influenza Virus Pathogenesis and Transmission. Viruses. 2010;2:1530–1563. doi: 10.3390/v20801530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lv J, Wang D, Hua YH, Pei SJ, Wang J, Hu WW, Wang XL, Jia N, Jiang QS. Pulmonary immune responses to 2009 pandemic influenza A (H1N1) virus in mice. BMC Infect Dis. 2014;14:197. doi: 10.1186/1471-2334-14-197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Glasser JR, Mallampalli RK. Surfactant and its role in the pathobiology of pulmonary infection. Microbes Infect. 2012;14:17–25. doi: 10.1016/j.micinf.2011.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Balakireva L, Schoehn G, Thouvenin E, Chroboczek J. Binding of adenovirus capsid to dipalmitoyl phosphatidylcholine provides a novel pathway for virus entry. J Virol. 2003;77:4858–4866. doi: 10.1128/JVI.77.8.4858-4866.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Guerra C, Koza RA, Walsh K, Kurtz DM, Wood PA, Kozak LP. Abnormal nonshivering thermogenesis in mice with inherited defects of fatty acid oxidation. J Clin Invest. 1998;102:1724–1731. doi: 10.1172/JCI4532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kurtz DM, Rinaldo P, Rhead WJ, Tian L, Millington DS, Vockley J, Hamm DA, Brix AE, Lindsey JR, Pinkert CA, O'Brien WE, Wood PA. Targeted disruption of mouse long-chain acyl-CoA dehydrogenase gene reveals crucial roles for fatty acid oxidation. Proc Natl Acad Sci U S A. 1998;95:15592–15597. doi: 10.1073/pnas.95.26.15592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Klein MS, Conn CA, Kluger MJ. Behavioral thermoregulation in mice inoculated with influenza virus. Physiol Behav. 1992;52:1133–1139. doi: 10.1016/0031-9384(92)90472-e. [DOI] [PubMed] [Google Scholar]
- 13.Schuler AM, Wood PA. Mouse models for disorders of mitochondrial fatty acid beta-oxidation. ILAR J. 2002;43:57–65. doi: 10.1093/ilar.43.2.57. [DOI] [PubMed] [Google Scholar]
- 14.Cox KB, Hamm DA, Millington DS, Matern D, Vockley J, Rinaldo P, Pinkert CA, Rhead WJ, Lindsey JR, Wood PA. Gestational, pathologic and biochemical differences between very long-chain acyl-CoA dehydrogenase deficiency and long-chain acyl-CoA dehydrogenase deficiency in the mouse. Hum Mol Genet. 2001;10:2069–2077. doi: 10.1093/hmg/10.19.2069. [DOI] [PubMed] [Google Scholar]
- 15.Houten SM, Herrema H, Te Brinke H, Denis S, Ruiter JP, van Dijk TH, Argmann CA, Ottenhoff R, Muller M, Groen AK, Kuipers F, Reijngoud DJ, Wanders RJ. Impaired amino acid metabolism contributes to fasting-induced hypoglycemia in fatty acid oxidation defects. Hum Mol Genet. 2013;22:5249–5261. doi: 10.1093/hmg/ddt382. [DOI] [PubMed] [Google Scholar]
- 16.Zhang D, Liu ZX, Choi CS, Tian L, Kibbey R, Dong J, Cline GW, Wood PA, Shulman GI. Mitochondrial dysfunction due to long-chain Acyl-CoA dehydrogenase deficiency causes hepatic steatosis and hepatic insulin resistance. Proc Natl Acad Sci U S A. 2007;104:17075–17080. doi: 10.1073/pnas.0707060104. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.