

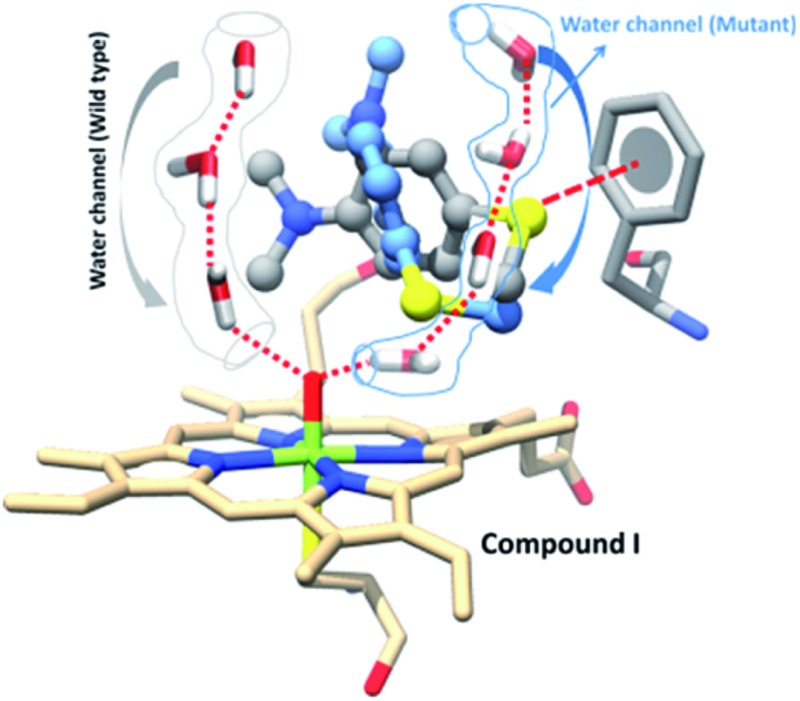
 A new water channel appears in the T268A mutant of P450BM3 and plays a role in the enzyme’s chemoselectivity.
A new water channel appears in the T268A mutant of P450BM3 and plays a role in the enzyme’s chemoselectivity.
Abstract
It is a long-standing mechanistic consensus that the mutation of the proton-shuttle mediator Threonine (T) in Cytochrome P450 enzymes severs the water channel and thereby quenches the formation of the active species: the high-valent iron(iv)-oxo porphyrin π-cation radical species, compound I (Cpd I). Using MD simulations and hybrid QM/MM calculations of P450BM3 we demonstrate that this is not the case. Thus, while the original water channel is disrupted in the T268A mutant of the enzyme, a new channel is formed that generates Cpd I. With this new understanding, we address the puzzling regiochemical and kinetic-isotope effect (KIE) results (Volz et al., J. Am. Chem. Soc., 2002, 124, 9724–9725) on the sulfoxidation and N-dealkylation of dimethyl-(4-methylsulfanyl-phenyl)-amine by wild type (WT) P450BM3 and its T268A vs. F87A mutants. We show that the observed variable ratio of S/Me oxidation for these enzymes, vis-à-vis the constant KIE, originates from Cpd I being the sole oxidant. Thus, while the conserved KIE probes the conserved nature of the transition state, the variable regiochemical S/Me ratio reflects the active-site reorganization in the mutants: the shifted location of the new water channel in T268A tightens the binding of the S-end by Cpd I and increases the S/Me ratio, whereas the absence of π-interaction with the S-end in F87A creates a looser binding that lowers the S/Me ratio. Our results match the experimental findings. As such, this study sheds light on puzzling experimental results, and may shift a central paradigm in P450 research. The broader implication on enzymatic research is that a single-site mutation is not a localised alteration but one that may lead to a profound change in the active site, sufficiently so as to change the chemoselectivity of catalyzed reactions.
1. Introduction
The mechanistic elucidation of reactions catalyzed by enzymes often raises enigmas that are inspired by considering simple chemical mechanistic schemes, when in fact these reactions proceed in highly complex and plastic active sites, with multiple residues, water channels, ions, etc. Variable chemoselectivity (e.g. regioselectivity and enantioselectivity) due to single site mutations in the protein is one such outcome, which often implies that the reactive species of the enzyme has changed its chemical identity/character or reaction mechanism in response to the mutation. In essence, however, what really changes is the active site, and hence it is essential to also consider the alternative possibility that the single-site mutation induces changes in the complexity of the active site and thereby imparts the observed changes in chemoselectivity. This paper addresses such a general conundrum by focusing on the enzyme P450BM3, which is a member of the superfamily of cytochrome P450 enzymes.1–9
It is generally thought that P450 enzymes use multiple oxidant species1,2,6,7,10 because single site mutations, which sever the proton shuttle pathways required to generate the principal oxidant species of the enzyme (2 in Scheme 1), cause a change in the regioselectivity of oxidation. One of these long-standing puzzles is the variable regioselectivity of sulfoxidation vs. N-dealkylation observed during the oxidation of dimethyl-(4-methylsulfanyl-phenyl) amine (Sub1 in Scheme 2), by wild-type (WT) P450BM3 and its T268A and F87A single-site mutants (MTs).6 Using molecular dynamics (MD) with sufficiently long timescales and hybrid QM/MM calculations, we address here the WT and the two MTs, as well as the variable oxidative regioselectivity and the constant KIE. As the story unfolds, our study will demonstrate that the selectivity alterations reflect major reorganization in the active sites, e.g., the formation of a new water channel,11 that affect the substrate juxtaposition vis-à-vis the active species of the enzyme. Single-site mutations are not merely localised alterations.
Scheme 1. A truncated catalytic cycle of P450.

Scheme 2. A summary of the regioselectivity and kinetic isotope effect (KIE) data concerning the substrates DMMSPA (Sub1) and p-DNNDMA (Sub2) by wild-type (WT) P450BM3 and its T268A and F87A mutants (MTs).

2. The mechanistic dilemma and chemical puzzles in P450BM3 and some of its mutants
Let us first present the mechanistic dilemma, and then proceed with the experimental background for invoking the two-oxidant scenario by Volz et al.6 A truncated, generic catalytic cycle of P450 is shown in Scheme 1, starting from the PorFeIII–OOH– species called compound 0 (Cpd 0), 1. Protonation of 1 on the distal oxygen (Od) leads to the liberation of a water molecule and the formation of the ultimate oxidant, 2, the porphyrin π-cation radical ferryl-species (Por˙+FeIV O), so-called Cpd I. Cpd I leads to the oxidation of the substrate (Sub), and is the consensus oxidant, responsible for the vast majority of the P450 catalyzed monooxygenation reactions.1–15 This process is referred7 to as “Coupling-I” because it couples the O2 activation to the substrate oxidation. This is also the native process of P450s.
However, a single mutation of the Thr residue to Ala (T → A) is thought to disrupt the proton-shuttle machinery of the enzyme and increase the amount/lifetime of Cpd 0 (ref. 7, 10, 11 and 16–19) that now undergoes protonation on the proximal oxygen (Op) of 1, leading to the formation of the ferric hydrogen peroxide complex, PorFeIII(O2H2) 3. Since the Fe–O bond in 3 is rather weak, much depends on the persistence of 3. Usually, 3 liberates hydrogen peroxide in a process called “uncoupling” which is an aborted oxidation. This leaves Cpd 0 (1) as a possible oxidant in the T → A mutants of P450. Therefore, the consideration of Cpd I and Cpd 0 as the potential oxidants in P450s and their T → A mutants (MTs) has become a mechanistic consensus in the field.
Let us turn now to Scheme 2 and inspect the intriguing mechanistic results of Volz et al.,6 which imply the presence of Cpd 0 alongside Cpd I. Thus, the WT enzyme sulfoxidates the sulfur site of Sub1 15 fold more than dealkylating the NMe2 site via the initial C–H hydroxylation. The MT T268A – thought to quench Cpd I formation and use Cpd 0 as an oxidant by the existing mechanistic paradigm – was found6 to increase the S/Me ratio to 60, whereas the MT F87A that is not thought to affect the proton-shuttle machinery actually decreases the S/Me ratio to below 15.
The mutations and changes in the S/Me reactivity-ratio do not affect, however, the intrinsic kinetic isotope effect KIEH/D (∼2), which is determined for the appropriately deuterated Sub1. Furthermore, to rule out the slow interchange of the two sites of Sub1, Volz et al. employed an analogous substrate Sub2, and observed that both the NMe2 and NMe2[D-6] (D-6 indicates the number of substituted deuterium atoms at the two methyl groups) sites of the latter undergo oxidation with a KIEH/D of ∼2, for both the WT and MT T268A enzymes. As such, all of these findings, taken together, were cautiously interpreted6 in a scenario whereby Cpd I and Cpd 0 co-exist in both the WT and the MT T268A enzymes, where Cpd I performs N-dealkylation whereas Cpd 0 performs sulfoxidation. As such, the S/Me ratio has to reflect the relative abundance of the two oxidants.
Similarly, Cryle and de Voss20a investigated the oxidation of fatty acids and the ω-2-thia-substituted ones by P450BM3 WT and MT T268A enzymes, and showed that C–H hydroxylation suffered high uncoupling in the MT T268A enzyme, whereas sulfoxidation was coupled in both the MT and WT enzymes. Nevertheless, both reactions conserved similar regio- and enantio-selectivities in the MT and WT enzymes. Thus, once again, this evidence seems to compellingly suggest that in both the WT and MT enzymes the two oxidants function in tandem; Cpd I is responsible for C–H hydroxylation, while Cpd 0 carries out the sulfoxidation.
We note that besides the two-oxidant scenario, Volz et al.6 considered that the same result could have been caused by changes in the structure of the active site due to mutations. Moreover, Cryle and de Voss20b showed more recently that the observed uncoupling – due to the presumed “loss” of Cpd I’s activity in favor of Cpd 0’s – was highly substrate dependent, while the information obtained from the radical clocks during C–H hydroxylation supported Cpd I as a single oxidant.21 Additionally, all known computational results for heme systems show that Cpd 0 is a very poor oxidant compared to Cpd I22,23 and it has extremely high sulfoxidation barriers.23 As such, Cpd 0 cannot really be responsible for the sulfoxidation of Sub1 in the presence of Cpd I. In fact, we7 and Altarsha et al.24 showed that in the MT T252A of P450cam, Cpd 0 is protonated very easily to form PorFeIII(O2H2), 3, and will not survive to carry out substrate oxidation.
In view of all of the intriguing results, Cpd I may well be the sole oxidant responsible for oxidation in the WT enzyme via the native coupling-I process. In the MT T268A of P450BM3, the formation of Cpd I may either occur via the coupling-I process or the recently found7 coupling-II pathway. However, the latter process requires a sufficiently large and tightly bound substrate that can barricade the H2O2 moiety.7 Otherwise, PorFeIII(O2H2) will lose H2O2, thus resulting in uncoupling (Scheme 1). As mentioned already, our results show that, in P450BM3, PorFeIII(O2H2) is unstable with respect to a loss of H2O2 (see ESI, Fig. S1†). We are therefore left with Cpd I as the only possible oxidant. But how does Cpd I actually form and how does it concoct the intriguing scenario described by Volz et al.?6 The present study answers these questions by means of extensive MD simulation sets of 100–200 ns of the species, combined with QM/MM calculations.25 First, we address the mechanisms of Cpd I formation in the T268A mutant of P450BM3, and then we explore the mechanism of Sub1 oxidation by P450BM3 and its mutants. Our findings will show that changes are required in some key mechanistic paradigms in P450 mechanistic research.
3. Computational methods and details
3.1. System preparation and setup
The substrate binding site in P450BM3 is well known, and therefore we started with a palmitic acid-bound P450BM3 (PDB no. ; 1FAG)8 and replaced the palmitic acid with Sub1 to use an experimentally verified binding site. Since the crystal structure of P450BM3 with Sub1 is not available, we used two different initial orientations of Sub1 to confirm the proper binding of the substrate; one where the N–(CH3)2 group is pointing towards the Fe O end and the other where the S–CH3 group points towards Fe O in the WT and T268A mutant.
Missing hydrogen atoms were added by the LEaP module of Amber 14.26 The study considered three of the P450BM3 species of interest (Cpd 0, Cpd I and Fe(H2O2)). The force field for Cpd I was taken from the literature,27 while the force fields for the Cpd 0 and Fe(H2O2) states were parameterized using “MCPB.py” modeling.28 The partial atomic charges and missing parameters for Sub1 were obtained by the RESP (restrained electrostatic potential) method,29,30 using HF/6-31G* calculations. A few Na+ ions were added to the protein surface to neutralize the total charge of the systems. Finally, the resulting system was solvated in a rectangular box of TIP3P31 water extending up to a minimum cutoff of 10 Å from the protein boundary. The Amber ff14SB force field was employed for the protein in all of the MD simulations.
3.2. MD simulations
After proper parameterizations and setup, the resulting system’s geometries were minimized (5000 steps for the steepest descent and 10 000 steps for the conjugate gradient) to remove the poor contacts and relax the system. The systems were then gently annealed from 10 to 300 K under a canonical ensemble for 50 ps with a weak restraint of 5 kcal mol–1 Å–2. Subsequently, the systems were maintained for 1 ns of density equilibration under an isothermal–isobaric ensemble at a target temperature of 300 K and a target pressure of 1.0 atm using a Langevin-thermostat32 and a Berendsen barostat33 with a collision frequency of 2 ps and a pressure-relaxation time of 1 ps with a weak restraint of 1 kcal mol–1 Å–2. This 1 ns of density equilibration is not identical with conformational equilibration, but is rather a weakly restrained MD in which one slowly relaxes the system to achieve a uniform density after heating dynamically under periodic boundary conditions. Thereafter, we removed all of the restraints applied during the heating and density dynamics and further equilibrated the systems for ∼3 ns to get well settled pressure and temperature for conformational and chemical analyses.
Finally, a productive MD run was conducted for each system for at least 100 ns. In general, 100 ns of MD sampling is sufficient for ligand binding and substrate recognition. In some cases, in order to ascertain the convergence of the MD, we proceeded with an additional 100 ns simulation, which did not change the substrate orientation that was obtained in the initial 100 ns. As such, we consider that this convergence test is sufficient for sampling the binding of Sub1 in the present study. The most populated structures were calculated by the clustering of MD trajectories by the CPPTRAJ module of Amber 14. During all of the MD simulations, the covalent bonds containing hydrogen were constrained using SHAKE,34 and particle mesh Ewald (PME)35 was used to treat the long-range electrostatic interactions. We used an integration step of 2 fs during the entire simulation. All of the MD simulations were performed with the GPU version of the Amber 14 package.36
3.3. QM/MM methodology
For the subsequent QM/MM calculations,37 we used representative snapshots extracted from the most populated structures within the clustering of the MD trajectories. This choice is statistically more rigorous than stochastically chosen snapshots. All of the water molecules beyond the sphere of the enzyme were removed, and the resulting enzyme was solvated with a 16 Å layer of TIP3P water molecules (yielding a total of ca. 45 600 atoms).
The propionate-truncated heme and the entire Sub1 were included in the QM region, whereas all of the protein residues and water within 8 Å of the heme and Sub1 were included in the active region. The atoms in the active region interact with the QM atoms through electrostatic and van der Waals interactions and were considered in the subsequent QM/MM calculations. As such, the QM/MM results also account for the interactions of the QM system with key residues e.g. Phe 87, Thr 268 and other water residues within 8 Å of the heme and Sub1. All of the QM/MM calculations were performed using ChemShell,38 by combining Turbomole39 for the QM part and DL_POLY40 for the MM part. The CHARMM27 force field was employed for the MM region. The electronic embedding scheme41 was used to account for the polarizing effect of the enzyme environment on the QM region. Hydrogen link atoms with the charge-shift model35,37a were applied to treat the QM/MM boundary.
During the QM/MM geometry optimizations, the QM region was treated by the hybrid UB3LYP42 functional with two basis sets. For the geometry optimization and frequency calculation, we used the polarized double-ζ basis set LACVP for iron and the 6-31G(d) basis set for all of the other atoms, collectively labeled as B1. The energies are further corrected with the large all-electron basis-set Def2-TZVP, labeled as B2. All of the QM/MM transition states (TSs) were located by relaxed potential energy surface (PES) scans followed by full TS optimizations using the P-RFO optimizer implemented in HDLC code43 and Dl-find.44 The QM/MM calculations used the hybrid UB3LYP functional. For Cpd I mediated reactions, the S = 3/2 and S = 1/2 states exhibit generally similar reactivities.7,45 As such, all of the reactions of Cpd I were studied in the S = 1/2 state.
4. Results and discussion
Let us start with the mechanisms of Cpd I formation and then proceed to the mechanisms of the oxidation of Sub 1 in the WT enzyme, and in the T268 and F87A mutants.
4.1. Cpd I formation in the T268A mutant of P450BM3
The anticipation of a second oxidant is based on the conjecture that the T268A mutant quenches Cpd I formation or decreases its production. In fact, we show here that T268A generates Cpd I efficiently via the appearance of a very intriguing new water channel due to the mutation. We first ran an MD simulation of 100 ns on Cpd 0 (see ESI, page S2, Fig. S1 and S2†), from which we took two snapshots from the two conformational basins we identified, and performed QM/MM calculations. Fig. 1a shows the results for the snapshot from the major conformer. It is seen that residue Glu267 is nicely connected to the Od of Cpd 0 through a few water molecules. The proton transfer from Glu267 to Od generates Cpd I in a single step, with a rather small barrier of 11.3 kcal mol–1. In the minor conformer (Fig. 1b), the channel leads to the protonation of the distal O of Cpd 0 and forms Fe(H2O2). To rule out the possibility that the coupling-II7 pathway operates in P450BM3, we ran 100 ns MD simulations on the as-formed Fe(H2O2). The simulations indicate that Sub1 is quite flexible in the pocket and is far away from Fe(H2O2) (see ESI, Fig. S3†). This in turn suggests that Sub1 is unable to barricade the H2O2 ligand so that Fe(H2O2) would lose H2O2 resulting in uncoupling (Scheme 1). Such a scenario seems to be consistent with experiments that show that the T268A mutant of BM3 always leads to a partial uncoupling in the presence of fatty acid substrates.11,20
Fig. 1. QM/MM (UB3LYP/B2//B1) energy profiles (in kcal mol–1) for Cpd I formation from Cpd 0 in the T268A mutant: (a) the coupling-I pathway for the major conformer leads to Cpd I formation and (b) protonation of Cpd 0 in the minor conformer leads to Fe(H2O2) formation. The energy barriers include ZPE corrections. Sub 1 is included in the MM part, and MD simulations show that it is located too far from Fe(H2O2) to barricade H2O2 against detachment (ESI, Fig. S3†). Thus, Cpd I formation from Fe(H2O2) (coupling-II7) can be ruled out in the T268A mutant of BM3.

4.2. MD simulation results as the preparation for Sub1 oxidation
To test the regiochemical preference of the binding sites of the S or N of Sub1 by Cpd I, we carried out MD simulations of the Cpd I/Sub1 complex within the active site. We used two different initial orientations of Sub1 called pose-1 and pose-2; pose-1 refers to the conformation where the N–(CH3)2 group points towards the Fe O end and pose-2 refers to the one where the S–CH3 group points towards Fe O. Starting the MD simulations from pose-1, Sub1 ended up moving away and remained far from the Fe O moiety during the entire simulation time. This was judged by the distance of the closest N–CH3 terminal of Sub1 to Fe O, which was >10 Å during the entire simulations (see Fig. S4 and Video VS1 in ESI†). To double-check this conclusion, we used molecular mechanical Poisson Boltzmann surface area (MMPBSA) calculations, and calculated the enthalpy changes (ΔH = ΔHcomplex – [ΔHprotein + ΔHSub1]) upon the binding of Sub1 for these two poses. The MMPBSA calculations also showed that the binding of Sub1 in pose-1 is significantly weaker than that in pose-2 (see Table S2† for details). Thus, both the MD simulations and MMPBSA calculations show that pose-1 does not represent an appropriate model for the experimental observations. As such, this orientation was dropped, and the MD simulations were done from pose-2 of Sub1, and are presented in Fig. 2.
Fig. 2. (a) The variation of the Fe O···H and Fe O···S distances vs. simulation time for the WT enzyme. Initially, the amine and sulfur ends of Sub1 are far away from iron-oxo, however, in the later stages both ends become accessible to Cpd I. However, the major basin of the snapshots places the sulfur end closest to Cpd I. (b) The structures in the initial snapshot and the one during the 80–100 ns of the MD trajectory are shown along with the noncovalent interactions of Sub1 with Phe87 and Cpd I. The green surfaces show the favorable noncovalent interactions and the area of the green patches indicates the interactions’ intensity. Note the increase in the intensity between the sulfur atom of Sub1 and Phe87 (highlighted by arrows) from the initial trajectory to the one in the 80–100 ns interval.

Fig. 2a shows the fluctuations of the oxidizable moieties of Sub1 (H in N–CH3and S in S–CH3) relative to Cpd I’s oxo atom. It is seen that the substrate is initially far away from Cpd I (>10 Å), but within 5 ns, Sub1 slides closer to Cpd I and achieves proper juxtapositions vis-à-vis the Fe O moiety. Looking at the distance vs. time plot in Fig. 2a, the 90–100 ns interval of the MD simulations shows that the NMe2 group, which was up to this point far away, now approached the Fe O moiety. This may raise the possibility that the NMe2 group may permanently settle close to the Fe O moiety, even at later times in the simulations. To test this possibility, we extended the MD simulations to 200 ns to check the persistence of the proximity of NMe2 to the Fe O end. The extended MD simulation in Fig. 2a shows that it is the sulfur terminal that remains closest to Cpd I for most of the 200 ns, while the NMe2 group is quite far away for most of the simulation time, with the exception of the 90–100 ns interval. To further characterize the substrate’s orientational distribution, we performed the clustering of the MD trajectories for the entire 200 ns of simulation into the related conformational basins (see Fig. S5,† page S5, for three typical snapshots during clustering). This enabled us to verify that in all of the clusters of the MD trajectory, the sulfur end is indeed closer to Cpd I and this is the most populated conformation, in agreement with Fig. 2a.
The reason for the closer binding of the S site of Sub1 to Cpd I was found to be the interaction of S with the Phe87 residue. The noncovalent interaction46 of Phe87 with Sub1 is shown in Fig. 2b for the initial snapshot and one taken from the 80–100 ns interval; the green patch signifies the S/π interaction. Judging from the intensity and area of these green patches, it is clear from the two snapshots that the phenyl ring of Phe87 intensifies its pull of the S–CH3 terminal of Sub1, which gets closer to Phe87 via this interaction. As such, the sulfur terminal is pulled away from Fe O while NMe2 slides momentarily closer to the Fe O end. This rearrangement creates a minor conformational basin in which NMe2 assumes an oxidizable distance to Fe O. After 100 ns or so, S restores its proximity to Fe O, such that the major conformational basin maintains a closer sulfur/Cpd I distance. As such, if the juxtaposition determines the fate of the complex, our MD results predict that sulfoxidation would lead to the major product while C–H activation (of the N–CH3 moiety) would lead to the minor one, unless the relative barriers predict otherwise. Furthermore, the orientation towards sulfur is preferentially S-prochiral, so provided the MD determines enantioselection the reaction should result in the S-enantioselective sulfoxide.
4.3. QM/MM results of the WT enzyme
To test these MD-based predictions, we performed QM/MM calculations on representative structures of two differently populated conformational basins; one corresponds to the major basin where the sulfur is closest to Cpd I, the other to a minority basin where the C–H bond is the closer moiety to Cpd I (Fig. S5a and c†). Fig. 3 displays the two reactant-complexes, 2RC(major) and 2RC(minor), corresponding to these basins. Analysis of the MD trajectories shows that the frequencies of visits in the two basins are 1866 (major) vs. 11 (minor). These relative-visit-frequencies plugged into the Boltzmann equation yield the relative energy of the RCs, which is 2.8 kcal mol–1 in favor of the major conformation at the experimental temperature of 303 K. The energy profiles for sulfoxidation and C–H activation, nascent from these RCs, are shown in Fig. 3, along with the structures of the lowest lying transition states for hydroxylation and sulfoxidation, where the energies are given relative to 2RC(major).
Fig. 3. The QM/MM (UB3LYP/B2//B1) energy profiles (in kcal mol–1) for WT P450BM3 (H-abstraction is to the left and sulfoxidation is to the right) nascent from the major and minor reactant-clusters (2RC(major) and 2RC(minor)) and going to the corresponding 2TSH and 2TSS. The energy barriers include ZPE corrections. The spin densities (ρ) on the substrate indicate that the oxidant in 2RC(major) corresponds to Cpd I while in 2RC(minor) it corresponds to Cpd II (see also Table S1†, page S5). Only the lowest TS structures are shown (the higher ones can be found in the ESI†), along with key distances (in Å).

Relative to 2RC(major) as the reference energy, the lowest QM/MM barriers (with ZPE corrections) are 10.1 kcal mol–1 for H-abstraction through the minor channel and 9.4 kcal mol–1 for sulfoxidation via the major channel. Using the barrier energy difference, 0.7 kcal mol–1, the predicted S/Me ratio at 303 K is 4 : 1, compared with 15 : 1 as reported by Volz et al.6 (corresponding to a 1.5 kcal mol–1 energy difference). Using another snapshot (see ESI, Fig. S6†), the corresponding barriers lead to the same conclusion that sulfoxidation is preferred over H-abstraction. This result is in qualitative agreement with experimental observations that sulfoxidation would lead to the major and C–H hydroxylation to the minor product in the WT enzyme.6
Looking at the other two barriers in Fig. 3, we can see that the reactions from the poorly juxtaposed RCs have high barriers. Thus, the sulfoxidation barrier in the minor basin is 14.6 kcal mol–1, whereas the H-abstraction barrier in the major basin, which highly disfavors N–CH3-juxtaposition, is huge at 26.5 kcal mol–1. These are much higher barriers than the ones discussed above and as such these pathways will clearly not contribute to the reaction.
To further articulate the reasons for these high barriers, we investigated the charge and spin densities of the species in the minor and major basins (see Fig. 3 and Table S1†). As seen from the spin density (ρ) on the substrate in 2RC(minor), there is a transfer of one electron from the substrate to the porphyrin ring of Cpd I, so that 2RC(minor) involves a single electron reduced Cpd I (so-called Cpd II) with Sub1+˙ that diminishes the reactivity in the minor basin, relative to 2RC(major) which involves a cluster of Cpd I with Sub1. Indeed, due to the decreased reactivity of Cpd II, the electronic contribution (EQM) to the energy barrier for the H-abstraction for 2RC(minor) (16.9 kcal mol–1) is higher than the electronic contribution to the energy barrier for 2RC(major) (11.2 kcal mol–1).
4.4. MD and QM/MM results of the T268A MT enzyme
The enhanced S/Me regioselectivity of the T268A MT enzyme6 with Sub1 raises an obvious question: how can a single mutation change the regio-selectivity of substrate oxidation? As written above, past explanations of this change6,20a assumed that Cpd 0 is present alongside Cpd I, and the fraction or lifetime of Cpd 0 increases in the T268A MT. Furthermore, it was assumed that Cpd 0 is the species responsible for sulfoxidation, whereas Cpd I is responsible for hydroxylation. However, since all computations show that Cpd 0 has high substrate-oxidation barriers,22,23 the assumption that Cpd 0 is responsible for the S/Me-regioselectivity enhancement is deemed questionable. Therefore, we performed MD simulations for the T268A MT to study the substrate reorientation caused by the mutation. As expected, this MD simulation shows completely different behavior to the WT simulation. Thus, in contrast to the WT enzyme, which showed the dynamic behavior of Sub1 (Fig. 2), in the MT Sub 1 adopts a stable orientation. And as seen in Fig. 4, now the sulfur end is always close to the oxo end of Cpd I, while the NMe2 end seldom approaches Cpd I. This is also well supported by the clustering of the trajectories, wherein no cluster represents an NMe2 end being closer to Cpd I than the sulfur end (see Fig. S7†). This clearly shows that the T268A mutation brings the sulfur end closer to Cpd I and pushes the NMe2 end away, hence causing enhanced sulfoxidation over H-abstraction.
Fig. 4. Comparison of Fe O···S and Fe O···H [the H in N(CH3)2] distances during the MD simulations of the T268A mutant.

Since, by analogy to T252 in P450cam,17 the primary role of T268 in P450BM3 is thought to be proton delivery via an organized water channel, we carefully monitored the water channel and permeability for the entire MD trajectories of the WT and T268A MT enzymes. Identical numbers of permeated water molecules were found for the WT and T268A MT enzymes (See ESI for details,† page S7). However, what completely changed for the T268A MT is the location of the water channel. As shown in Fig. 5a, throughout the MD simulations of the WT enzyme the Thr268 residue mediates a very organized water chain (see ESI videos VS2 for the WT and VS3 for the T268A mutant†). The T268A mutation disrupts this channel and instead provides a new aqueduct where the substrate acts as a “wall” to direct the water flow. As a result, the substrate is reoriented and the S terminal gets closer to Cpd I. As shown in Fig. 5b (left item), the water molecules of the new channel, jointly with channel residues, barricade the substrate and fix the S-end close to Cpd I, thus enhancing sulfoxidation. Indeed, as seen from the energy profiles in Fig. 5b (right item), the sulfoxidation barrier is lowered (compared to Fig. 2 for the WT), while the H-abstraction barrier is raised, which is in qualitative agreement with experimental data.6 The unique role of the substrate is in line with the observations of Cryle and de Voss.20b Furthermore, here we can see how a major mechanistic paradigm for P450 must be revised in view of these findings. Thus, the T268A mutation in P450BM3 destroys the “old” proton shuttle pathway and recreates a new one, which leads to the formation of Cpd I (Fig. 1a). Meanwhile, because the new water channel is located to the far right of the WT’s channel (Fig. 5a, right-hand drawing), the T268A mutation changes the reorientation of the substrate and thus impacts the regioselectivity of the oxidation of Sub1 by tightly juxtaposing the S end towards Cpd I.
Fig. 5. (a) The water permeation routes were calculated using Pore Walker and are shown near Cpd I in the WT and the T268A MT enzymes. On the right side the two enzymes (the WT is in yellow and the MT is in grey) are overlapped and their water routes are drawn by color-coded arrows; a bluish arrow for the WT and a reddish arrow for the MT. Note that the substrate is acting as a guiding wall for the water stream in T268A. (b) The left item shows water molecules jointly with channel residues barricading the substrate and fixing its S-end near Cpd I. The right hand drawing shows the QM/MM energies and TS structures for H-abstraction (2TSH-MT) and sulfoxidation (2TSS-MT).

4.5. MD results for the F87A MT enzyme
As discussed above, the F87A mutation decreases the amount of the sulfoxidation product.6 Therefore, we also performed MD simulations for the F87A MT. As expected, Fig. 6 shows that now the S-terminal in the F87A MT (red color) is farther away from the oxo ligand of Cpd I, in comparison to that in the T268A MT (and also in the WT, see Fig. 2a). As already discussed, Phe87 forms a good S/π interaction that is crucial for the substrate orientation (Fig. 2b). Therefore, mutating Phe87 to Ala eliminates this interaction and causes a different orientation of the S-terminal in the substrate, which might be responsible for the decreased sulfoxidation as seen in experiments.
Fig. 6. The Fe O···S distance vs. the MD simulation time for T268A (black) and F87A (red) mutants. Note that the Fe O···S distance is longer in the F87A MT enzyme.

A closer inspection of the MD trajectories for the F87A mutant (see ESI video VS4†) demonstrates that the routes for the water channel are similar to those for the WT enzyme, but different compared to those found for the T268A MT. Clearly, the T268A MT has a profound impact on the regioselectivity and reactivity of P450BM3. The T268A MT completely alters the water channel and its location in the active site. With the new channel, there is also a new mechanism for Cpd I formation (Fig. 1). Additionally, the shifted location of the water channel alters the binding conformation of the substrate (Fig. 5a and b), and thereby increases the S/Me regioselectivity ratio. On the other hand, F87A conserves the water pathways as in the WT, but it lacks the π/S glue to bring the S terminal as close to Cpd I, thus lowering the S/Me regioselectivity. Our findings strongly support the notion that what causes the changes in regioselectivity are the modifications of the active site in response to the specific mutations. Meanwhile, Cpd I is conserved in the three enzymes, and its S/Me regioselectivity is determined by the modifications in the substrate binding exerted by the T268A and F87A mutations. We further verified that the KIE values in WT and MT T268A are both close to ∼2 (see ESI,† Page S8) and reflect the constant nature of the H-abstraction TS of the Cpd I species.
5. Conclusions
In conclusion, all of the above findings are consistent with a scenario whereby sulfoxidation and hydroxylation are both mediated by the active species Cpd I, in the WT as well as the MT enzymes. The T → A mutation of P450BM3 generates a newly located water channel, guided by Glu267, thus highlighting the flexibility of the enzyme active sites in enzymatic catalysis.9,47 This new water channel forms Cpd I very efficiently (Fig. 1a) but this is in competition with uncoupling to H2O2 (Fig. 1b) which is consistent with experimental observations.11,20b The variable S/Me ratio originates from variations of the active site in response to the mutations. Thus, since Phe87 confers a π/S interaction that brings the S terminal of the substrate close to Cpd I in the WT enzyme, the lack of Phe87 due to the F87A mutation is a reason behind the decreased yield of sulfoxidation. In the T268A MT, the S/Cpd I proximity increases due to the relocated water channel, and the role of the substrate as a wall for the aqueduct. In contrast, the constant KIE (see ESI,† Page S8) is in line with a “conserved” transition state due to the presence of Cpd I as the sole oxidant in both the WT and T268A.
The very efficient formation of Cpd I in the T268 mutant, along with the mutation-induced active site reorganization that causes an altered water route and an altered orientation of substrate, provides a new paradigm for understanding the regio- and stereo-selectivity issues in Cpd I-mediated reactions. Finally, the study leads to an additional modification of a tacitly assumed mechanistic paradigm that a single site mutation is a localised alteration that helps the mechanistic chemist to decipher the chemical mechanism. Thus, much like the T268A mutation, other single-site mutations are not localised alterations but ones that lead to a major change in the complexity of the active site, so as to change the chemoselectivity of reactions.
Supplementary Material
Acknowledgments
S. S. acknowledges support by the Israel Science Foundation (ISF 1183/13 grant). B. W. and K. D. D. acknowledge the Planning and Budgeting Committee (PBC) of the Council for Higher Education in Israel for the Post Doctoral Fellowships.
Footnotes
†Electronic supplementary information (ESI) available: The RMS deviation of the protein during 100 ns (and 200 ns) of simulation and the total QM/MM and ZPE energies, as well as the Cartesian coordinates of all of the computed species. See DOI: 10.1039/c7sc01932g
References
- Montellano P. O. d., Cytochrome P450: Strcture, mechanism and biochemistry, Plenum Press, New York and London, 2nd edn, 1995. [Google Scholar]
- Sono M., Roach M. P., Coulter E. D., Dawson J. H. Chem. Rev. 1996;96:2841–2881. doi: 10.1021/cr9500500. [DOI] [PubMed] [Google Scholar]
- Ortiz de Montellano P. R. Chem. Rev. 2010;110:932–948. doi: 10.1021/cr9002193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groves J. T. Nat. Chem. 2014;6:89–91. doi: 10.1038/nchem.1855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Eldik R. Chem. Rev. 2005;105:1917–1922. doi: 10.1021/cr050571f. [DOI] [PubMed] [Google Scholar]
- Volz T. J., Rock D. A., Jones J. P. J. Am. Chem. Soc. 2002;124:9724–9725. doi: 10.1021/ja026699l. [DOI] [PubMed] [Google Scholar]
- Wang B., Li C., Dubey K. D., Shaik S. J. Am. Chem. Soc. 2015;137:7379–7390. doi: 10.1021/jacs.5b02800. [DOI] [PubMed] [Google Scholar]
- Li H., Poulos T. L. Nat. Struct. Biol. 1997;4:140–146. doi: 10.1038/nsb0297-140. [DOI] [PubMed] [Google Scholar]
- For a recent intriguing enigma, regarding the consensus view in P450cam, see: Prasad B., Mah D. J., Lewis A. R., Plettner E., PLoS One, 2013, 8 , e61897 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- (a) Meunier B., Visser S. P., Shaik S. Chem. Rev. 2004;104:3947–3980. doi: 10.1021/cr020443g. [DOI] [PubMed] [Google Scholar]; (b) Vaz A. D. N., McGinnity D. F., Coon M. J. Proc. Natl. Acad. Sci. U. S. A. 1998;95:3555–3560. doi: 10.1073/pnas.95.7.3555. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Newcomb M., Toy P. H. Acc. Chem. Res. 2000;33:449–455. doi: 10.1021/ar960058b. [DOI] [PubMed] [Google Scholar]
- Yeom H., Sligar S. G., Li H., Poulos T. L., Fulco A. J. Biochemistry. 1995;34:14733–14740. doi: 10.1021/bi00045a014. [DOI] [PubMed] [Google Scholar]
- Hasemann C. A., Ravichandran K. G., Peterson J. A., Deisenhofer J. J. Mol. Biol. 1994;236:1169–1185. doi: 10.1016/0022-2836(94)90019-1. [DOI] [PubMed] [Google Scholar]
- Cupp-Vickery J., Poulos T. L. Nat. Struct. Biol. 1995;2:144–153. doi: 10.1038/nsb0295-144. [DOI] [PubMed] [Google Scholar]
- Shimizu H., Park S. Y., Gomi Y., Arakawa H., Nakamura H., Adachi S.-I., Obayashi E., Iizuka T., Shoun H., Shiro Y. J. Biol. Chem. 2000;275:4816–4826. doi: 10.1074/jbc.275.7.4816. [DOI] [PubMed] [Google Scholar]
- Yano J. K., Koo L. S., Schuller D. J., Li H., Ortiz de Montellano P. R., Poulos T. L. J. Biol. Chem. 2000;275:31086–31092. doi: 10.1074/jbc.M004281200. [DOI] [PubMed] [Google Scholar]
- Modi A. R., Dawson J. H. Adv. Exp. Med. Biol. 2015;851:63–75. doi: 10.1007/978-3-319-16009-2_2. [DOI] [PubMed] [Google Scholar]
- Jin S., Makris T. M., Bryson T. A., Sligar S. G., Dawson J. H. J. Am. Chem. Soc. 2003;125:3406–3407. doi: 10.1021/ja029272n. [DOI] [PubMed] [Google Scholar]
- Clark J. P., Miles C. S., Mowat C. G., Walkinshaw M. D., Reid G. A., Daff S. N., Chapman S. K. J. Inorg. Biochem. 2006;100:1075–1090. doi: 10.1016/j.jinorgbio.2005.11.020. [DOI] [PubMed] [Google Scholar]
- Davydov R., Perera R., Jin S., Yang T. C., Bryson T. A., Sono M., Dawson J. H., Hoffman B. M. J. Am. Chem. Soc. 2005;127:1403–1413. doi: 10.1021/ja045351i. [DOI] [PubMed] [Google Scholar]
- (a) Cryle M. J., De Voss J. J. Angew. Chem., Int. Ed. 2006;45:8221–8223. doi: 10.1002/anie.200603411. [DOI] [PubMed] [Google Scholar]; (b) Cryle M. J., De Voss J. J. ChemBioChem. 2008;9:261–266. doi: 10.1002/cbic.200700537. [DOI] [PubMed] [Google Scholar]
- (a) Kumar D., Visser S. P., Sharma P. K., Cohen S., Shaik S. J. Am. Chem. Soc. 2004;126:1907–1920. doi: 10.1021/ja039439s. [DOI] [PubMed] [Google Scholar]; (b) Kumar D., Visser S. P., Shaik S. J. Am. Chem. Soc. 2003;125:13024–13025. doi: 10.1021/ja036906x. [DOI] [PubMed] [Google Scholar]
- (a) Ogliaro F., de Visser S. P., Cohen S., Sharma P. K., Shaik S. J. Am. Chem. Soc. 2002;124:2806–2817. doi: 10.1021/ja0171963. [DOI] [PubMed] [Google Scholar]; (b) Kamachi T., Shiota Y., Ohta T., Yoshizawa K. Bull. Chem. Soc. Jpn. 2003;76:721–732. [Google Scholar]; (c) Sharma P. K., de Visser S. P., Shaik S. J. Am. Chem. Soc. 2003;125:8698–8699. doi: 10.1021/ja035135u. [DOI] [PubMed] [Google Scholar]
- (a) Li C., Zhang L., Zhang C., Hirao H., Wu W., Shaik S. Angew. Chem., Int. Ed. 2007;46:8168–8170. doi: 10.1002/anie.200702867. [DOI] [PubMed] [Google Scholar]; (b) de Visser S. P. Coord. Chem. Rev. 2009;253:754–768. [Google Scholar]
- Altarsha M., Benighaus T., Kumar D., Thiel W. J. Am. Chem. Soc. 2009;131:4755–4763. doi: 10.1021/ja808744k. [DOI] [PubMed] [Google Scholar]
- Senn H. M., Thiel W. Angew. Chem., Int. Ed. 2009;48:1198–1229. doi: 10.1002/anie.200802019. [DOI] [PubMed] [Google Scholar]
- Case D., Berryman J. T., Betz D. S. S. R. M., Cheatham T. E., Darden T. A., Duke R. E., Giese T. J., Gohlke H., Goetz A. W., Homeyer N., Izadi S., Janowski P., Kaus J., Kovalenko A., Lee T. S., LeGrand S., Li P., Luo R., Madej B., Merz K. M., Monard G., Needham P., Nguyen H., Nguyen H. T., Omelyan I., Onufriev A., Roe D. R., Roitberg A., Salomon-Ferrer R., Simmerling C. L., Smith W., Swails J., Walker R. C., Wang J., Wolf R. M., Wu X., York D. M. and Kollman P. A., Amber 14, University of California, San Francisco, 2015.
- Shahrokh K., Orendt A., Yost G. S., Cheatham T. E. J. Comput. Chem. 2012;33:119–133. doi: 10.1002/jcc.21922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li P., Merz Jr K. M. J. Chem. Inf. Model. 2016;56:599–604. doi: 10.1021/acs.jcim.5b00674. [DOI] [PubMed] [Google Scholar]
- Bayly C. I., Cieplak P., Cornell W., Kollman P. A. J. Phys. Chem. 1993;97:10269–10280. [Google Scholar]
- Cornell W. D., Cieplak P., Bayly C. I., Kollmann P. A. J. Am. Chem. Soc. 1993;115:9620–9631. [Google Scholar]
- Jorgensen W. L., Chandrasekhar J., Madura J. D., Impey R. W., Klein M. L. J. Chem. Phys. 1983;79:926–935. [Google Scholar]
- Izaguirre J. A., Catarello D. P., Wozniak J. M., Skeel R. D. J. Chem. Phys. 2001;114:2090–2098. [Google Scholar]
- Berendsen H. J. C., Postma J. P. M., van Gunsteren W. F., DiNola A., Haak J. R. J. Chem. Phys. 1984;81:3684–3690. [Google Scholar]
- Ryckaert J.-P., Ciccotti G., Berendsen H. J. C. J. Comput. Phys. 1977;23:327–341. [Google Scholar]
- Darden T., York D., Pedersen L. J. Chem. Phys. 1993;98:10089–10092. [Google Scholar]
- Salomon-Ferrer R., Götz A. W., Poole D., Le Grand S., Walker R. C. J. Chem. Theory Comput. 2013;9:3878–3888. doi: 10.1021/ct400314y. [DOI] [PubMed] [Google Scholar]
- (a) Senn H. M., Thiel W. Curr. Opin. Chem. Biol. 2007;11:182–187. doi: 10.1016/j.cbpa.2007.01.684. [DOI] [PubMed] [Google Scholar]; (b) Lin H., Truhlar D. G. Theor. Chem. Acc. 2007;117:185–199. [Google Scholar]; (c) Ryde U. Curr. Opin. Chem. Biol. 2003;7:136–142. doi: 10.1016/s1367-5931(02)00016-9. [DOI] [PubMed] [Google Scholar]; (d) Olah J., Mulholland A. J., Harvey J. N. Proc. Natl. Acad. Sci. U. S. A. 2011;108:6050–6055. doi: 10.1073/pnas.1010194108. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Chung L. W., Sameera W. M. C., Ramozzi R., Page A. J., Hatanaka M., Petrova G. P., Harris T. V., Li X., Ke Z., Li H., Ding L., Morokuma K. Chem. Rev. 2015;115:5678–5796. doi: 10.1021/cr5004419. [DOI] [PubMed] [Google Scholar]
- (a) Sherwood P., de Vries A. H., Guest M. F., Schreckenbach G., Catlow C. R. A., French S. A., Sokol A. A., Bromley S. T., Thiel W., Turner A. J., Billeter S., Terstegen F., Thiel S., Kendrick J., Rogers S. C., Casci J., Watson M., King F., Karlsen E., Sjøvoll M., Fahmi A., Schäfer A., Lennartz C. J. Mol. Struct. 2003;632:1–28. [Google Scholar]; (b) Metz S., Kästner J., Sokol A. A., Keal T. W., Sherwood P. Wiley Interdiscip. Rev.: Comput. Mol. Sci. 2014;4:101–110. [Google Scholar]
- Ahlrichs R., Bär M., Häser M., Horn H., Kölmel C. Chem. Phys. Lett. 1989;162:165–169. [Google Scholar]
- Smith W., Forester T. R. J. Mol. Graphics. 1996;14:136–141. doi: 10.1016/s0263-7855(96)00043-4. [DOI] [PubMed] [Google Scholar]
- Bakowies D., Thiel W. J. Phys. Chem. 1996;100:10580–10594. [Google Scholar]
- Becke A. D. J. Chem. Phys. 1993;98:5648–5652. [Google Scholar]
- Billeter S. R., Turner A. J., Thiel W. Phys. Chem. Chem. Phys. 2000;2:2177–2186. [Google Scholar]
- Kästner J., Carr J. M., Keal T. W., Thiel W., Wander A., Sherwood P. J. Phys. Chem. A. 2009;113:11856–11865. doi: 10.1021/jp9028968. [DOI] [PubMed] [Google Scholar]
- (a) Shaik S., de Visser S., Kumar D., Altun A., Thiel W. Chem. Rev. 2005;105:2279–2326. doi: 10.1021/cr030722j. [DOI] [PubMed] [Google Scholar]; (b) Shaik S., Cohen S., Wang Y., Chen H., Kumar D., Thiel W. Chem. Rev. 2010;110:949–1017. doi: 10.1021/cr900121s. [DOI] [PubMed] [Google Scholar]
- Johnson E. R., Keinan S., Mori-Sanchez P., Contreras-García J., Cohen A. J., Yang W. J. Am. Chem. Soc. 2010;132:6498–6506. doi: 10.1021/ja100936w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peracchi A. Trends Biochem. Sci. 2010;26:497–503. doi: 10.1016/s0968-0004(01)01911-9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.