

Summary
Blood-brain barrier (BBB) disruption alters the composition of the brain microenvironment by allowing blood proteins into the CNS. However, whether blood-derived molecules serve as extrinsic inhibitors of remyelination is unknown. Here we show that the coagulation factor fibrinogen activates the bone morphogenetic protein (BMP) signaling pathway in oligodendrocyte progenitor cells (OPCs) and suppresses remyelination. Fibrinogen induces phosphorylation of Smad 1/5/8 and inhibits OPC differentiation into myelinating oligodendrocytes (OLs), while promoting an astrocytic fate in vitro. Fibrinogen effects are rescued by BMP type I receptor inhibition using dorsomorphin homologue 1 (DMH1) or CRISPR/Cas9 activin A receptor type I (ACVR1) knockout in OPCs. Fibrinogen and the BMP target Id2 are increased in demyelinated multiple sclerosis (MS) lesions. Therapeutic depletion of fibrinogen decreases BMP signaling and enhances remyelination in vivo. Targeting fibrinogen may be an upstream therapeutic strategy to promote the regenerative potential of CNS progenitors in diseases with remyelination failure.
Graphical abstract
Extrinsic inhibitors contribute to remyelination failure in neurological diseases. Petersen et al. identify the blood coagulation factor fibrinogen as an activator of BMP receptor signaling in oligodendrocyte progenitor cells that can be targeted therapeutically to promote remyelination.
Introduction
Remyelination is critical for recovery in several diseases, such as MS, neonatal white matter injury (NWMI) and stroke (Franklin and Ffrench-Constant, 2008; Rosenzweig and Carmichael, 2015). In these conditions, OPCs often fail to differentiate into mature OLs required for myelin repair (Fancy et al., 2011a). However, molecules in the lesion environment that activate pathways in OPCs to inhibit their differentiation are not fully known (Gallo and Deneen, 2014). OPCs are closely associated with the perivascular niche, which is altered when increased BBB permeability allows blood proteins into the CNS (Tsai et al., 2016; Zlokovic, 2008). The contribution of blood-derived signals to OPC dysfunction is unknown. Without treatments to promote remyelination, identifying upstream blood-derived signals that dysregulate the progenitor niche may open novel therapeutic strategies for remyelination.
Fibrinogen, a blood coagulation protein, is deposited in many CNS diseases with BBB disruption and myelin abnormalities, including MS, stroke, traumatic brain injury, and Alzheimer's disease (Bardehle et al., 2015; Davalos et al., 2012). Fibrinogen is found in the progressive MS cortex and in active and chronic MS lesions (Vos et al., 2005; Yates et al., 2017). BBB disruption and fibrinogen deposition occur early in MS and precede demyelination (Marik et al., 2007). Progressive MS cases with severe cortical fibrinogen deposition have perturbed fibrinolysis and reduced neuronal density (Yates et al., 2017). Fibrinogen is not just a marker of BBB disruption, but a driver of neuropathology (Bardehle et al., 2015). It promotes neuroinflammation and glial scar formation by direct effects on microglia, astrocytes, and neurons (Adams et al., 2007; Davalos et al., 2012; Schachtrup et al., 2010). Since fibrinogen regulates functions of CNS cells and is found in MS lesions, we hypothesized that fibrinogen triggers inhibitory signaling to control differentiation of OPCs and remyelination.
Results
Fibrinogen Inhibits OPC Differentiation and Myelination
To test effects of fibrinogen on myelination, we treated primary cultures of rat cortical OPCs with fibrinogen. Fibrinogen inhibited OPC differentiation into mature OLs shown by decreased myelin basic protein (MBP)+ OLs and MBP gene and protein expression (Figure 1A,B). Fibrinogen did not affect OPC apoptosis or proliferation (Figure S1A-C). In OPC/dorsal root ganglion (DRG) myelinating co-cultures, fibrinogen inhibited axonal myelination, reducing mature, myelinating OLs by 60% (Figure 1C). Although some fibrinogen-treated OPCs differentiated to MBP+ OLs, many did not form myelin sheaths, suggesting impaired axonal wrapping (Figure 1C, arrows). In a neuron-free, nanofiber myelinating culture system (Lee et al., 2012), fibrinogen-coated nanofibers inhibited OPC differentiation and myelination compared to control poly(L-lysine)-coated or albumin-coated nanofibers (Figure 1D, S1D-F). In fibrinogen-coated nanofiber cultures, the few OPCs that matured to MBP+ cells had a marked deficit in wrapping nanofibers and formed large membrane sheets rather than myelin-like segments (Figure 1D). Fibrinogen is a potent activator of microglia and macrophages (Adams et al., 2007; Ryu et al., 2015). OPCs treated with conditioned media from fibrin-primed macrophages reduced differentiation to MBP+ cells (Figure S1G), suggesting that fibrinogen exerts immune-mediated and cell-autonomous inhibition of OPC differentiation (Figure 1A-D). In human MS lesions and NWMI, OPCs expressing the high activity Wnt marker RNF43 (Fancy et al., 2014) were associated with leaky blood vessels and fibrinogen deposits (Figure 1E).
Figure 1. Fibrinogen Inhibits OPC Differentiation and Myelination in Vitro.
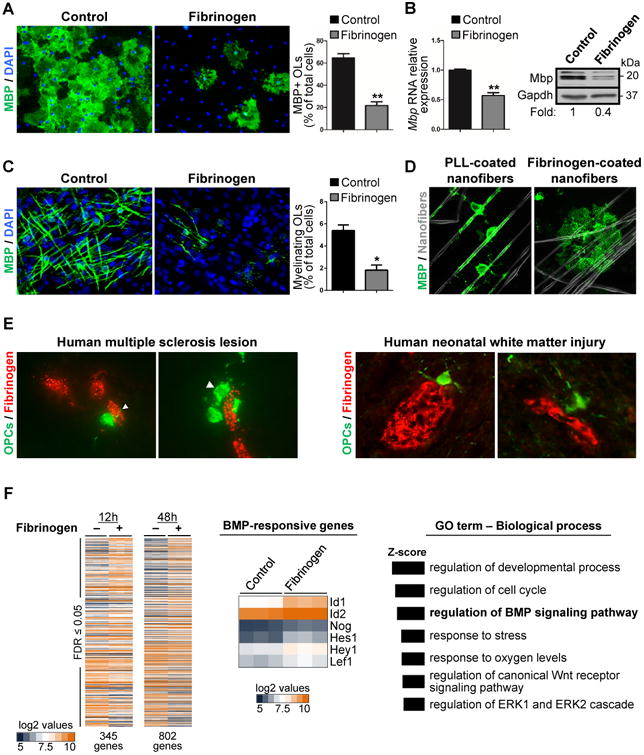
, (A) MBP immunostaining (green) indicates mature OLs in control or fibrinogen-treated primary rat OPC cultures after 4 days. Nuclei are stained with DAPI (blue). Representative images from n = 3 independent experiments. Scale bar: 50 μm. Values are mean ± s.e.m., **p < 0.01 (unpaired t-test).
(B) Mbp gene (left) and protein (right) expression analysis from control or fibrinogen-treated primary rat OPCs. Values are mean ± s.e.m. from n = 3 independent experiments. **p < 0.01 (unpaired t-test). Representative immunoblot from n = 2 independent experiments.
(C) MBP immunostaining (green) indicating mature OLs in OPC-DRG myelinating co-cultures at 7 days in vitro. Nuclei are stained with DAPI (blue). Arrows: MBP+ OLs without formed myelin segments. Scale bar: 25 μm. Representative images from n = 3 independent experiments. Values are mean ± s.e.m., *p < 0.05 (unpaired t-test).
(D) MBP+ mature OLs (green) in myelinating cultures of OPCs on control PLL- or fibrinogen-coated nanofibers at 4 days in vitro. Representative images from n = 2 independent experiments. Arrowheads: myelin-like segments. Scale bar: 25 μm.
(E) Immunostaining of fibrinogen (red) and RNF43 (green) in the chronic active border of ahuman MS lesion and the subcortical white matter of an infant with NWMI from hypoxic-ischemic encephalopathy. Arrowheads: RNF43+ cells with bipolar OPC morphology in contactwith fibrinogen (red). Scale bar: 10 μm.
(F) Affymetrix microarray gene expression analysis of controls and fibrinogen-stimulated ratprimary OPCs after 12 and 48 h of treatment. Heatmap and GO analysis of differentiallyexpressed genes; 345 genes (12 h) and 802 genes (48 h) (false-discovery rate (FDR) ≤0.05). Heatmap of BMP-responsive genes shown at 12 h of fibrinogen treatment. Log2 values are represented on the heatmaps.
To determine the mechanism mediating fibrinogen's inhibitory effect on OPC differentiation, we used whole-genome microarray of fibrinogen-treated OPCs. Gene ontology (GO) analysis revealed that fibrinogen upregulates the BMP pathway, a major suppressor of OPC differentiation (Gallo and Deneen, 2014) (Figure 1F, Table S1). Fibrinogen increased expression of several BMP-responsive genes (e.g., Id1, Id2, Nog, Hes1, Hey1, and Lef1) (Figure 1F) that are associated with impaired OPC differentiation and upregulated in some MS patients (Pedre et al., 2011; Wu et al., 2012). We evaluated brain autopsy samples of patients with different types of MS lesions by immunostaining for fibrinogen with MBP or the BMP target Id2. We compared active lesions, characterized by florid parenchymal inflammation and ongoing demyelination, chronic lesions of demyelinated areas with absent or few inflammatory cells, and remyelinated lesions (Absinta et al., 2016; Han et al., 2008). Active lesions had high levels of Id2 in areas of fibrinogen deposition and demyelination (Figure 2A-C). Perivascular fibrinogen was seen in chronic MS lesions but was minimal in remyelinated lesions and absent in normal white matter (Figure 2A,B). Id2 expression was reduced in the chronic lesions and similar to controls in remyelinated lesions (Figure 2A,B). These results suggest that fibrinogen may increase BMP signaling at sites of increased BBB permeability.
Figure 2. Fibrinogen Activates BMP Signaling in OPCs.
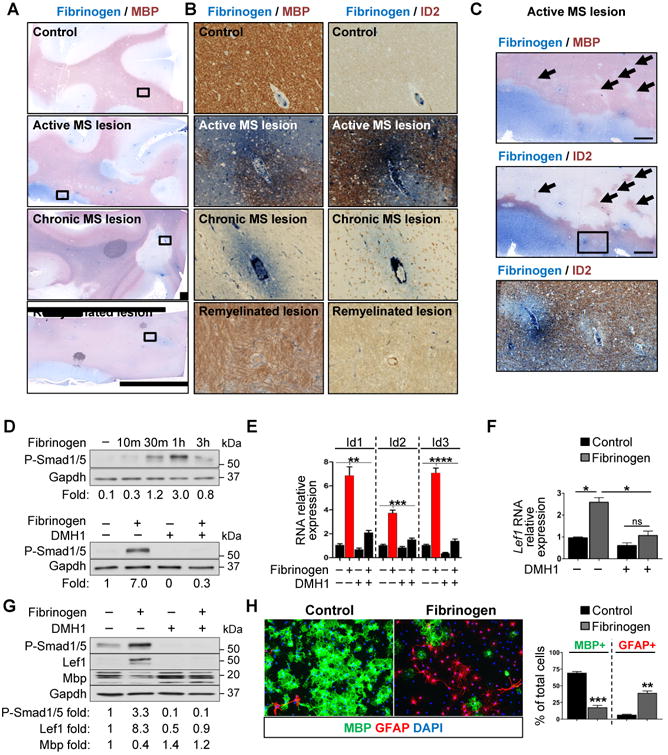
, (A-C) Immunohistochemistry for fibrinogen (blue) and either MBP (red) or Id2 (red) in active, chronic, and remyelinating MS lesions. (A) Boxes show location of images in B. Scale bar: 2 mm. (B) Scale bar: 100 μm (C) Box shows location of bottom image. Arrows indicate areas of demyelination with increased Id2 immunoreactivity. Scale bar: 1 mm (top images), 100 μm (bottom).
(D) Top: P-Smad1/5 levels in primary rat OPCs treated with fibrinogen for the indicated time. Bottom: P-Smad1/5 levels in control or fibrinogen-treated primary rat OPCs in the presence of the BMP receptor inhibitor DMH1. Values are mean of n = 2 independent experiments.
(E) Id1-3 in primary rat OPCs treated with fibrinogen for 3 h and DMH1. Values are mean ±s.e.m. from n = 3 independent experiments. **p < 0.01, ***p < 0.001, ****p < 0.0001 (two-wayANOVA with Bonferroni).
(F) Lef1 in primary rat OPCs treated with fibrinogen for 48 h and DMH1. Values are mean ±s.e.m. from n = 2 independent experiments. ns = not significant, *p < 0.05 (two-way ANOVAwith Bonferroni).
(G) P-Smad1/5, Lef1, and MBP in primary rat OPCs treated with fibrinogen and DMH1 for 4 days. Representative immunoblot and densitometry from n = 2 independent experiments.
(H) Immunofluorescence for MBP (green) and GFAP (red) in primary rat OPCs treated with fibrinogen or control. Nuclei are stained with DAPI. Representative images from n = 3 independent experiments. Scale bar: 50 μm. Values are mean ± s.e.m., **p < 0.01, ***p < 0.001 (unpaired t-test).
Fibrinogen Activates BMP Receptor Signaling in OPCs
In cultured rat OPCs, fibrinogen increased phosphorylation of the BMP signal transducers Smad1/5 and induced Id1-3 expression (Figure 2D,E), indicating activation of BMP downstream signaling. DMH1, a dorsomorphin analogue that inhibits the BMP type I receptor ACVR1 (Alk2) (Hao et al., 2010), blocked fibrinogen-induced phosphorylation of Smad1/5 and suppressed the Id genes (Figure 2D,E). Fibrinogen induced RNA and protein expression of LEF1 (Figure 2F,G), which is regulated by ACVR1 and associated with arrested OPC maturation (Choe et al., 2013; Fancy et al., 2014). DMH1 blocked fibrinogen-induced LEF1 expression and increased MBP expression (Figure 2F,G), indicating that fibrinogen activates ACVR1 signal transduction to inhibit myelin production.
A striking effect of BMP signaling in OPCs is differentiation to GFAP+ astrocyte-like cells instead of mature OLs in vitro (Mabie et al., 1997). Similarly, fibrinogen increased GFAP+ cells in OPC cultures (Figure 2H). To test whether GFAP+ cells in fibrinogen-treated cultures derived from OPCs, we traced the cell-fate of OPCs from NG2-CreER™:Rosa-tdTomato mice, allowing tamoxifen-induced expression of a red fluorescent protein, tdTomato, in nerve/glial antigen-2 (NG2)+ OPCs and their progeny (Figure S2A). Fibrinogen reduced formation of mature MBP+ OLs from genetically labeled NG2+ OPCs and increased the proportion of GFAP+ cells in culture (Figure S2B). Chronic infusion of fibrinogen into brains of NG2-CreER™:Rosa-tdTomato mice increased the percentage of tdTomato+ cells expressing GFAP (Figure S2C), suggesting fibrinogen induces the same BMP-like effect in vivo.
Fibrinogen Disrupts OPC Differentiation by Activating ACVR1
Noggin, a secreted BMP inhibitor, is a key homeostatic regulator of BMP that binds to extracellular BMPs to antagonize receptor binding (Groppe et al., 2002). Noggin rescued OPCs from the inhibitory effects of BMPs in vitro, but failed to block fibrinogen (Figure 3A, B). By ELISA, fibrinogen had no free BMP-2, -4, or -7 (not shown). Unlike noggin, DMH1 blocks BMP signaling by antagonizing the intracellular kinase domain of ACVR1 (Hao et al., 2010). DMH1 rescued OPCs from the inhibitory effects of BMP-7, an ACVR1 ligand, but not BMP-4, which does not act through ACVR1, showing the selectivity of DMH1 for ACVR1 (Figure 3A). In fibrinogen-treated OPCs, DMH1 increased the number of mature OLs, decreased the GFAP+ cells, and suppressed fibrinogen-induced Id1 gene expression (Figure 3A,B). Knockout of ACVR1 in primary OPCs by CRISPR/Cas9 reduced fibrinogen-induced nuclear accumulation of phosphorylated Smad1/5 and Id1 expression and enhanced formation of mature MBP+ OLs after fibrinogen treatment (Figure 3C, S3A-C). In the HAP1 human cell line, ACVR1 CRISPR/Cas9 knockout suppressed fibrinogen-induced Id1 (Figure S3D). Lipid rafts regulate BMP receptor signaling and progenitor cell differentiation (North et al., 2015). Pre-treating OPCs with the lipid raft disrupting methyl-β-cyclodextrin reduced fibrinogen-induced phospho-Smad1/5 levels by ∼45% (Figure S3E), suggesting fibrinogen enhances ACVR1 receptor association in lipid rafts to activate BMP signaling. These results suggest fibrinogen overcomes the endogenous homeostatic mechanisms that scavenge free BMPs and inhibits myelination by BMP ligand-independent activation of ACVR1.
Figure 3. Fibrinogen Disrupts OPC Differentiation through BMP Ligand-Independent Activation of ACVR1.
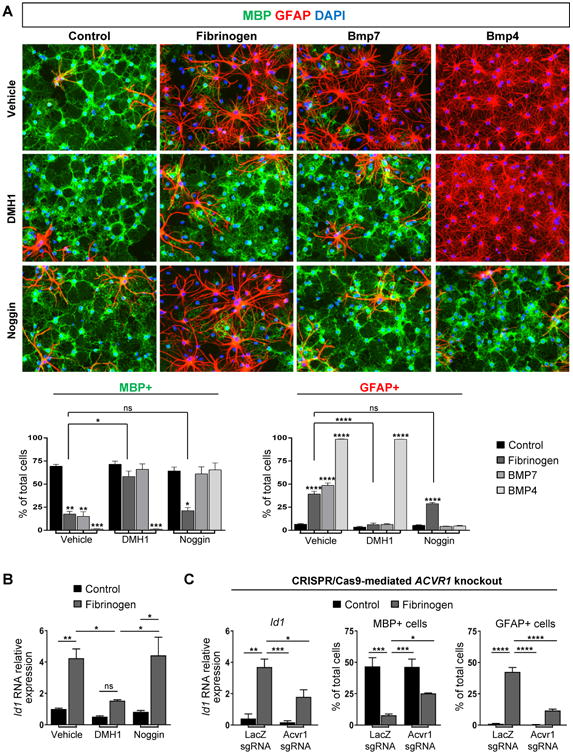
(A) Immunofluorescence for MBP (green) and GFAP (red) in primary rat OPCs treated with fibrinogen, BMP7, or BMP4, and DMH1, noggin, or vehicle control. Nuclei are stained with DAPI. Data are mean ± s.e.m. from n = 2-3 independent experiments. ns = not significant, *p < 0.05, **p < 0.01, ***p<0.001, ****p < 0.0001 (two-way ANOVA with Bonferroni). Scale bar: 50 μm.
(B) Id1 in primary rat OPCs treated with fibrinogen and DMH1, noggin, or vehicle control. Values are mean ± s.e.m. from n = 4–7 wells from 2-3 independent experiments. ns = not significant, *p < 0.05, **p < 0.01 (two-way ANOVA with Bonferroni).
(C) Analysis of primary rat OPCs transfected with a Cas9 expression plasmid containing single-guide RNA (sgRNA) for either LacZ (control) or Acvr1. Left: Id1 after 2h fibrinogen treatment, n = 3 independent experiments. Right: Quantification of MBP+ and GFAP+ cells after 3 day fibrinogen treatment, n = 4 wells from 2 independent experiments. Values are mean ± s.e.m. *p < 0.05, **p < 0.01, ***p<0.001, ****p < 0.0001 (two-way ANOVA with Holm-Sidak).
Therapeutically Depleting Fibrinogen Increases Remyelination in Vivo
To assess OPC differentiation and remyelination in vivo, we used the lysolecithin (LPC) focal demyelination model (Fancy et al., 2014). Fibrinogen was deposited in LPC lesions, suggesting BBB disruption (Figure 4A), that was cleared by 10–14 days post lesion (d.p.l.), when remyelination begins (Figure 4A). To therapeutically deplete fibrinogen in the LPC model, we administered the defibrinogenating agent ancrod at 3 d.p.l. (Adams et al., 2007). Fibrinogen depletion reduced phospho-Smad1/5/8 in demyelinated lesions (Figure 4B), suggesting reduction of BMP receptor pathway activation. Fibrinogen-depleted mice had more mature PLP-expressing OLs within LPC lesions (Figure 4C) and enhanced remyelination (Figure 4D), as indicated by a reduced proportion of axons that remained demyelinated and a decreased G-ratio of remyelinated axons that indicates thicker myelin. Ancrod alone did not affect OPC differentiation in vitro (Figure S4F), suggesting the pro-myelinating effect of ancrod is due to fibrinogen depletion.
Figure 4. Therapeutically Depleting Fibrinogen Increases Remyelination in Vivo.
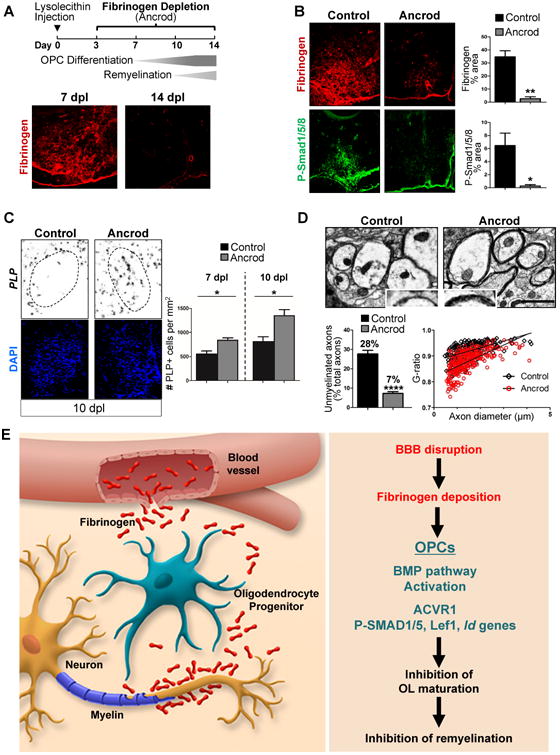
(A) Top: LPC injections with fibrinogen depletion using Ancrod. Bottom: Immunostaining for fibrinogen (red) in LPC-induced demyelinated lesions in ventral spinal cord of mice at 7 and 14 dpl. Dotted lines: lesion border. Representative images are shown. Scale bar: 100 μm.
(B) Immunostaining for fibrinogen (red) and P-Smad1/5/8 (green) in LPC lesions in ventral spinal cord of mice treated with ancrod or vehicle control at 7 dpl. Dotted lines: lesion border. Quantification of fibrinogen and P-Smad1/5/8 immunoreactivity from n = 3 mice per group. Values are mean ± s.e.m. *p<0.05, **p<0.01 (unpaired t-test). Scale bar: 100 μm.
(C) In situ hybridization for PLP mRNA (black) labeling mature OLs in LPC lesions in ventral spinal cord of mice treated with ancrod or vehicle control at 10 dpl. Nuclei are stained with DAPI (blue). Dotted lines: lesion border. Quantification of PLP+ cells within LPC lesions at 7 and 10 dpl from n = 3–4 mice per group. Values are mean ± s.e.m. *p < 0.05 (unpaired t-test). Scale bar: 100 μm.
(D) Electron microscopy of remyelinating axons within LPC lesions at 14 dpl in ancrod-treated and control mice. Inset image (blue box) shows close-up of myelin sheath (black). Quantification of axons within LPC lesions that remained unmyelinated at 14 dpl from n = 4 mice per group. Values are mean ± s.e.m. **** p <0.0001 (unpaired t-test). G-ratios between controls (G ratio mean = 0.92, s.e.m. = 0.002) and the ancrod group (G ratio mean = 0.88, s.e.m. = 0.003). n = ∼75 axons counted per mouse, 4 mice per group. ****p < 0.0001 (unpaired t-test). Scale bar: 1 μm.
(E) Working model of fibrinogen in CNS remyelination. Fibrinogen enters the CNS after BBB disruption and has pleiotropic functions as an inducer of inflammatory responses and inhibitor of regeneration.
Discussion
We found that fibrinogen is a potent extrinsic inhibitor of OPC differentiation and remyelination. Fibrinogen activates the ACVR1 receptor on OPCs to induce downstream signaling and BMP target gene expression and inhibit remyelination (Figure 4E). Fibrinogen may be beneficial in acute CNS injuries by acting as a “damage signal” that delays regeneration until the extracellular environment is conducive to repair. But, in chronic diseases with excessive fibrinogen deposition or inadequate fibrinogen clearance, this mechanism may be deleterious and lead to a state akin to a non-healing wound with aberrant activation of signaling pathways that block regeneration. Indeed, persistent fibrin deposition inhibits wound repair and is a feature of MS lesions with impaired fibrinolysis (Bugge et al., 1996; Gveric et al., 2003; Yates et al., 2017). Endogenous homeostatic mechanisms that scavenge free BMPs do not block fibrinogen's inhibitory effects. Thus, targeting fibrinogen therapeutically may tip the balance from a dysregulated environment to one that promotes repair.
Fibrinogen may contribute to a hostile environment in demyelinating diseases by activating BMP receptor signaling in OPCs (this study) and as an upstream driver of inflammation and reactive gliosis (Bardehle et al., 2015). At sites of BBB disruption, fibrinogen is converted to proinflammatory fibrin, which induces reactive oxygen species (ROS) release and M1-like activation of microglia and macrophages (Davalos et al., 2012; Ryu et al., 2015). M1-like polarization of innate immune cells and ROS are toxic to OPCs and hinder remyelination (Back et al., 1998; Lehnardt et al., 2002). A switch from an M1- to an M2-dominant response is essential for remyelination (Miron et al., 2013). Conditioned media from fibrin-treated macrophages hindered OPC differentiation. Fibrin-induced M1-like polarization of microglia and macrophages (Davalos et al., 2012; Ryu et al., 2015) may hinder the reparative inflammatory response for remyelination, which would agree with failed repair of MS lesions where BBB leakage at the lesion edge is prolonged (Absinta et al., 2016). Fibrinogen depletion with ancrod decreases inflammation and demyelination in animal MS models (Adams et al., 2007; Akassoglou et al., 2004; Paterson, 1976; Ryu et al., 2015). In the LPC model, active demyelination occurs at 1–3 d.p.l., and initial recruitment and activation of inflammatory cells at 12 h–3 d.p.l. (Mei et al., 2014; Ousman and David, 2000). In our study, ancrod was administered therapeutically starting at 3 d.p.l. to avoid interference with the initial demyelination and inflammation. Ancrod decreased BMP pathway activation and increased synthesis of myelin genes, but we cannot exclude ancrod effects in fibrin-driven inflammation and demyelination after 3 d.p.l. Future studies will determine the relative contributions of direct effects of fibrinogen on OPCs and its indirect effects mediated by inflammation in inhibiting CNS remyelination.
Given their pleiotropic functions, fibrinogen and fibrin may affect multiple pathological mechanisms that alter repair. Fibrinogen inhibits Schwann cell differentiation and inhibits neurite outgrowth (Akassoglou et al., 2002; Schachtrup et al., 2007). Fibrinogen induces production of other extrinsic remyelination inhibitors like chondroitin sulfate proteoglycans (CSPGs) in astrocytes and endothelin-1 in endothelial cells (Schachtrup et al., 2010; Sen et al., 2009). Thus, fibrinogen may be an apical signal that orchestrates the molecular composition of the inhibitory extracellular environment. An effect of fibrinogen in OPCs consistent with activation of BMP signaling was formation of GFAP+, astrocyte-like cells. OPC multipotency in vivo and potential lineage switches in disease are controversial (Richardson et al., 2011). Future studies will determine the expression of BMP activation markers in OPCs and astrocytes in acute and chronic MS lesions. In acute animal models of demyelination, such as LPC, the switch of OPCs to astrocytes is a rare event (∼3%) (Zawadzka et al., 2010); thus, fibrinogen's inhibitory effect on remyelination in the LPC model is more likely related to inhibiting final OL maturation. Since the LPC model has no permanent remyelination blockade, fibrinogen depletion likely accelerates remyelination. It is possible that fibrinogen-induced BMP signaling drives cell fate decisions in other models with large or more hemorrhagic insults. Indeed, fate-mapping studies after spinal cord injury show that ∼25% of NG2 cells mature into astrocytes (Hackett et al., 2016). Thus, in large traumatic injuries, OPC differentiation to GFAP+ cells may be more pronounced than in acute LPC injection. Fibrinogen is abundantly deposited in the spinal cord after injury (Schachtrup et al., 2007), and chronic fibrinogen infusion is sufficient to increase the number of NG2-derived GFAP+ cells in vivo (this study). Future studies will determine if fibrinogen is required for the generation of astrocyte-like cells by OPCs and stem/progenitor cell fate determination at sites of vascular damage in animal models with chronic insults.
Our study suggests targeting fibrinogen may overcome the inhibitory environment that persists in chronically demyelinated lesions. Although screens of FDA-approved drugs identified those that promote myelination (Mei et al., 2014; Najm et al., 2015), it is unknown if these drugs will overcome inhibition by extrinsic factors in the demyelinated lesions. OPC differentiation drugs failed to rescue inhibition of remyelination by extracellular CSPGs (Keough et al., 2016), suggesting a need for new approaches to target extrinsic inhibition of remyelination. With the many functions of fibrinogen as an OPC differentiation inhibitor with pro-inflammatory and pro-fibrotic functions, anti-coagulant therapies that inhibit fibrin formation or inhibition of fibrinogen binding to immune cell receptors or growth factors might be beneficial for tissue repair. Counteracting inhibitory BMP signaling in OPCs by BMP receptor inhibitors like DMH-1 (our study) or pro-myelinating ligands like activin B (Dutta et al., 2014) may alleviate the inhibitory effects of fibrinogen on remyelination. Novel inhibitors may overcome the hostile, dysregulated environment that persists in chronically demyelinated lesions and open new therapeutic strategies to promote regeneration in neurological diseases with fibrin deposition.
Star Methods
Contact for Reagent and Research Sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Katerina Akassoglou (kakassoglou@gladstone.ucsf.edu).
Experimental Model and Subject Details
Animals
Sprague-Dawley timed-pregnant females or females with litters were purchased from Simonsen Laboratories or Charles River. E15 and P1–P7 rats of both sexes were used for DRG and OPC isolations, respectively. C57BL/6, NG2-CreER™ (Zhu et al., 2011), and Rosa-tdTomato (Madisen et al., 2010) mice were purchased from the Jackson Laboratory. Heterozygous, male and female NG2-CreER™:Rosa-tdTomato P7 mice were used for in vitro OPC fate-mapping studies and 8–12-week-old mice for in vivo fate-mapping. All LPC experiments were performed on 8–12-week-old male C57BL/6 mice. Mice were housed in groups of five per cage under standard vivarium conditions and a 12-h light/dark cycle. All animal protocols were approved by the Committee of Animal Research at the University of California, San Francisco, and in accord with the National Institutes of Health guidelines.
Primary OPC cultures
Rat cortical OPCs were isolated with immunopanning as previously described (Dugas and Emery, 2013) with the following modifications. Cerebral cortices from P1-7 Sprague-Dawley rats were dissected and then digested in papain (Worthington) in DPBS at 35°C for 30-45 minutes. After trituration, cells were incubated sequentially on three immunopanning dishes: RAN-2 (negative selection), O1 (negative selection), and O4 (positive selection). OPCs were released from the final panning dish with trypsin (Worthington) and seeded onto polyethyleneimine (Sigma-Aldrich)-coated tissue-culture plates or 8-well Permanox-chamber slides (Thermo Scientific Nunc) at an initial density of approximately 15,000 cells per cm2. OPCs were maintained in a 5% CO2 incubator at 37°C, first in proliferation media for 2 days, then in differentiation media for up to 4 additional days with a 50% media change after 2 days. The chemically defined base media was DMEM (4.5g/L glucose, +pyruvate, +glutamine; Thermo Fisher Scientific), 1× B27 (Thermo Fisher Scientific), 1× N2 (Thermo Fisher Scientific), 1% penicillin-streptomycin (Thermo Fisher Scientific), and 50 ng ml−1 NT3 (Peprotech). Proliferation media consisted of the base media supplemented with 10-20 ng ml−1 PDGF-AA (Peprotech). Differentiation media contained 20 ng ml−1 CNTF (Peprotech) and 40 ng ml−1 triiodothyronine (Sigma-Aldrich) with no PDGF-AA. Experiments were performed after the initial 2 days of proliferation.
OPC-DRG cultures
OPC-DRG co-cultures were prepared as previously described (Lee et al., 2012). Briefly, DRG neurons from E15 Sprague-Dawley rats were dissociated and plated (150,000 cells per collagen-coated 25-mm coverslip) with 100 ng ml–1 NGF (AbD Serotec). Neurons were maintained in MEM (Thermo Fisher Scientific) supplemented with 10% FBS (Hyclone) and 25 mM glucose. Proliferating cells were selected against with three treatments of 2 mM FDU/Uridine (Sigma-Aldrich). Neurons were maintained for 3 weeks and washed extensively with DMEM (Thermo Fisher Scientific) to remove any residual NGF before seeding OPCs. A2B5+ OPCs were isolated from P7-8 Sprague-Dawley rat brain cortices using immunopanning, and 250,000 cells were plated onto each DRG cover glass. Co-cultures were grown in chemically defined medium composed of DMEM (Thermo Fisher Scientific) supplemented with B27 (Thermo Fisher Scientific), N2 (Thermo Fisher Scientific), penicillin-streptomycin (Thermo Fisher Scientific), N-acetyl-cysteine (Sigma-Aldrich), and forskolin (Sigma-Aldrich).
OPC-nanofiber cultures
OPC-nanofiber cultures were prepared as previously described (Lee et al., 2012). Briefly, electron-spun polystyrene fibers (J. M. Corey, Univ. of Michigan) were aligned and secured to 12-mm coverslips with silicone adhesive (Dow Corning), sterilized with 70% ethanol, and coated with 100 μg ml−1 poly(L-lysine). Immunopanned O4+ OPCs (200,000 cells per coverslip) were plated onto the coated fibers and maintained in DMEM (Thermo Fisher Scientific) supplemented with B27 (Thermo Fisher Scientific), N2 (Thermo Fisher Scientific), penicillin-streptomycin (Thermo Fisher Scientific), N-acetyl-cysteine (Sigma-Aldrich), and forskolin (Sigma-Aldrich). For the first 2-3 days in vitro, the media was supplemented with 12.5 ng ml−1 PDGF-AA (Peprotech) to induce proliferation. Thereafter, the media was without PDGF-AA to promote OPC differentiation and myelination.
Macrophage conditioned media
Bone marrow–derived macrophages (BMDMs) were isolated from tibia and femur of adult Sprague-Dawley rats as described (Ryu et al., 2015). Bone-marrow cells were cultured in RPMI-1640 medium with 10% heat-inactivated FBS (Invitrogen), 1% penicillin-streptomycin (Corning), and 10 ng/ml rat M-CSF (Peprotech) for 7 d in vitro before use. On day 7, adherent BMDMs were dissociated from the plate and re-plated on uncoated or fibrin-coated 24 wells in OPC culture medium. Fibrin coating was prepared as previously described (Ryu et al., 2015). After 24 hours of stimulation, macrophage-conditioned media was collected, clarified by centrifugation, and added to OPC cultures.
HAP1 cell cultures
HAP1 cells edited by CRISPR/Cas to contain a 2bp insertion in a coding exon of ACVR1 (HZGHC000195c001) and parental HAP1 control cells (C631) were purchased from Horizon (Cambridge, UK) and cultured in Iscove's Modified Dulbecco's Medium (IMDM) supplemented with 10% FBS and 1% penicillin-streptomycin according to manufacturer's instructions. Cells were serum-starved for 5 hours prior to fibrinogen stimulation.
Human MS and neonatal HIE tissue
All human tissue was collected after informed consent and after institutional approval. Brain autopsy samples were collected from patients with different clinical subtypes of MS (Absinta et al., 2016; Fancy et al., 2014; Fancy et al., 2011b; Han et al., 2008). Lesions with florid parenchymal and perivascular inflammatory cell infiltration, abundant astroglial hypertrophy, myelin fragmentation, edema and ongoing demyelination with indistinct margins were classified as active lesions (Han et al., 2008). Chronic lesions had areas of demyelination with well-demarcated borders and abundant astrogliosis, but few or no inflammatory cells (Han et al., 2008). Remyelinated lesions were defined as discrete focal areas of pale Luxol fast blue staining but near-normal PLP/myelin immunostaining, with few or no inflammatory cells (Patrikios et al., 2006). Age-matched control brain samples were analysed similarly and were devoid of CNS abnormalities. Human HIE tissue was collected after informed consent and in accordance with guidelines established by the UCSF Committee on Human Research (H11170-19113-07), as previously described (Fancy et al., 2011b). Tissue was processed and evaluated by UCSF neuropathology staff as described (Fancy et al., 2014).
Method Details
Fibrinogen and pharmacologic inhibitors
Fibrinogen (EMD Millipore) was used at 2.5 mg ml−1, which is its physiological concentration in plasma (Davalos and Akassoglou, 2012). For signaling experiments, OPCs were factor-starved for 6–15 h before experimental treatments. For analysis of differentiation, OPCs were treated with 2.5 mg ml−1 fibrinogen, 50 ng ml−1 BMP4 (Peprotech), or 50 ng ml−1 BMP7 (Peprotech) when changed to differentiation media. For inhibitor studies, OPCs were pre-treated with 1 μM DMH1 (Tocris), 500–1000 ng ml−1 recombinant human noggin (Peprotech), or vehicle (control) 1 h before treatments. In noggin experiments, the inhibitors were also incubated with the vehicle (control)-, fibrinogen-, or BMP-containing differentiation media at 37°C for 1 h before being added to the OPCs. Inhibitors were replaced with subsequent media changes.
Myelinating cultures
In OPC-DRG cultures, 2.5 mg ml−1 fibrinogen was added when OPCs were plated onto DRGs and with media changes every 3 days. In OPC-nanofiber cultures, poly(L-lysine)-coated fibers were additionally coated with fibrinogen (Calbiochem), albumin (A4161, Sigma-Aldrich), fibrinogen-Alexa Fluor 594 or 647 conjugate (Thermo Fisher Scientific), or albumin-Alexa Fluor 594 or 647 conjugate (Thermo Fisher Scientific) at 50 μg ml−1 for 2 h at 37° before adding OPCs. Cultures were fixed and processed for immunostaining at 4–6 DIV for OPC-nanofibers and 7–9 DIV for OPC-DRGs.
Cell fate mapping studies
For in vitro fate-mapping studies, mouse cortical OPCs were isolated from NG2-CreER™:Rosa-tdTomato mice by immunopanning as previously described (Emery and Dugas, 2013) with the exception that papain digestion occurred in DPBS at 35°C for 30-45 minutes. Cells were incubated sequentially on three immunopanning dishes: BSL1 × 2 (negative selection) and PDGFRα (positive selection). Mouse OPC proliferation and differentiation media has been described previously (Emery and Dugas, 2013). To induce Cre-mediated recombination, OPCs were treated with 1 μM 4-hydroxytamoxifen (Sigma-Aldrich) in proliferation media for 48 hours. Cultures were then changed to control or fibrinogen-containing differentiation media and processed for immunostaining 3 days after the media change. For in vivo fate-mapping, 8- to 12-wk-old NG2-CreER™:Rosa-tdTomato mice were injected intraperitoneally with 1 mg tamoxifen (Sigma-Aldrich) daily for four days. Experiments were performed 3-4 weeks after tamoxifen administration.
CRISPR/Cas9-mediated knockout of Acvr1 in primary OPCs
Experimental overview
One to five days after initial isolation, primary rat OPCs were transfected with a Cas9 expression plasmid expressing EGFP and sgRNA for either LacZ (control) or Acvr1. To determine genome targeting efficiency, transfected OPCs underwent T7 endonuclease I assays three days after transfection. To determine changes in gene expression, cultures 2-4 days after transfection were fibrinogen-treated (2.5 mg ml−1 for 2 hours) and then underwent FACS to select the transfected cells for RNA isolation and analysis. In some experiments, OPCs were sorted two days after transfection, re-plated into proliferation media and allowed to recover for two days before fibrinogen treatment and RNA isolation. To assess differentiation, OPCs were transfected at 5 DIV, underwent FACS two days after transfection, re-cultured for two days in proliferation media, and then fibrinogen-treated when changed to differentiation media. Cultures were fixed and processed for immunostaining after 3 days of differentiation.
Plasmids
The plasmid peSpCas9(1.1)(BB) encoding SpCas9 (with improved specificity) was a gift from Dr. Feng Zhang (Addgene plasmid # 71814) (Slaymaker et al., 2016), peSpCas9(1.1)-fG(BB) was constructed by fusing the sequence encoding P2A-farnesylated EGFP to Cas9 in peSpCas9(1.1)(BB). pCMV-Cas9-fG(BB) was constructed by replacing the CBh promoter in peSpCas9(1.1)-fG(BB) with the CMV promoter. The annealed oligonucleotides for the sgRNA guide sequence were ligated to BbsI-digested peSpCas9(1.1)-fG(BB) or pCMV-Cas9-fG(BB). sgRNA guide sequences are listed in Table S2.
OPC transfection
Primary OPCs cultured in proliferation medium were transfected by adding the plasmid, Lipofectamine LTX and Plus reagent mixed in OPTI-MEM (Thermo Fisher Scientific) according to the manufacturer's instructions. Two μg of plasmids, 2 μl of Plus and 4μl of Lipofectamine LTX were used to transfect OPCs cultured on a 3.5-cm dish; double the amount was used for cells cultured on 6-cm dishes. After overnight incubation, the medium was replaced.
T7 endonuclease I assays
Three days after transfection of cultured OPCs, the cells were lysed in lysis buffer (100 mM NaCl, 10 mM Tris-Cl pH 8.0, 25 mM EDTA, 0.5% SDS, and 0.1 mg/ml proteinase K) at 56°C for 1 hr. Genomic DNA was precipitated by adding double the volume of 100% ethanol and centrifuging at 16100 × g for 5 min at room temperature. After washing twice with 70% ethanol and drying, the DNA pellet was dissolved in TE buffer (10 mM Tris-Cl pH 8.0 and 1 mM EDTA) at 56°C for 10 min. The genome editing events were detected by using the EnGen Mutation Detection Kit (New England BioLabs) and 25-65 ng of genomic DNA according to the manufacturer's instructions. The primer pairs used for PCR amplification of the edited loci are listed in Table S2.
FACS of transfected OPCs
Two and four days after transfection, cultured OPCs were rinsed twice with Hanks' balanced salt solution without calcium and magnesium (Thermo Fisher Scientific), and then trypsinized with 0.25% trypsin (Thermo Fisher Scientific) in the incubator for 5 min. Cells were resuspended by pipetting and trypsinization was stopped by adding an equal volume of DPBS without calcium and magnesium (Thermo Fisher Scientific) containing 0.25% ovomucoid (Worthington) and 0.25% bovine serum albumin (Sigma-Aldrich). After centrifugation at 300 × g for 5 min, the cell pellet was resuspended in 0.4 ml of FACS buffer (DPBS without calcium and magnesium containing 1 × B-27, 1 × N-2, 2 mM EDTA, 5.56 mM glucose, and 25 mM HEPES-Na pH 7.0) and kept on ice. Cell suspension was filtered through a 40 μm cell strainer and EGFP+ cells (MFI cut-off 103) were sorted using the FACSAria II (BD Biosciences). EGFP+ cells were collected into 300 μl FACS buffer at 4°C for RNA isolation, or in 300 μl proliferation medium containing 25 mM HEPES-Na pH 7.0 at room temperature for re-culture experiments. For RNA isolation, sorted cell suspension was centrifuged for 8 minutes at 300 × g at 4°C and the cell pellet lysed in 350 μl RLT buffer with 1% 2-mercaptoethalnol (Sigma). For re-culture experiments, sorted cells were re-plated into proliferation media and allowed to recover for two days before fibrinogen treatment.
RNA Isolation and Quantitative PCR
Total RNA was extracted from OPC cultures with the RNeasy kit (QIAGEN) according to the manufacturer's instructions. cDNA was generated and real-time (RT)-PCR performed as previously described (Ryu et al., 2015). Data are expressed as 2−ΔΔCT for the experimental gene-of-interest normalized against the housekeeping gene and presented as the fold change versus the relevant control. Primers are listed in Table S2.
Gene-Expression Profiling by Microarray Analysis
Microarray analysis was performed on triplicate samples of primary rat OPCs treated with 2.5 mg ml−1 fibrinogen for 12 h and 48 h in differentiation media. Total RNA was isolated using RNeasy Mini kit (QIAGEN) according to the manufacturer's instruction. Probes were prepared using NuGEN Ovation Pico WTA V2 kit and NuGEN Encore Biotin Module, and hybridized to Rat Gene 1.0 ST GeneChip arrays (Affymetrix). Arrays were scanned using an Affymetrix GCS3000 scanner and Affymetrix Command Console software, and data were normalized using the RMA algorithm in Affymetrix Expression Console. Microarrays were normalized for array-specific effects using Affymetrix's ‘Robust Multi-Array’ normalization. Normalized array values were reported on a log2 scale. For statistical analyses, we removed all array probe sets where no experimental groups had an average log2 intensity >3.0. This is a standard cutoff, below which expression is indistinguishable from background noise. Linear models, moderated t-statistics, fold change, false discovery rate (FDR)-adjusted values, and the associated P values were calculated as described (Ryu et al., 2015).
Immunoblots
Cells were lysed in cell lysis buffer supplemented with protease/phosphatase inhibitor cocktails (Calbiochem) and lysates were cleared by centrifuging at 13,000×g for 15 minutes at 4°C. Equal amounts of protein were loaded in 4-12% or 8-16% gels and analyzed by Western blotting. Bands were visualized with HRP-conjugated secondary antibodies (Cell Signaling Technology or Santa Cruz Biotechnology). Densitometry was performed using ImageJ Software (NIH) with values for each band normalized to GAPDH loading controls from the same membrane.
Immunocytochemistry, Immunohistochemistry, and in situ hybridization
Immunocytochemistry (ICC) was performed as previously described (Lee et al., 2012). Briefly, cultures were fixed with 4% PFA for 15 min at room temperature, permeabilized, and blocked by incubation with 10-20% goat or donkey serum (Sigma-Aldrich or Jackson ImmunoResearch) and 0.1–0.2% Triton X-100 (Sigma-Aldrich) in PBS. Primary antibody incubations were performed overnight at 4°C. Fluorescent secondary antibodies (Jackson ImmunoResearch or Thermo Fisher Scientific) were incubated for 1–2 h at room temperature. DAPI was used to detect cell nuclei.
For mouse immunohistochemistry (IHC), mice were transcardially perfused with 4% PFA under deep avertin anesthesia. Tissue was removed, post-fixed overnight in 4% PFA, cryoprotected in 30% sucrose/PBS, frozen in Neg-50 media (Thermo Scientific), cryosectioned into 12-μm sections, and placed on ColorFrost Plus microscope slides (Fisher Scientific). Sections were permeabilized in 0.1% Triton X-100, blocked with 5% BSA or 5% normal donkey serum, and incubated with primary antibodies overnight at 4°C and then fluorescent secondary antibodies for 1–2 h at room temperature. Slides were coverslipped with Prolong Gold or SlowFade Gold antifading agent with DAPI (Thermo Fisher Scientific). In situ labeling of the mature oligodendrocyte marker PLP was performed as previously described (Fancy et al., 2011b).
For human IHC, formalin-fixed paraffin embedded tissue sections were first deparaffinized then rehydrated. Heat-mediated antigen retrieval was performed with Target Retrieval Solution, Low pH (Dako) for 1 hour in 95° water bath. Endogenous alkaline phosphatase and horseradish peroxidase activity was blocked with Bloxall (Vector). Sections were permeabilized in 0.3% Triton X-100, blocked with 2.5% normal horse serum, and incubated with primary antibodies overnight at 4°C. Sequential labeling of multiple antigens was performed using the ImmPRESS™-AP Anti-Mouse IgG (alkaline phosphatase) and ImmPRESS™ VR Anti-Rabbit IgG HRP Kits (Vector) and developed with the Vector Blue or NovaRED Substrate Kits (Vector) according to manufacturer's instruction. Slides were coverslipped in VectaMount AQ (Vector).
Images were acquired with an Axioplan II epifluorescence microscope (Carl Zeiss) equipped with dry Plan-Neofluar objectives (10× 0.3 NA, 20× 0.5 NA, or 40× 0.75 NA), an Axiocam HRc CCD camera, and the Axiovision image analysis software; the BIOREVO BZ-9000 inverted fluorescence microscope (Keyence) equipped with a Nikon CFI 60 Series infinite optical system and Keyence imaging software; or Olympus Fluoview confocal microscope equipped with 20× NA1.0 objective. All images were processed and analyzed in ImageJ. For ICC, cell counts from multiple wells (5–10 images per well) from two or more independent experiments were quantified for each test group. For IHC in the brain, cell counts from multiple 200μm × 200μm randomly selected regions of interest within 250μm of the cannula tract were quantified on two or more nonadjacent sections per mouse. N = 6-8 mice per group. In the spinal cord, quantification was performed on thresholded images. N = 3-4 mice per group. For ISH, PLP-positive cells were counted within lesions on three or more nonadjacent sections per mouse with N = 3-4 mice per group.
The following primary antibodies were used: cleaved caspase-3 (ICC: 1:200, rabbit polyclonal, #9661, Cell Signaling Technology); fibrinogen (mouse IHC: 1:1000, rabbit polyclonal, gift from J. Degen, Cincinnati; human IHC: 1:500, mouse monoclonal, #ab58207, Abcam); GAPDH (WB: 1:1000, rabbit monoclonal, #2118, Cell Signaling Technology); GFAP (ICC/IHC: 1:200, WB: 1:1000, rabbit monoclonal, #12389, Cell Signaling Technology; or rat monoclonal, #13-0300, Thermo Fisher Scientific); phospho-histone H3 (ICC: 1:200, rabbit monoclonal, #3377, Cell Signaling Technology); Id2 (human IHC: 1:1000, rabbit monoclonal, # M213, CalBioreagents); LEF1 (ICC: 1:200, WB: 1:1000, rabbit monoclonal, #2230, Cell Signaling Technology); MBP (ICC/IHC: 1:500, WB: 1:1000, mouse monoclonal, SMI-94, BioLegend; or rat monoclonal, #MAB395, EMD Millipore); Olig2 (ICC/IHC: 1:200, rabbit polyclonal, #ab9610, EMD Millipore); RNF43 (IHC: 1:200, rabbit polyclonal, #ab84125, Abcam); phospho-Smad1/5 (WB: 1:1000, rabbit monoclonal, #9516, Cell Signaling Technology); phospho-Smad1/5/9 (WB: 1:1000, ICC: 1:200, rabbit monoclonal, #13820, Cell Signaling Technology); and phospho-Smad1/5/8 (IHC: 1:100, goat polyclonal, #sc-12353, Santa Cruz Biotechnology).
Chronic fibrinogen infusion
Fibrinogen was infused continuously into the brains of tamoxifen-induced NG2-CreER™:Rosa-tdTomato mice using mini-osmotic pumps (ALZET model 2002, 0.5 μl/h for 14 d) and the brain infusion kit (ALZET Brain infusion kit 3) according to the manufacturer's instruction. Pumps were filled with either fibrinogen (2 mg ml-1) or artificial CSF and primed for 24 h at 37 °C. The sterile brain infusion cannula (30-gauge) was stereotactically implanted at the following coordinates: 0.5 mm rostral to the bregma, 1 mm lateral to the midline, and 2 mm deep from the skull surface. The cannula was glued to the skull using Metabond (Parkell) and dental cement, while the pump was inserted subcutaneously on the back. Mice were euthanized for histological analysis after 14 day infusion.
Lysolecithin-induced focal demyelination
Demyelinated lesions were chemically induced by focal injection of lysolecithin into the spinal cord ventrolateral white matter of 8- to 12-wk-old C57Bl/6 mice as previously described (Mei et al., 2014) with modifications highlighted in the following experimental description. Animals were anesthetized via intraperitoneal injection of ketamine (100 mg/kg) and xylazine (15 mg/kg). A midline skin incision was made over the upper lumbar regions of spinal cord, and the spinal column was secured with mouse vertebral clamps fixed in a stereotaxic frame. The epidural space was exposed by disruption of the L1-L2 interspinous ligament without laminectomy as described (Ryu et al., 2015). A pulled-glass micropipette secured to a Hamilton syringe was prefilled with 1% lysolecithin (L-α-lysophosphatidylcholine, Sigma-Aldrich) and inserted into the spinal cord 0.3 mm lateral to the spinal midline and a depth of 0.9 mm from the spinal cord surface. Then, 1 μL of 1% lysolecithin was injected (0.2 μl/min) and the glass micropipette remained in place for 5 min before being slowly withdrawn. After surgery, skin was sutured, and mice were allowed to recover.
Pharmacologic depletion of fibrinogen after lysolecithin injection
Mice were depleted of fibrinogen with ancrod at 3 dpl as described (Adams et al., 2007). The mice received 2.75 U ancrod/day by mini-osmotic pump (Alzet). In control animals, buffer-filled mini-pumps were implanted.
Electron microscope analysis of remyelination
At 14 days post-lysolecithin injection, mice were transcardially perfused with 2% PFA/2.5% glutaraldehyde in 0.1 M sodium cacodylate buffer with 2 mM CaCl2. Spinal cords were processed and EM performed with a FEI Spirit transmission electron microscope, according to standard procedures (Davalos et al., 2012). We examined ∼300 axons (∼75 axons per mouse, N = 4 mice per group) within the rim of active remyelination at the lesion edge, and calculated g-ratios as the diameter of the axon divided by the diameter of the axon plus the surrounding myelin sheath. Unmyelinated axons were excluded from G-ratio graph and analysis.
Quantification and Statistical Analysis
Data are presented as mean ± standard error of the mean. Differences between experimental conditions were analyzed with GraphPad Prism software. Data distribution was assumed to be normal, but this was not formally tested. No statistical methods were used to predetermine sample size, but our sample sizes are similar to those reported (Fancy et al., 2014). Mice and cells were divided into experimental groups in an unbiased manner. No randomization was used to assign groups or to collect data. All animals survived until the end of the study, and all data points were included in analysis. Analyses of in vitro inhibitor, CRISPR, and conditioned media experiments were performed by a blinded observer. The unpaired Student's t-test was used for isolated pairs, and two-way analysis of variance (ANOVA) with a Bonferroni or Holm-Sidak post-test was used for multiple comparisons. P < 0.05 was considered significant.
Data and Software Availability
The raw data files from the microarrays have been deposited in the Gene Expression Omnibus under GEO: GSE104450.
Supplementary Material
Highlights.
Fibrinogen is a blood-derived inhibitor of oligodendrocyte differentiation
Fibrinogen activates the ACVR1 BMP receptor on OPCs
The ACVR1 inhibitor DMH1 blocks fibrinogen's inhibitory effects on OPCs
Depletion of fibrinogen promotes remyelination in the CNS
Acknowledgments
We thank Reshmi Tognatta for discussions, Belinda Cabriga, Yun-An Shen, Marielle Cavrois, Nandhini Raman for expert technical assistance, John Lewis for graphics, Gary Howard for editorial. The Gladstone FACS core was supported by NIH P30 AI027763, NIH S10 RR028962, Department of Defence (DoD) W81XWH-11-1-0562 and the Gladstone Institutes from a National Center for Research Resources Grant RR18928. This work was supported by NIH/NICHD K12-HD072222 grant and Pediatric Scientist Development Program fellowship (March of Dimes 4-FY10-461 and NIH/NICHD K12-HD000850) to M.A.P; A Race to Erase MS Young Investigator Award and American Heart Association Scientist Development Grant to J.K.R; National Multiple Sclerosis Society (NMSS) fellowship grants FG-2093-A-1 to M.A. and FG-1507-05195 to K.J.C.; the Marilyn Hilton Bridging Award for Physician-Scientists to M.A., the NINDS Intramural Research Program to D.S.R.; NIH R01 NS027177, R01 GM086197, P41 GM103412 to M.H.E. for support of the National Center for Microscopy and Imaging Research; the NMSS RG5203, NIH/NINDS R01 NS062796, and the Rachleff Endowment to J.R.C.; and the NIH/NINDS R35 NS097976, NMSS RG4985, DoD MS160082, and Conrad N. Hilton Foundation grants to K.A. K.A. is a co-founder of MedaRed, Inc and K.A., J.K.R. and A.M.F. are named inventors on patents and patent applications related to fibrin. Their interests are managed by the Gladstone Institutes in accordance with its conflict of interest policy.
Footnotes
Author Contributions: M.A.P. designed, performed experiments, and analyzed data; J.K.R. performed in vivo studies; K.J.C. performed CRISPR studies; A.E. performed in vitro studies; S.B. performed cell sorting, histology, and quantification. A.S.M performed in vitro studies and quantification. W.K.D performed in vitro studies and animal care; S.P.J.F performed histology; A.T. and E.A.B. performed electron microscopy; B.B.R. performed immunoblots; C.A.S., M.D.W., P.E.R.C. performed image analysis and quantification; A.M.F. performed in vitro experiments and ELISAs; E.J.H. procured human neonatal brain tissue; M.H.H., M.A, and D.S.R. provided human MS tissue. S.Y, J.K.L., L.P, C.S., H.L. analysed and interpreted data. M.H.E., D.H.R., and J.R.C. designed experiments and interpreted data; K.A. and M.A.P. designed the study, analysed and interpreted data, wrote the manuscript with input from all authors.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Absinta M, Sati P, Schindler M, Leibovitch EC, Ohayon J, Wu T, Meani A, Filippi M, Jacobson S, Cortese IC, et al. Persistent 7-tesla phase rim predicts poor outcome in new multiple sclerosis patient lesions. J Clin Invest. 2016;126:2597–2609. doi: 10.1172/JCI86198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams RA, Bauer J, Flick MJ, Sikorski SL, Nuriel T, Lassmann H, Degen JL, Akassoglou K. The fibrin-derived gamma377-395 peptide inhibits microglia activation and suppresses relapsing paralysis in central nervous system autoimmune disease. J Exp Med. 2007;204:571–582. doi: 10.1084/jem.20061931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akassoglou K, Adams RA, Bauer J, Mercado P, Tseveleki V, Lassmann H, Probert L, Strickland S. Fibrin depletion decreases inflammation and delays the onset of demyelination in a tumor necrosis factor transgenic mouse model for multiple sclerosis. Proc Natl Acad Sci U S A. 2004;101:6698–6703. doi: 10.1073/pnas.0303859101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akassoglou K, Yu WM, Akpinar P, Strickland S. Fibrin inhibits peripheral nerve remyelination by regulating Schwann cell differentiation. Neuron. 2002;33:861–875. doi: 10.1016/s0896-6273(02)00617-7. [DOI] [PubMed] [Google Scholar]
- Back SA, Gan X, Li Y, Rosenberg PA, Volpe JJ. Maturation-dependent vulnerability of oligodendrocytes to oxidative stress-induced death caused by glutathione depletion. J Neurosci. 1998;18:6241–6253. doi: 10.1523/JNEUROSCI.18-16-06241.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bardehle S, Rafalski VA, Akassoglou K. Breaking boundaries—Coagulation and fibrinolysis at the neurovascular interface. Front Cell Neurosci. 2015;9 doi: 10.3389/fncel.2015.00354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bugge TH, Kombrinck KW, Flick MJ, Daugherty CC, Danton MJ, Degen JL. Loss of fibrinogen rescues mice from the pleiotropic effects of plasminogen deficiency. Cell. 1996;87:709–719. doi: 10.1016/s0092-8674(00)81390-2. [DOI] [PubMed] [Google Scholar]
- Choe Y, Kozlova A, Graf D, Pleasure SJ. Bone morphogenic protein signaling is a major determinant of dentate development. J Neurosci. 2013;33:6766–6775. doi: 10.1523/JNEUROSCI.0128-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davalos D, Akassoglou K. Fibrinogen as a key regulator of inflammation in disease. Semin Immunopathol. 2012;34:43–62. doi: 10.1007/s00281-011-0290-8. [DOI] [PubMed] [Google Scholar]
- Davalos D, Kyu Ryu J, Merlini M, Baeten KM, Le Moan N, Petersen MA, Deerinck TJ, Smirnoff DS, Bedard C, Hakozaki H, et al. Fibrinogen-induced perivascular microglial clustering is required for the development of axonal damage in neuroinflammation. Nat Commun. 2012;3:1227. doi: 10.1038/ncomms2230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dugas JC, Emery B. Purification of oligodendrocyte precursor cells from rat cortices by immunopanning. Cold Spring Harb Protoc. 2013;2013:745–758. doi: 10.1101/pdb.prot070862. [DOI] [PubMed] [Google Scholar]
- Dutta DJ, Zameer A, Mariani JN, Zhang J, Asp L, Huynh J, Mahase S, Laitman BM, Argaw AT, Mitiku N, et al. Combinatorial actions of Tgfbeta and Activin ligands promote oligodendrocyte development and CNS myelination. Development. 2014;141:2414–2428. doi: 10.1242/dev.106492. [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]
- Emery B, Dugas JC. Purification of oligodendrocyte lineage cells from mouse cortices by immunopanning. Cold Spring Harb Protoc. 2013;2013:854–868. doi: 10.1101/pdb.prot073973. [DOI] [PubMed] [Google Scholar]
- Fancy SP, Chan JR, Baranzini SE, Franklin RJ, Rowitch DH. Myelin regeneration: a recapitulation of development? Annu Rev Neurosci. 2011a;34:21–43. doi: 10.1146/annurev-neuro-061010-113629. [DOI] [PubMed] [Google Scholar]
- Fancy SP, Harrington EP, Baranzini SE, Silbereis JC, Shiow LR, Yuen TJ, Huang EJ, Lomvardas S, Rowitch DH. Parallel states of pathological Wnt signaling in neonatal brain injury and colon cancer. Nat Neurosci. 2014;17:506–512. doi: 10.1038/nn.3676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fancy SP, Harrington EP, Yuen TJ, Silbereis JC, Zhao C, Baranzini SE, Bruce CC, Otero JJ, Huang EJ, Nusse R, et al. Axin2 as regulatory and therapeutic target in newborn brain injury and remyelination. Nat Neurosci. 2011b;14:1009–1016. doi: 10.1038/nn.2855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franklin RJ Ffrench-Constant, C. Remyelination in the CNS: from biology to therapy. Nat Rev Neurosci. 2008;9:839–855. doi: 10.1038/nrn2480. [DOI] [PubMed] [Google Scholar]
- Gallo V, Deneen B. Glial development: the crossroads of regeneration and repair in the CNS. Neuron. 2014;83:283–308. doi: 10.1016/j.neuron.2014.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groppe J, Greenwald J, Wiater E, Rodriguez-Leon J, Economides AN, Kwiatkowski W, Affolter M, Vale WW, Izpisua Belmonte JC, Choe S. Structural basis of BMP signalling inhibition by the cystine knot protein Noggin. Nature. 2002;420:636–642. doi: 10.1038/nature01245. [DOI] [PubMed] [Google Scholar]
- Gveric D, Herrera B, Petzold A, Lawrence DA, Cuzner ML. Impaired fibrinolysis in multiple sclerosis: a role for tissue plasminogen activator inhibitors. Brain. 2003;126:1–9. doi: 10.1093/brain/awg167. [DOI] [PubMed] [Google Scholar]
- Hackett AR, Lee DH, Dawood A, Rodriguez M, Funk L, Tsoulfas P, Lee JK. STAT3 and SOCS3 regulate NG2 cell proliferation and differentiation after contusive spinal cord injury. Neurobiol Dis. 2016;89:10–22. doi: 10.1016/j.nbd.2016.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han MH, Hwang SI, Roy DB, Lundgren DH, Price JV, Ousman SS, Fernald GH, Gerlitz B, Robinson WH, Baranzini SE, et al. Proteomic analysis of active multiple sclerosis lesions reveals therapeutic targets. Nature. 2008;451:1076–1081. doi: 10.1038/nature06559. [DOI] [PubMed] [Google Scholar]
- Hao J, Ho JN, Lewis JA, Karim KA, Daniels RN, Gentry PR, Hopkins CR, Lindsley CW, Hong CC. In vivo structure-activity relationship study of dorsomorphin analogues identifies selective VEGF and BMP inhibitors. ACS Chem Biol. 2010;5:245–253. doi: 10.1021/cb9002865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keough MB, Rogers JA, Zhang P, Jensen SK, Stephenson EL, Chen T, Hurlbert MG, Lau LW, Rawji KS, Plemel JR, et al. An inhibitor of chondroitin sulfate proteoglycan synthesis promotes central nervous system remyelination. Nat Commun. 2016;7:11312. doi: 10.1038/ncomms11312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Leach MK, Redmond SA, Chong SY, Mellon SH, Tuck SJ, Feng ZQ, Corey JM, Chan JR. A culture system to study oligodendrocyte myelination processes using engineered nanofibers. Nat Methods. 2012;9:917–922. doi: 10.1038/nmeth.2105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehnardt S, Lachance C, Patrizi S, Lefebvre S, Follett PL, Jensen FE, Rosenberg PA, Volpe JJ, Vartanian T. The toll-like receptor TLR4 is necessary for lipopolysaccharide-induced oligodendrocyte injury in the CNS. J Neurosci. 2002;22:2478–2486. doi: 10.1523/JNEUROSCI.22-07-02478.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mabie PC, Mehler MF, Marmur R, Papavasiliou A, Song Q, Kessler JA. Bone morphogenetic proteins induce astroglial differentiation of oligodendroglial-astroglial progenitor cells. J Neurosci. 1997;17:4112–4120. doi: 10.1523/JNEUROSCI.17-11-04112.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madisen L, Zwingman TA, Sunkin SM, Oh SW, Zariwala HA, Gu H, Ng LL, Palmiter RD, Hawrylycz MJ, Jones AR, et al. A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat Neurosci. 2010;13:133–140. doi: 10.1038/nn.2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marik C, Felts PA, Bauer J, Lassmann H, Smith KJ. Lesion genesis in a subset of patients with multiple sclerosis: a role for innate immunity? Brain. 2007;130:2800–2815. doi: 10.1093/brain/awm236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mei F, Fancy SP, Shen YA, Niu J, Zhao C, Presley B, Miao E, Lee S, Mayoral SR, Redmond SA, et al. Micropillar arrays as a high-throughput screening platform for therapeutics in multiple sclerosis. Nat Med. 2014;20:954–960. doi: 10.1038/nm.3618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miron VE, Boyd A, Zhao JW, Yuen TJ, Ruckh JM, Shadrach JL, van Wijngaarden P, Wagers AJ, Williams A, Franklin RJ, et al. M2 microglia and macrophages drive oligodendrocyte differentiation during CNS remyelination. Nat Neurosci. 2013;16:1211–1218. doi: 10.1038/nn.3469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Najm FJ, Madhavan M, Zaremba A, Shick E, Karl RT, Factor DC, Miller TE, Nevin ZS, Kantor C, Sargent A, et al. Drug-based modulation of endogenous stem cells promotes functional remyelination in vivo. Nature. 2015;522:216–220. doi: 10.1038/nature14335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- North HA, Pan L, McGuire TL, Brooker S, Kessler JA. beta1-Integrin alters ependymal stem cell BMP receptor localization and attenuates astrogliosis after spinal cord injury. J Neurosci. 2015;35:3725–3733. doi: 10.1523/JNEUROSCI.4546-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ousman SS, David S. Lysophosphatidylcholine induces rapid recruitment and activation of macrophages in the adult mouse spinal cord. Glia. 2000;30:92–104. [PubMed] [Google Scholar]
- Paterson PY. Experimental allergic encephalomyelitis: role of fibrin deposition in immunopathogenesis of inflammation in rats. Fed Proc. 1976;35:2428–2434. [PubMed] [Google Scholar]
- Patrikios P, Stadelmann C, Kutzelnigg A, Rauschka H, Schmidbauer M, Laursen H, Sorensen PS, Bruck W, Lucchinetti C, Lassmann H. Remyelination is extensive in a subset of multiple sclerosis patients. Brain. 2006;129:3165–3172. doi: 10.1093/brain/awl217. [DOI] [PubMed] [Google Scholar]
- Pedre X, Mastronardi F, Bruck W, Lopez-Rodas G, Kuhlmann T, Casaccia P. Changed histone acetylation patterns in normal-appearing white matter and early multiple sclerosis lesions. J Neurosci. 2011;31:3435–3445. doi: 10.1523/JNEUROSCI.4507-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson WD, Young KM, Tripathi RB, McKenzie I. NG2-glia as multipotent neural stem cells: fact or fantasy? Neuron. 2011;70:661–673. doi: 10.1016/j.neuron.2011.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenzweig S, Carmichael ST. The axon-glia unit in white matter stroke: mechanisms of damage and recovery. Brain Res. 2015;1623:123–134. doi: 10.1016/j.brainres.2015.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryu JK, Petersen MA, Murray SG, Baeten KM, Meyer-Franke A, Chan JP, Vagena E, Bedard C, Machado MR, Rios Coronado PE, et al. Blood coagulation protein fibrinogen promotes autoimmunity and demyelination via chemokine release and antigen presentation. Nat Commun. 2015;6:8164. doi: 10.1038/ncomms9164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schachtrup C, Lu P, Jones LL, Lee JK, Lu J, Sachs BD, Zheng B, Akassoglou K. Fibrinogen inhibits neurite outgrowth via beta3 integrin-mediated phosphorylation of the EGF receptor. Proc Natl Acad Sci U S A. 2007;104:11814–11819. doi: 10.1073/pnas.0704045104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schachtrup C, Ryu JK, Helmrick MJ, Vagena E, Galanakis DK, Degen JL, Margolis RU, Akassoglou K. Fibrinogen triggers astrocyte scar formation by promoting the availability of active TGF-beta after vascular damage. J Neurosci. 2010;30:5843–5854. doi: 10.1523/JNEUROSCI.0137-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen U, Tyagi N, Patibandla PK, Dean WL, Tyagi SC, Roberts AM, Lominadze D. Fibrinogen-induced endothelin-1 production from endothelial cells. Am J Physiol Cell Physiol. 2009;296:C840–847. doi: 10.1152/ajpcell.00515.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slaymaker IM, Gao L, Zetsche B, Scott DA, Yan WX, Zhang F. Rationally engineered Cas9 nucleases with improved specificity. Science. 2016;351:84–88. doi: 10.1126/science.aad5227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai HH, Niu J, Munji R, Davalos D, Chang J, Zhang H, Tien AC, Kuo CJ, Chan JR, Daneman R, et al. Oligodendrocyte precursors migrate along vasculature in the developing nervous system. Science. 2016;351:379–384. doi: 10.1126/science.aad3839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vos CM, Geurts JJ, Montagne L, van Haastert ES, Bo L, van der Valk P, Barkhof F, de Vries HE. Blood-brain barrier alterations in both focal and diffuse abnormalities on postmortem MRI in multiple sclerosis. Neurobiol Dis. 2005;20:953–960. doi: 10.1016/j.nbd.2005.06.012. [DOI] [PubMed] [Google Scholar]
- Wu M, Hernandez M, Shen S, Sabo JK, Kelkar D, Wang J, O'Leary R, Phillips GR, Cate HS, Casaccia P. Differential modulation of the oligodendrocyte transcriptome by sonic hedgehog and bone morphogenetic protein 4 via opposing effects on histone acetylation. J Neurosci. 2012;32:6651–6664. doi: 10.1523/JNEUROSCI.4876-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yates RL, Esiri MM, Palace J, Jacobs B, Perera R, DeLuca GC. Fibrin(ogen) and neurodegeneration in the progressive multiple sclerosis cortex. Ann Neurol. 2017 doi: 10.1002/ana.24997. [DOI] [PubMed] [Google Scholar]
- Zawadzka M, Rivers LE, Fancy SP, Zhao C, Tripathi R, Jamen F, Young K, Goncharevich A, Pohl H, Rizzi M, et al. CNS-resident glial progenitor/stem cells produce Schwann cells as well as oligodendrocytes during repair of CNS demyelination. Cell Stem Cell. 2010;6:578–590. doi: 10.1016/j.stem.2010.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu X, Hill RA, Dietrich D, Komitova M, Suzuki R, Nishiyama A. Age-dependent fate and lineage restriction of single NG2 cells. Development. 2011;138:745–753. doi: 10.1242/dev.047951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zlokovic BV. The blood-brain barrier in health and chronic neurodegenerative disorders. Neuron. 2008;57:178–201. doi: 10.1016/j.neuron.2008.01.003. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.