

Abstract
Measuring molecular binding to membrane proteins is critical for understanding cellular functions, validating biomarkers and screening drugs. Despite the importance, developing such a capability has been a difficult challenge, especially for small molecule binding to membrane proteins in their native cellular environment. Here we show that the binding of both large and small molecules to membrane proteins can be quantified on single cells by trapping single cells with a microfluidic device, and detecting binding-induced cellular membrane deformation on nanometer-scale with label-free optical imaging. We develop a thermodynamic model to describe the binding-induced membrane deformation, validate the model by examining the dependence of membrane deformation on cell stiffness, membrane protein expression level and binding affinity, and study four major types of membrane proteins, including glycoproteins, ion channels, G-protein coupled and tyrosine kinase receptors. The single cell detection capability reveals the importance of local membrane environment on molecular binding, and variability in the binding kinetics of different cell lines, and heterogeneity of different cells within the same cell line.
Keywords: single cell analysis, binding kinetics, membrane proteins, label-free detection, and microfluidics
Graphical abstract
Membrane proteins are responsible for relaying signals between a cell and its external environment, moving ions and molecules across the membrane, and allows it to recognize other cells and attach to a surface, all vital for the survival of living organisms.1–3 Membrane proteins are also important for disease diagnosis and treatment, accounting for over a half of all drug targets.4,5 For both studying the cellular functions and discovering new drugs, it is necessary to measure the binding of membrane proteins with various ligand and drug candidate molecules, and quantify the binding kinetics in order to determine how strong and fast the binding is3,6 and evaluate drug potency and efficacy.6–8
Despite the importance, quantifying the binding kinetics of membrane proteins on whole cells has been a difficult task. A common approach to study molecular interactions is to use radioactive9–12 or fluorescent labels.13–17 The latter includes total internal reflection fluorescence (TIRF) microscopy18,19 and super-resolution microscopy.20,21 TIRF is limited to imaging of the bottom of cells only because the evanescent wave penetration length is ~200 nm from the surface on which the cells are attached. Super-resolution microscopy takes several minutes to acquire one image frame, making it difficult to track binding kinetics in real time. To determine the kinetics, the current mainstream practice is to isolate membrane proteins from the cell membranes and measure the binding with a label-free detection technology, such as surface plasmon resonance (SPR), quartz crystal microbalance (QCM) and Bio-layer interferometry.22–27 The isolation procedure involves extraction of membrane proteins from cells, purification and immobilization of them on a sensing surface, which is laborious, and often difficult because of the low solubility and low expression levels of many membrane proteins. Furthermore, the isolated membrane proteins may lose their native structures and functions outside the lipid environment. Studying membrane proteins in the native cellular membranes is thus important.28–30 A significant advance is the study of membrane proteins on the surfaces of unilamellar vesicles with a backscattering interferometry technology.31 The technology is still an endpoint assay.
All the above label-free technologies are mass based detection, and their signals diminish with the molecular weight, making them difficult to measure small molecule binding to proteins. Small molecules account for more than 90% FDA approved drugs, and play essential roles in various cellular functions. We have recently shown that molecular binding to a cell adhered on a surface causes it to deform.29 However, the molecular binding-induced cellular deformation is sensitive to the detail of the cell-surface adhesion, and varies from one region of the cell to another, which complicates the interpretation of the data.
Here we report a method to trap single cells onto an array of micro-holes with a microfluidic device, and measure the molecular binding to membrane proteins by tracking the cellular mechanical deformation with sub-nm precision using an optical imaging system. This label-free method offers a capability of measuring both large and small molecule binding to membrane proteins on single cells in real time. We demonstrate this capability by measuring the binding kinetics of major types of transmembrane proteins, including glycoproteins, ion channels, G-protein coupled receptors (GPCRs) and tyrosine kinase receptors on single whole cells. We establish the basic principle of the method by developing a model to express the binding-induced nanometer-scale cellular deformation in terms of molecular interactions, and cellular mechanical properties, and carrying out experiments to validate the model.
RESULTS
Detection Principle
Thermodynamics predicts that molecular binding to a surface changes the surface tension. This phenomenon was manifested in microcantilever biosensors, where molecular binding to a microcantilever causes it to bend, and the amount of bending increases with decreasing thickness of the microcantilever.32 Cell membranes are a few nanometer thick, much thinner than typical microcantilevers (a few microns), so we expect to observe cell membrane deformation upon molecular binding to the membrane proteins on a cell surface. By tracking the membrane deformation in real time, we can thus quantify the kinetics of molecular binding taking place on the surface of a trapped cell as shown in Figure 1a. The change in the diameter of the trapped cell (ΔD) can be obtained by minimizing the free energy of the cell surface,33 including membrane bending energy, surface tension, membrane lipid-membrane protein interactions, and entropy change associated with molecular binding, which is given by (Supporting Information S-1),
| (1) |
where Λ1 and Λ2 are the interaction strengths between the membrane lipids and the membrane proteins (receptors) with and without bound molecules (ligands), respectively, H is the mean curvature, κ is the bending modulus, C0 is the spontaneous curvature of the cell, γ is the surface tension, and ϕ is the fraction of the receptors with bound ligands. Eq. 1 shows that the molecular binding induced membrane deformation is proportional to the number of ligands bound to the receptors.34–36 According to this model, the membrane deformation depends on the nature of ligand-receptor interactions, but it is not directly related to the masses of the ligands. So the present method works for both large and small molecule ligands, as long as the binding changes the interactions of the receptors with the membrane.
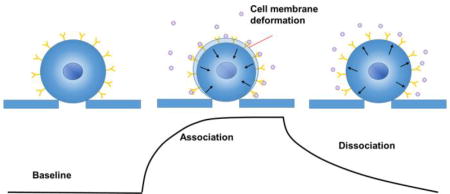
Figure 1. Principle and setup for measuring binding of small and large molecules to membrane proteins on trapped cells.

(a) Schematic illustration of the experimental setup consisting of a microfluidic system for trapping single cells onto micro-holes, and for introducing ligand molecules at different concentrations for binding kinetics measurement, and an optical imaging and signal processing system for tracking the cell deformation associated with the binding in real time. (b) Flow design of the cell trapping microfluidic chip and optical images of trapped cells with 40× phase contrast objectives. (c) Schematics of a binding kinetic curve determined from the cell deformation. Insets: Cell edge positions before binding (i), during binding (association) (ii), and during dissociation (iii), where the blue and red boxes indicate a region of interest (ROI) used in a differential optical tracking algorithm of the cell deformation. (d) Differential image intensity vs. cell edge position (inset), where the two vertical dashed lines mark a linear region used in the differential optical tracking algorithm. (e) Calibration curve plotting differential image intensity vs. cell deformation (edge movement distance).
We used a microfluidic chip consisting of two parallel fluidic channels separated with a thin wall with micro-holes (diameter of 10 µm) to trap single cells for measurement. Channel 1 had an inlet and outlet to allow sample and buffer solutions to flow in and out, and channel 2 had a lower pressure than channel 1 (Figure 1a, and Supporting Information S-2). We flew cells along channel 1 while maintaining a lower pressure in channel 2, which resulted in trapping of the cells onto the individual micro-holes (Figure 1b).37 We then introduced ligands from channel 1, and studied binding of the ligands to the membrane protein receptors on each of the trapped cells by measuring the binding-induced mechanical deformation of the cell as stated in Eq. 1.
To measure the small binding induced cell deformation, we used a differential optical tracking method (Figure 1c). First, we imaged the trapped cells with phase contrast microscopy, which clearly revealed the edge of each cell. We then selected a rectangular region of interest (ROI) such that the cell edge passed through the center of the ROI, and then divided the ROI into two equal halves, one was inside the cell (red), and the other half fell outside of the cell (blue, Figure 1c inset). When the cell deformed, the image intensity in one half increased, and the other half decreased. The differential image intensity of the two halves was defined as, (I1−I2)/(I1+I2), where I1 and I2 are the intensities of the first and second halves, respectively, which was proportional to cell deformation (Figure S2). We calibrated this differential deformation-tracking algorithm by shifting the ROIs over different numbers of pixels in the direction normal to the cell edge (Figure 1d, inset). The differential image intensity was linearly proportional to the cell deformation within a certain range (dashed vertical lines, Figure 1d). Knowing the pixel size, we obtained the calibration factor (slope of Figure 1e). The differential optical detection method subtracted the common noise (e.g., light intensity and mechanical perturbation) in the optical system, thus providing precise tracking of subtle cell deformation associated with the molecular binding. The standard deviation of the cell deformation averaged over a cell was as small as ~0.4 nm, which was a key for the success of the method (Supporting Information Figure S2f). This detection limit was mainly due to the mechanical instability of the cells (Supporting Information Figure S9).
Binding Kinetics of Four Major Types of Cell Surface Membrane Proteins
To demonstrate the capability of the method, we studied different transmembrane proteins, glycoproteins, ion channel proteins, G-protein coupled receptors (GPCRs) and tyrosine kinase receptors, each representing a major membrane protein type. These membrane proteins are involved in most of the cellular signaling pathways,38,39 and determining the binding kinetics of ligand molecules with them are important for cell biology research, biomarker discovery and drug development. We expressed these membrane proteins in cell lines, cultured and then harvested them as cell suspensions, followed by briefly fixing and re-suspending the cells in 1× PBS buffer. We injected the cell suspension of each membrane protein into channel 1 of the microfluidic chip, trapped the individual cells onto the micro-holes by applying a small negative pressure, and then injected the corresponding ligands to measure the binding kinetics by tracking the edge movement (cell deformation) averaged over an entire cell with the differential tracking algorithm.
Glycoprotein
Glycoproteins are critical for cell recognition and communication,2, 40 and lectins are carbohydrate-binding proteins that recognize and bind to the specific sugar groups of glycoproteins on cell surfaces for regulation of cell adhesion and synthesis of glycoproteins. We studied wheat germ agglutinin (WGA; molecular weight, 35 kDa), a lectin that recognizes N-acetylglucosamine (GlcNAc) and sialic acid groups on the surface of brain neuroblastoma SH-EP1 cells. First, 1× PBS buffer was injected in channel to flow over the trapped cells (Figure 2a, inset) with a flow rate of 300 µL/min for 50 s to obtain a baseline. Then, WGA (in 1× PBS buffer) solution was introduced for 100 s to allow the binding (association) of WGA with the glycoproteins on the cells. After the association process, the flow was switched back to 1× PBS buffer for another 100 s to allow dissociation of WGA from the cells. The entire procedure was repeated for three times with different WGA concentrations, starting from low to high concentrations (70, 140 and 280 nM). The cell contracted gradually during the association process, and expanded back during the dissociation process. By tracking cell deformation vs. time, we obtained binding curves, shown in Figure 2a (black curves), presenting up to 225 nm cellular deformation. Both the rate of association and the amount of cell deformation increased with WGA concentration, which is expected for first order kinetics. By globally fitting the binding curves obtained at different concentrations with a 1:1 binding kinetics model, the association rate constant (kon), dissociation rate constant (koff), and dissociation constant (KD) were found to be kon = 1.8 ± 0.02 × 105 M−1 s−1, koff = 5.6 ± 0.01 × 10−3 s−1 and KD = 31 ± 0.5 nM. These results are consistent with those obtained with SPR imaging method.28
Figure 2. Representative binding kinetics curves of different molecular ligands to four major types of membrane proteins on cells. The cells contracted upon molecular binding, and the absolute edge movements were used to obtain binding kinetics.

(a) WGA binding to glycoprotein on SH-EP1 cells (inset). (b) Acetylcholine binding to nicotinic acetylcholine receptors (ion channel) on SH-EP1- α4β2 cells (inset). (c) AMD3100 binding to CXCR-4 receptors (GPCR) on A549 cells (inset). (d) Insulin binding to insulin receptors (tyrosine kinase receptor) on Hep G2 cells (inset).
Ion channel protein
Ion channels are membrane proteins that establish electrical signals (action potentials) in cells, and mediate communication across the synapses in nerve systems. We studied here nicotinic acetylcholine receptor (nAChR), an ion channel protein that plays a critical role in neurotransmission and drug addiction, and measured its binding to acetylcholine, a neurotransmitter. The neurotransmitter has a small mass (182 Da), which is difficult to study with traditional label-free detection methods. We transfected brain neuroblastoma SH-EP1 cells with human α4β2 receptor (SH-EP1- α4β2) for binding kinetics measurement (Figure 2b). The estimated expression level of the receptor is ~110 nAChRs/µm2 (~0.8 million per cell).41 Following a similar procedure as described above for the glycoproteins, we measured the binding of acetylcholine at different concentrations to nAChR (Figure 2b) and obtained obvious membrane deformation signals and the maximum deformation was around 70 nm. The red lines are the global fitting to the binding curves (black) using the 1:1 binding kinetics model. The association (kon) and dissociation (koff) rate constants were found to be 2.9 ± 0.01 × 104 M−1 s−1 and 2.0 ± 0.02 × 10−3 s−1, respectively, from which the dissociation constant (KD) was determined to be 69 ± 0.4 nM. Both the kinetic rate and equilibrium constants are consistent with those reported for adherent cells.29
G-protein coupled receptor
GPCRs are a major type of drug targets,5 accounting for 25% of all current drugs. We studied C-X-C chemokine receptor type 4 (CXCR-4), a common GPCR broadly expressed in central nervous and immune cells. CXCR-4 is responsible for leukocyte trafficking, human immunodeficiency virus type 1 (HIV-1) infection and the proliferation and metastasis of various carcinomas.42 To inhibit the activation of CXCR-4 in cancer treatment, various antagonists are used to block the binding pocket of the receptor, among which AMD3100 (Plerixafor, a small molecule drug, 794 Da) has been approved for clinic use.43,44 Quantification of binding kinetics between CXCR-4 and the antagonist is essential to evaluate the therapeutic effect. We measured the binding of AMD3100 to CXCR-4 on the surface of human lung carcinoma cells (A549). The A549 cells (Figure 2c, inset) are smaller and more rigid than the SH-EP1 cells (Figure 2b, inset), and the binding induced membrane deformation was smaller with a maximum value of 46 nm. We obtained the binding curves for CXCR-4 at different AMD3100 concentrations (black curves), and performed global fitting of the binding curves (red curves) with the first order binding kinetics model and obtained, kon = 3.8 ± 0.05 × 103 M−1 s−1, koff = 7.7 ± 0.07 × 10−4 s−1 and KD = 0.2 ± 0.003 µM. The affinity constant is in good consistence with the literature value (0.1 µM) measured by the radioligand assay.45
Tyrosine kinases Receptor
Kinases are enzyme-linked receptors, which are responsible for the regulation of normal cells and also for the initiation and progression of various types of cancers.46 An important example is insulin receptor that binds with insulin, which is a critical step in regulating glucose homeostasis, a functional process that is directly relevant to diabetes and cancer. Determining the binding between insulin and the receptor is thus needed for understanding type 2 diabetes, and discovering related drugs. Studying the binding interaction to the insulin receptor with the existing technologies has been a challenge because of its low expression levels and difficulty in purification. We measured the binding kinetics of insulin with insulin receptor on the surface of human liver cancer cells (Hep G2) (black curves, Figure 2d) and performed global fitting of the data (red curves) with binding kinetic constants of kon = 7.5 ± 0.20 × 103 M−1 s−1, koff = 2.5 ± 0.04 × 10−4 s−1 and KD = 33 ± 0.8 nM. The binding induced deformation is smaller but detectable, presenting about 17 nm signal for high concentration ligand interaction. The affinity constant is within the literature reported range,23,47 but the kinetic constants on intact cells have not been reported before due to the lack of a suitable technology.
Validation of the Binding Kinetic Measurements with Control Experiments
To verify the cell deformation is originated from the binding of the ligands to the corresponding membrane protein receptors, we measured acetylcholine binding to wild type SH-EP1 cells (had no nAChRs expressed on their surfaces), and did not observe cell deformation (Figure 3a). This control experiment shows that the deformation in the engineered SH-EP1- α4β2 cells was indeed due to the binding of acetylcholine to the nAChRs. To examine if the cell deformation was resulted from a cell downstream effect triggered by the binding, we measured ligand binding to CXCR-4 receptors by blocking the downstream pathway with pertussis toxin (PTX). PTX is known to block the downstream events of many GPCRs, including CXCR4, by binding to the G protein, while leaving the GPCR outer membrane-binding sites available for the ligand to bind. We treated A549 cells with 100 ng/mL pertussis toxins in the cell culture medium for 2 hours to block CXCR-4 downstream events,48,49 and then measured binding of the ligands to the cells with and without PTX treatment. The binding curves are similar (Figures 3b and 3c) with kinetic constants of kon=4.5 ± 0.03 × 103 M−1 s−1, koff=1.1 ± 0.01 × 10−3 s−1 and KD= 0.25 ± 0.002 µM without PTX blocking and kon =2.9 ± 0.02 × 103 M−1 s−1, koff=0.7 ± 0.01 × 10−3 s−1 and KD =0.24 ± 0.002 µM after downstream blocking, showing little difference in the binding kinetics. This finding indicates that the cell membrane deformation is a direct response of the cell to ligand binding, rather than due to an indirect downstream event.
Figure 3. Control experiments.

(a) Cell deformation (cell edge displacement) of two wild-type SH-EP1 cells to high dose (500 nM) acetylcholine. Since the wild-type cells do not express nicotinic acetylcholine receptors, the lack of cell deformation indicates the observed cell deformation in cell lines with the receptors is due to the molecular binding. (b) and (c) Binding kinetics curves of untreated (b) and pertussis toxin-treated (c) A549 cells to AMD3100 with concentrations of 100, 200, and 500 nM. Pertusis toxin binds to G proteins and blocks their downstream processes. The similarity in the binding kinetics of cells with and without blocking the downstream processes shows that the cell deformation signal is not from a downstream event. A549 cells contracted upon binding, and the absolute cell deformation values (edge movements) were used to extract binding kinetics.
Binding of Ligands with Different Functions to Membrane Protein Receptors
After establishing that our method can be applied to different types of membrane proteins, we examined the binding of ligands with different functions to a given membrane protein receptor, such as CXCR-4 receptor. Stromal-derived-factor-1 (SDF-1) is a natural peptide agonist, which binds to CXCR-4 and activates it. In contrast, AMD3100 is a competitive antagonist, which inhibits the activation by blocking the binding site of the receptor. Although both SDF-1 and AMD3100 bind to the same pocket of the receptor, their functions are completely different.45 We performed binding kinetics measurements of the two ligands to A549 cells. The results (Figure 4a) show that both the agonist and antagonist bind to the CXCR-4 receptor on A540 cells, but the binding kinetics are significantly different with kon= 1.2 ± 0.03 × 104 M−1 s−1, koff= 9.4 ± 0.1 × 10−4 s−1 and KD=80 ± 2 nM for SDF-1, and kon=4.5 ± 0.03 × 103 M−1 s−1, koff=1.1 ± 0.01 × 10−3 s−1 and KD=250 ± 2 nM for AMD3100.
Figure 4. Variability of binding kinetics of different ligands to the same cell line, and same ligands to different cell lines.

(a) Binding of an agonist and antagonist at different concentrations to CXCR-4 receptor on the same cell line (A549). Upper panel: Binding kinetic curves of antagonist AMD3100 (100, 200, and 500 nM) to CXCR-4. Bottom panel: Binding kinetic curves of agonist SDF-1 (5, 10, and 50 nM) to CXCR-4. (b) Binding of anti-EGFR at different concentrations to EGFR on different cell lines. Upper panel: Binding kinetic curves of anti-EGFR to EGFR on A431 cell surface. Bottom panel: Binding kinetic curves of anti-EGFR to EGFR on HeLa cell surface. The cells upon binding contracted in a), and expanded in b), but the absolute cell deformation values (edge movements) were used to extract binding kinetics.
Effect of Cellular Microenvironment on Membrane Protein Binding Kinetics
An advantage of the present single cell-based assay is its ability to evaluate the effect of the membrane protein microenvironment on the molecular binding kinetics. Because different cell lines provide different lipid microenvironments to membrane protein receptors, we studied anti-EGFR binding to EGFR receptor expressed on two different cell lines, A431 cells and HeLa (Figure 4b), and determined binding kinetic constants of kon=8.2 ± 0.07 × 104 M−1 s−1, koff = 3.0 ± 0.04 × 10−4 s−1 and KD= 3.6 ± 0.03 nM for A431, and kon= 4.3 ± 0.04 × 104 M−1 s−1, koff= 2.0 ± 0.05 × 10−4 s−1 and KD=4.6 ± 0.04 nM for HeLa. The differences in the kinetic constants are small but detectable for the two cell lines. This finding underscores the importance of studying membrane proteins in their native cellular environment in order to obtain accurate and physiologically relevant ligand-receptor binding kinetics.
Heterogeneity of Different Cells, and Different Regions of a Cell
The present method measures binding kinetics on single cells, which offers an opportunity to measure cell-to-cell variability in the binding kinetics. Before the analysis of cell-to-cell variability, we first tested the repeatability of our method to evaluate the measurement errors. The binding kinetic measurement on one cell was repeated for three times and the standard deviation of the kinetic constants were within 10% (Supporting Information Figure S3 and Table S1). Then, we measured the binding of WGA to the glycoprotein receptor on two SH-EP1 cells trapped on the same microfluidic chip simultaneously for cell-cell variation study (Supporting Information Figures S4a and S4b). The results revealed ~2 fold difference in the dissociation rate constant for the two cells. To further validate the finding, additional cells were tested and statistical analysis of four different membrane proteins is presented in Supporting Information Table S2. The high spatial resolution of the method also allows analysis of region-to-region variability within a cell. We examined the local variability in the binding kinetics by tracking local cell deformation along the edge of the cell (Supporting Information Figure S4c) and determined binding kinetic curves (Supporting Information Figure S4d), and observed local variations in the binding kinetics. A possible reason for this local variability is the heterogeneous membrane protein distribution on the cell surface, which is known to play an important role in cell communication and recognition.50
Membrane Deformation Dependence on Cell Stiffness
The thermodynamic model (Eq. 1) predicts that ligand binding to membrane proteins causes the cell to deform, and the deformation magnitude decreases with the stiffness (bending modulus) of the cell. To examine the effect of cell mechanical stiffness on the binding-induced cell deformation, we first determined the cell stiffness by measuring cell edge movement associated with a change in the osmotic pressure created by decreasing 300 mOsm (1× PBS) to 270 mOsm (0.9× PBS). In isotonic buffer solution (300 mOsm), the cell preserves the normal shape, and it is expected to expand in the hypotonic solution (270 mOsm) as the water is transported into the cell to balance the osmotic pressure difference. We tracked the osmotic pressure-induced cell membrane expansion and used the average expansion to determine the membrane stiffness. The cell used in this study was A431 cells expressed with EGFR. We then studied the binding of anti-EGFR to EGFR receptors on the cell surface, and determined the associated cell deformation (Supporting Information Figure S5). Our result indicates that the binding-induced cell deformation in soft cells is greater than that in the stiff cells, and the mean correlation ratio is 0.96 with the standard deviation of 0.04 (Supporting Information Table S3, n=3). Despite the dependence of the deformation magnitude on cell stiffness, the binding kinetic constants are not affected by the cell mechanical properties (Supporting Information Figure S5).
Membrane Deformation Dependence on Receptor Density
The thermodynamic model also predicts that the binding-induced cell deformation is proportional to the density of the expressed membrane proteins on a cell surface. We examined this prediction by measuring the binding of anti-EGFR with EGFR on different cell lines, which are known to express EGFR with different densities.30 These cell lines include A431 cell, HeLa cell, A549 cell and Hek 293 cell (Figure 5), and the EGFR densities are 1.2, 0.53, 0.28, and 0.02 million per cell, respectively.16, 30 The binding curves for these cell lines were calibrated and normalized with the osmotic pressure-induced cell deformation (using 0.9× PBS, 270 mOsm). The data shows that the cell deformation increases with EGFR density, which is in agreement with the thermodynamic model (Eq. 1). We analyzed multiple cells and plotted the distribution vs. the receptor density (Supporting Information Figure S6), and observed a clear correlation between the binding-induced cell deformation and receptor density. We also examined possible dependence of the cell deformation on the binding kinetic constants (Supporting Information Figure S7) but found no obvious correlation between them. This observation is also consistent with the thermodynamic model.
Figure 5. Dependence of cell deformation on receptor density.

Optical images of trapped cells (insets), and Anti-EGFR –EGFR binding curves for (a) A431, (b) HeLa, (c) A549, and (d) Hek 293 cell lines. The cells expanded upon anti-EGFR-EGFR binding, and the absolute edge movements were used to extract binding kinetics.
Comparison of Fixed and Live Cells
In this work, we mainly focused on the fixed cells for binding kinetic measurements, because it minimizes cell micro-motion (Supporting Information S-10 and Figure S8a). The fixation used 4% paraformaldehyde (PFA), the most common fixation that has been widely used for various immune-assays using fluorescence and other detection technologies. This procedure may crosslink proteins at certain epitopes, and could thus affect the binding kinetics. However, it has been shown that only several amino acids (e.g., lysine and tryptophan side chains) can be easily cross-linked by the fixation, and even for these amino acids, the crosslinking rate is between 3–22%51–54 after 48 h fixation. In the present work our fixation time is brief (10 mins) at 4% PFA, which should significantly reduce the chance of crosslinking in the binding pockets of the target membrane proteins. Additionally, the formaldehyde cross-links are reversible in buffer solutions,55 so a majority of the membrane proteins, especially their binding pockets, are expected to remain intact. To further examine the possible fixation effect, we carried out the binding kinetic measurements on living cells (Figure 6 and Supporting Information S-10). Despite of the increased noise and difference in the absolute amount of cell deformation in the living cells, the binding kinetic constants are similar within the experimental uncertainty for fixed and living cells examined here.
Figure 6. Comparison of fixed and live cells.

WGA- glycoprotein binding kinetic curves on (a) fixed and (b) live SH-EP1 cells (WGA concentration=200 nM). The SH-EP1 cells contracted upon WGA binding, and the absolute edge movements were used to extract binding kinetics.
DISCUSSION
Molecular binding to membrane proteins is critical for both cellular function study and drug development, which has motivated researchers to develop various detection technologies. The current detection technologies fall into two categories: labeled and label-free methods. The former includes fluorescence methods (e.g., TIRF), which are typically end-point assays, and thus not for kinetics. The latter includes SPR and other optical and mechanical methods. These methods allow real time tracking of binding kinetics, but often face challenges of detecting small molecules (low molecular mass ligands). The method presented here measures molecular binding by tracking binding-induced cell deformation at sub-nm with an optical tracking method. It is label-free, yet its detection sensitivity does not diminish with molecular mass, which is thus capable of studying both large and small molecules. Furthermore, by integrating the sensitive optical cell edge tracking method with a microfluidic device, single cells can be trapped and analyzed, which is particularly attractive for studying suspension cells. The basic principle of the present detection method, as described with a thermodynamic model, is universal. Predicting the actual amount of the molecular binding-induced cell deformation (contraction or expansion) with the model requires details of the ligand-membrane receptor interactions, and cell mechanical properties. However, the binding kinetics measures how fast a ligand binds to its receptor, which is determined by the time evolution of the cell deformation, rather than by the amount of cell deformation.
CONCLUSION
We described here a label-free optical detection method coupled with a microfluidic device to measure the kinetics of both large and small ligands binding with membrane protein receptors on whole cells. The method uses a microfluidic chip to trap individual suspended cells onto microfabricated holes, and measure the molecular binding induced-cell membrane deformation of the trapped cells with sub-nanometer precision. Using the method, we studied the molecular interactions of large and small molecule ligands, and agonists and antagonists with four major types of membrane proteins (glycoprotein, nAChR, CXCR-4 and insulin receptors), demonstrating the capability of measuring both large and small molecule binding kinetics with membrane proteins on whole cells. We developed a thermodynamic model to describe the basic mechanism of the method, and validated the model by changing the cell stiffness, receptor density and binding kinetics. The single-cell detection capability revealed variability in the binding kinetics of a membrane protein receptor expressed on different cell lines, demonstrating the importance of the cellular microenvironment in membrane protein functions. The method offers single cell detection capability, which enabled the study of heterogeneity in the membrane protein binding within the same cell line. The microfluidic design allowed trapping of the individual cells, measuring the binding kinetics, and then releasing the trapped cells, thus potentially compatible with high throughput need for studying multiple cells.
METHODS
Materials
Wheat germ agglutinin (WGA), acetylcholine chloride, AMD3100 octahydrochloride hydrate, SDF-1 α and insulin molecules were purchased from Sigma-Aldrich (St. Louis, MO). Anti-EGFR monoclonal antibody was purchased from EMD Millipore (Billerica, MA). Pertussis Toxin was purchased from List Biological Labs. All solutions used in the experiments were prepared with 1× PBS. All reagents were analytical grade from Sigma-Aldrich, except those stated.
Cell Culture
SH-EP1, SH-EP1_ α4β2, A549, Hep G2, A431, HeLa and HEK293 cell lines were obtained from the American Type Culture Collection (Rockville, MD). All the cells were cultured in a humidified incubator at 37°C with 5% CO2 and 70% relative humidity. SH-EP1, SH-EP1_ α4β2, Hep G2, A431, HeLa and HEK293 cells were grown in Dulbecco Modified Eagle Medium (DMEM) (Invitrogen, Carlsbad, California) with 10% Fetal Bovine Serum (FBS) and 1% penicillin and streptomycin. A549 cells were cultured in F-12K with 10% FBS and 1% penicillin and streptomycin. Cells were passaged with 0.25% trypsin and 0.02% ethylenediaminetetraacetic acid in Hank’s balanced salt solution (Sigma-Aldrich, St Louis, Missouri) when they were approximately 75% confluent.
Before experiments, suspension cells in 1× PBS buffer solution were prepared. First, the cells were trypsinized from the flask to round up, and centrifuged for 5 mins to discard the old medium. Then, the centrifuged cell piles were suspended in 4% paraformaldehyde solution for 10 mins fixation. Finally, the fixed floating cells were centrifuged again to discard the fixatives and transferred to the 1× PBS. Overnight recovery was performed after cell fixation to improve cell elasticity and deformability.
For living cell binding kinetic measurements, the cells were also trypsinized from the flask and centrifuged for 5 min to discard the old medium. Then, the centrifuged cells were suspended in live cell imaging solution (Life Technologies) and immediately used for kinetic measurements to maintain cell viability.
Optical Tracking of Cell Deformation
An inverted microscope (Olympus IX-81) equipped with a top illuminating white light, a phase contrast condenser and 40× phase contrast objective was used as the imaging device. The trapped cells within the microfluidic plate were imaged from bottom with a CCD camera (AVT Pike F032B) at a frame rate of 7 frames per second (fps). The spatial resolution of the system was ~0.5 µm. The binding-induced cell deformation was determined with nm-precision with a differential detection algorithm (Supporting Information Figure S2). First, the edge of the cell was manually chosen, and the center of the cell was determined (Supporting Information Figure S2a). Then, the calibration curve at each location along the cell edge was obtained by shifting the ROI (1.11 × 2.22 µm) from outside to inside of the cell, in the direction perpendicular to the cell edge (Figures S2, d and e). Using the calibration curves, the cell deformation (edge movement) at each point along the cell edge was determined, and the cell deformation averaged over the entire cell was used to determine the molecular binding kinetics. Depending on the cell lines, both contraction and expansion were observed, but the binding kinetics was determined from the absolute values of the cell deformation vs. time.
Microfluidic Device for Cell Trapping and Binding Kinetics Measurement
As described in the Supporting Information (Figure S1), we used a homemade and modified commerical (AX150, AXIS™ Axon Isolation Device from Millipore) PDMS-microfluidic device to trap single cells. The fabrication used the standard soft lithography. The homemade microfluidic plate had two parallel channels (10.6 mm long, 1500 µm wide and 40 µm high), separated with a barrier. The barrier had 60 holes (each hole is 150 µm long, 10 µm wide, separation between two holes is 50 µm) to trap cells. Sample solution containing suspended cells was flowed in one channel while keeping a lower pressure in the second channel. This allowed trapping of one cell in each hole for binding kinetics measurement (~5 mins). The optics allowed imaging of two cells simutaneously. When one measurement was completed, the cells were released from the trapping holes, and new cells were trapped for repeated measurement. The sample and buffer flows were controlled with a drug perfusion system (SF-77B, Warner Instruments, Connecticut). The typical transition time between different solutions was about 1–2 seconds.
Supplementary Material
Acknowledgments
Financial support from NIH (5R01GM107165-04 and 1R01GM124335-01) and Moore foundation is acknowledged.
Footnotes
ASSOCIATED CONTENT
Supporting Information Available: Derivation of Eq. 1; Cell trapping microfluidic chip; Calibration of the differential intensity and cell edge movement; Measurement error evaluation; Cell-to-cell variability; Statistical analysis of cell-to-cell variation; Cell stiffness vs. cell deformation amplitude; Membrane receptor density vs. cell deformation amplitude; Binding kinetic constants vs. Cell deformation amplitude; Binding kinetics of fixed and living cells; Noise analysis.
This Supporting Information is available free of charge on the ACS Publications website.
The authors declare no competing financial interest.
References
- 1.Escriba PV, Nicolson GL. Membrane Structure and Function: Relevance of Lipid and Protein Structures in Cellular Physiology, Pathology and Therapy Preface. Bba-Biomembranes. 2014;1838:1449–1450. doi: 10.1016/j.bbamem.2014.03.008. [DOI] [PubMed] [Google Scholar]
- 2.DiNitto JP, Cronin TC, Lambright DG. Membrane Recognition and Targeting by Lipid-Binding Domains. Sci STKE. 2003;2003:re16. doi: 10.1126/stke.2132003re16. [DOI] [PubMed] [Google Scholar]
- 3.Cho W, Stahelin RV. Membrane-Protein Interactions in Cell Signaling and Membrane Trafficking. Annu Rev Biophys Biomol Struct. 2005;34:119–51. doi: 10.1146/annurev.biophys.33.110502.133337. [DOI] [PubMed] [Google Scholar]
- 4.Ehrlich P. Address in Pathology on Chemotherapeutics : Scientific Principles, Methods, and Results. Lancet. 1913;2:445–451. [Google Scholar]
- 5.Hopkins AL, Groom CR. The Druggable Genome. Nat Rev Drug Discov. 2002;1:727–730. doi: 10.1038/nrd892. [DOI] [PubMed] [Google Scholar]
- 6.Swinney DC. The Role of Binding Kinetics in Therapeutically Useful Drug Action. Curr Opin Drug Disc. 2009;12:31–39. [PubMed] [Google Scholar]
- 7.Copeland RA, Pompliano DL, Meek TD. Opinion - Drug-Target Residence Time and Its Implications for Lead Optimization. Nat Rev Drug Discov. 2006;5:730–739. doi: 10.1038/nrd2082. [DOI] [PubMed] [Google Scholar]
- 8.Copeland RA. The Dynamics of Drug-Target Interactions: Drug-Target Residence Time and Its Impact on Efficacy and Safety. Expert Opin Drug Dis. 2010;5:305–310. doi: 10.1517/17460441003677725. [DOI] [PubMed] [Google Scholar]
- 9.Hellmuth A, Calderón Villalobos LIA. Radioligand Binding Assays for Determining Dissociation Constants of Phytohormone Receptors. In: Lois LM, Matthiesen R, editors. Plant Proteostasis: Methods and Protocols. Springer New York; New York, NY: 2016. pp. 23–34. [DOI] [PubMed] [Google Scholar]
- 10.Dong C, Liu Z, Wang F. Radioligand Saturation Binding for Quantitative Analysis of Ligand-Receptor Interactions. Biophys Rep. 2015;1:148–155. doi: 10.1007/s41048-016-0016-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhen J, Antonio T, Ali S, Neve KA, Dutta AK, Reith MEA. Use of Radiolabeled Antagonist Assays for Assessing Agonism at D2 and D3 Dopamine Receptors: Comparison with Functional Gtpγs Assays. J Neurosci Methods. 2015;248:7–15. doi: 10.1016/j.jneumeth.2015.03.028. [DOI] [PubMed] [Google Scholar]
- 12.Glickman JF, Schmid A, Ferrand S. Scintillation Proximity Assays in High-Throughput Screening. Assay Drug Dev Techn. 2008;6:433–455. doi: 10.1089/adt.2008.135. [DOI] [PubMed] [Google Scholar]
- 13.Owicki JC. Fluorescence Polarization and Anisotropy in High Throughput Screening: Perspectives and Primer. J Biomol Screen. 2000;5:297–306. doi: 10.1177/108705710000500501. [DOI] [PubMed] [Google Scholar]
- 14.Lohse MJ, Nuber S, Hoffmann C. Fluorescence/Bioluminescence Resonance Energy Transfer Techniques to Study G-Protein-Coupled Receptor Activation and Signaling. Pharmacol Rev. 2012;64:299–336. doi: 10.1124/pr.110.004309. [DOI] [PubMed] [Google Scholar]
- 15.Veiksina S, Kopanchuk S, Mazina O, Link R, Lille A, Rinken A. Homogeneous Fluorescence Anisotropy-Based Assay for Characterization of Ligand Binding Dynamics to Gpcrs in Budded Baculoviruses: The Case of Cy3b-Ndp-Alpha-Msh Binding to Mc4 Receptors. Methods Mol Biol. 2015;1272:37–50. doi: 10.1007/978-1-4939-2336-6_3. [DOI] [PubMed] [Google Scholar]
- 16.Ayoub MA, Trebaux J, Vallaghe J, Charrier-Savournin F, Al-Hosaini K, Gonzalez Moya A, Pin JP, Pfleger KD, Trinquet E. Homogeneous Time-Resolved Fluorescence-Based Assay to Monitor Extracellular Signal-Regulated Kinase Signaling in a High-Throughput Format. Front Endocrinol (Lausanne) 2014;5:94. doi: 10.3389/fendo.2014.00094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Oueslati N, Hounsou C, Belhocine A, Rodriguez T, Dupuis E, Zwier JM, Trinquet E, Pin JP, Durroux T. Time-Resolved Fret Strategy to Screen Gpcr Ligand Library. Methods Mol Biol. 2015;1272:23–36. doi: 10.1007/978-1-4939-2336-6_2. [DOI] [PubMed] [Google Scholar]
- 18.Mashanov GI, Tacon D, Peckham M, Molloy JE. The Spatial and Temporal Dynamics of Pleckstrin Homology Domain Binding at the Plasma Membrane Measured by Imaging Single Molecules in Live Mouse Myoblasts. J Biol Chem. 2004;279:15274–15280. doi: 10.1074/jbc.M312140200. [DOI] [PubMed] [Google Scholar]
- 19.Parhamifar L, Moghimi SM. Total Internal Reflection Fluorescence (Tirf) Microscopy for Real-Time Imaging of Nanoparticle-Cell Plasma Membrane Interaction. Methods Mol Biol. 2012;906:473–82. doi: 10.1007/978-1-61779-953-2_38. [DOI] [PubMed] [Google Scholar]
- 20.Nickerson A, Huang T, Lin LJ, Nan XL. Photoactivated Localization Microscopy with Bimolecular Fluorescence Complementation (Bifc-Palm) for Nanoscale Imaging of Protein-Protein Interactions in Cells. Plos One. 2014;9 doi: 10.1371/journal.pone.0100589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hu CD, Chinenov Y, Kerppola TK. Visualization of Interactions among Bzip and Rel Family Proteins in Living Cells Using Bimolecular Fluorescence Complementation. Mol Cell. 2002;9:789–798. doi: 10.1016/s1097-2765(02)00496-3. [DOI] [PubMed] [Google Scholar]
- 22.Pei ZC, Saint-Guirons J, Kack C, Ingemarsson B, Aastrup T. Real-Time Analysis of the Carbohydrates on Cell Surfaces Using a Qcm Biosensor: A Lectin-Based Approach. Biosens Bioelectron. 2012;35:200–205. doi: 10.1016/j.bios.2012.02.047. [DOI] [PubMed] [Google Scholar]
- 23.Subramanian K, Fee CJ, Fredericks R, Stubbs RS, Hayes MT. Insulin Receptor-Insulin Interaction Kinetics Using Multiplex Surface Plasmon Resonance. J Mol Recognit. 2013;26:643–652. doi: 10.1002/jmr.2307. [DOI] [PubMed] [Google Scholar]
- 24.Navratilova I, Besnard J, Hopkins AL. Screening for Gpcr Ligands Using Surface Plasmon Resonance. Acs Med Chem Lett. 2011;2:549–554. doi: 10.1021/ml2000017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bieri C, Ernst OP, Heyse S, Hofmann KP, Vogel H. Micropatterned Immobilization of a G Protein-Coupled Receptor and Direct Detection of G Protein Activation. Nat Biotechnol. 1999;17:1105–1108. doi: 10.1038/15090. [DOI] [PubMed] [Google Scholar]
- 26.Nirschl M, Reuter F, Voros J. Review of Transducer Principles for Label-Free Biomolecular Interaction Analysis. Biosensors (Basel) 2011;1:70–92. doi: 10.3390/bios1030070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Concepcion J, Witte K, Wartchow C, Choo S, Yao DF, Persson H, Wei J, Li P, Heidecker B, Ma WL, Varma R, Zhao LS, Perillat D, Carricato G, Recknor M, Du K, Ho H, Ellis T, Gamez J, Howes M, Phi-Wilson J, Lockard S, Zuk R, Tan H. Label-Free Detection of Biomolecular Interactions Using Biolayer Interferometry for Kinetic Characterization. Comb Chem High T Scr. 2009;12:791–800. doi: 10.2174/138620709789104915. [DOI] [PubMed] [Google Scholar]
- 28.Wang W, Yang YZ, Wang SP, Nagaraj VJ, Liu Q, Wu J, Tao NJ. Label-Free Measuring and Mapping of Binding Kinetics of Membrane Proteins in Single Living Cells. Nat Chem. 2012;4:846–853. doi: 10.1038/nchem.1434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Guan Y, Shan X, Zhang F, Wang S, Chen HY, Tao N. Kinetics of Small Molecule Interactions with Membrane Proteins in Single Cells Measured with Mechanical Amplification. Sci Adv. 2015;1:e1500633. doi: 10.1126/sciadv.1500633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang FN, Wang SP, Yin LL, Yang YZ, Guan Y, Wang W, Xu H, Tao NJ. Quantification of Epidermal Growth Factor Receptor Expression Level and Binding Kinetics on Cell Surfaces by Surface Plasmon Resonance Imaging. Anal Chem. 2015;87:9960–9965. doi: 10.1021/acs.analchem.5b02572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Baksh MM, Kussrow AK, Mileni M, Finn MG, Bornhop DJ. Label-Free Quantification of Membrane-Ligand Interactions Using Backscattering Interferometry. Nat Biotechnol. 2011;29:357–60. doi: 10.1038/nbt.1790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bergese P, Oliviero G, Alessandri I, Depero LE. Thermodynamics of Mechanical Transduction of Surface Confined Receptor/Ligand Reactions. J Colloid Interf Sci. 2007;316:1017–1022. doi: 10.1016/j.jcis.2007.08.048. [DOI] [PubMed] [Google Scholar]
- 33.Helfrich W. Elastic Properties of Lipid Bilayers - Theory and Possible Experiments. Z Naturforsch C. 1973;C 28:693–703. doi: 10.1515/znc-1973-11-1209. [DOI] [PubMed] [Google Scholar]
- 34.Zimmerberg J, Kozlov MM. How Proteins Produce Cellular Membrane Curvature. Nat Rev Mol Cell Biol. 2006;7:9–19. doi: 10.1038/nrm1784. [DOI] [PubMed] [Google Scholar]
- 35.McMahon HT, Boucrot E. Membrane Curvature at a Glance. J Cell Sci. 2015;128:1065–70. doi: 10.1242/jcs.114454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Callan-Jones A, Bassereau P. Curvature-Driven Membrane Lipid and Protein Distribution. Curr Opin Solid State Mater Sci. 2013;17:143–150. [Google Scholar]
- 37.Ionescu-Zanetti C, Shaw RM, Seo JG, Jan YN, Jan LY, Lee LP. Mammalian Electrophysiology on a Microfluidic Platform. P Natl Acad Sci USA. 2005;102:9112–9117. doi: 10.1073/pnas.0503418102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Purves D. Neuroscience. 2. Sinauer Associates; Sunderland, Mass. ; [Great Britain]: 2001. [Google Scholar]
- 39.Cooper GM, Hausman RE. The Cell : A Molecular Approach. 6. Sinauer Associates; Sunderland, MA: 2013. [Google Scholar]
- 40.Lemmon MA. Membrane Recognition by Phospholipid-Binding Domains. Nat Rev Mol Cell Biol. 2008;9:99–111. doi: 10.1038/nrm2328. [DOI] [PubMed] [Google Scholar]
- 41.Eaton JB, Peng JH, Schroeder KM, George AA, Fryer JD, Krishnan C, Buhlman L, Kuo YP, Steinlein O, Lukas RJ. Characterization of Human Alpha 4 Beta 2-Nicotinic Acetylcholine Receptors Stably and Heterologously Expressed in Native Nicotinic Receptor-Null Sh-Ep1 Human Epithelial Cells. Mol Pharmacol. 2003;64:1283–1294. doi: 10.1124/mol.64.6.1283. [DOI] [PubMed] [Google Scholar]
- 42.Zou YR, Kottmann AH, Kuroda M, Taniuchi I, Littman DR. Function of the Chemokine Receptor Cxcr4 in Haematopoiesis and in Cerebellar Development. Nature. 1998;393:595–9. doi: 10.1038/31269. [DOI] [PubMed] [Google Scholar]
- 43.Hatse S, Princen K, Bridger G, De Clercq E, Schols D. Chemokine Receptor Inhibition by Amd3100 Is Strictly Confined to Cxcr4. Febs Lett. 2002;527:255–262. doi: 10.1016/s0014-5793(02)03143-5. [DOI] [PubMed] [Google Scholar]
- 44.Domanska UM, Timmer-Bosscha H, Nagengast WB, Munnink THO, Kruizinga RC, Ananias HJK, Kliphuis NM, Huls G, De Vries EGE, de Jong IJ, Walenkamp AME. Cxcr4 Inhibition with Amd3100 Sensitizes Prostate Cancer to Docetaxel Chemotherapy. Neoplasia. 2012;14:709–718. doi: 10.1593/neo.12324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhang WB, Navenot JM, Haribabu B, Tamamura H, Hiramatu K, Omagari A, Pei G, Manfredi JP, Fujii N, Broach JR, Peiper SC. A Point Mutation That Confers Constitutive Activity to Cxcr4 Reveals That T140 Is an Inverse Agonist and That Amd3100 and Alx40-4c Axe Weak Partial Agonists. J Biol Chem. 2002;277:24515–24521. doi: 10.1074/jbc.M200889200. [DOI] [PubMed] [Google Scholar]
- 46.Zwick E, Bange J, Ullrich A. Receptor Tyrosine Kinase Signalling as a Target for Cancer Intervention Strategies. Endocr-Relat Cancer. 2001;8:161–173. doi: 10.1677/erc.0.0080161. [DOI] [PubMed] [Google Scholar]
- 47.Knudsen L, De Meyts P, Kiselyov VV. Insight into the Molecular Basis for the Kinetic Differences between the Two Insulin Receptor Isoforms. Biochem J. 2011;440:397–403. doi: 10.1042/BJ20110550. [DOI] [PubMed] [Google Scholar]
- 48.Schneider OD, Weiss AA, Miller WE. Pertussis Toxin Signals through the Tcr to Initiate Cross-Desensitization of the Chemokine Receptor Cxcr4. J Immunol. 2009;182:5730–5739. doi: 10.4049/jimmunol.0803114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mangmool S, Kurose H. G(I/O) Protein-Dependent and -Independent Actions of Pertussis Toxin (Ptx) Toxins. 2011;3:884–899. doi: 10.3390/toxins3070884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sengupta P, Jovanovic-Talisman T, Skoko D, Renz M, Veatch SL, Lippincott-Schwartz J. Probing Protein Heterogeneity in the Plasma Membrane Using Palm and Pair Correlation Analysis. Nat Methods. 2011;8:969–975. doi: 10.1038/nmeth.1704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Klockenbusch C, Kast J. Optimization of Formaldehyde Cross-Linking for Protein Interaction Analysis of Non-Tagged Integrin Beta1. J Biomed Biotechnol. 2010;2010:927585. doi: 10.1155/2010/927585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Metz B, Kersten GF, Hoogerhout P, Brugghe HF, Timmermans HA, de Jong A, Meiring H, ten Hove J, Hennink WE, Crommelin DJ, Jiskoot W. Identification of Formaldehyde-Induced Modifications in Proteins: Reactions with Model Peptides. J Biol Chem. 2004;279:6235–43. doi: 10.1074/jbc.M310752200. [DOI] [PubMed] [Google Scholar]
- 53.Metz B, Kersten GF, Baart GJ, de Jong A, Meiring H, ten Hove J, van Steenbergen MJ, Hennink WE, Crommelin DJ, Jiskoot W. Identification of Formaldehyde-Induced Modifications in Proteins: Reactions with Insulin. Bioconjug Chem. 2006;17:815–22. doi: 10.1021/bc050340f. [DOI] [PubMed] [Google Scholar]
- 54.Sutherland BW, Toews J, Kast J. Utility of Formaldehyde Cross-Linking and Mass Spectrometry in the Study of Protein-Protein Interactions. J Mass Spectrom. 2008;43:699–715. doi: 10.1002/jms.1415. [DOI] [PubMed] [Google Scholar]
- 55.Thavarajah R, Mudimbaimannar VK, Elizabeth J, Rao UK, Ranganathan K. Chemical and Physical Basics of Routine Formaldehyde Fixation. J Oral Maxillofac Pathol. 2012;16:400–5. doi: 10.4103/0973-029X.102496. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.