

Key Points
IMiD compounds cause selective ubiquitination and degradation of IKZF1 in CD34+ cells by the CRBN E3 ubiquitin ligase.
Loss of IKZF1 is associated with a decrease of PU.1, critical for the development and maturation of neutrophils.
Abstract
We have previously shown that immunomodulatory drug (IMiD) compounds induce a shift into immature myeloid precursors with a maturational arrest and subsequent neutropenia. The mechanism of action is unknown. Here we found that IMiD compounds cause selective ubiquitination and degradation of the transcription factor IKZF1 in CD34+ cells by the Cereblon (CRBN) E3 ubiquitin ligase. Loss of IKZF1 is associated with a decrease of the IKZF1-dependent transcription factor PU.1, critical for the development and maturation of neutrophils. Using a thalidomide analog bead pull-down assay, we showed that IMiD compounds directly bind CRBN in CD34+ cells. Knockdown of CRBN in CD34+ cells resulted in resistance to POM-induced IKZF1 downregulation and reversed the POM-induced lineage shift in colony-formation assays, suggesting that the POM-induced degradation of IKZF1 in CD34+ cells requires CRBN. Chromatin immunoprecipitation assays revealed that IKZF1 binds to the promoter region of PU.1, suggesting that PU.1 is a direct downstream target of IKZF1 in CD34+ cells. POM failed to induce IKZF1 degradation in IKZF1-Q146H-OE CD34+ cells, indicating that CRBN binding to IKZF1 and subsequent IKZF1 ubiquitination is critical in this process. Using the NOD/SCID/γ-c KO mouse model, we confirmed the induction of myeloid progenitor cells by IMiD compounds at the expense of common lymphoid progenitors. These results demonstrate a novel mechanism of action of IMiD compounds in hematopoietic progenitor cells, leading to selective degradation of transcription factors critical for myeloid maturation, and explain the occurrence of neutropenia associated with treatment by IMiD compounds.
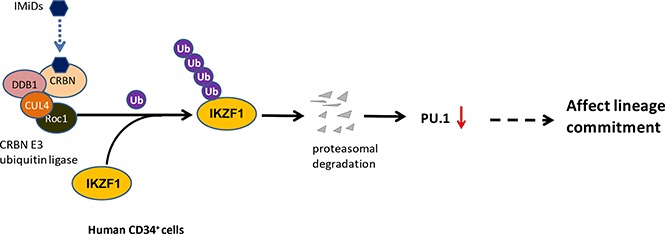
Visual Abstract
Introduction
Sixty years ago, thalidomide was used to treat nausea, particularly morning sickness in pregnant women, but it was subsequently banned because of its teratogenicity. Since then, thalidomide derivatives including lenalidomide (LEN) and pomalidomide (POM) have been developed and used for the treatment of multiple myeloma (MM), resulting in significantly improved overall survival in patients with myeloma.1-5 Immunomodulatory drug (IMiD) compounds inhibit myeloma cell growth, block cytokine production, impair angiogenesis, and enhance T-cell stimulation and proliferation, leading to MM cell death.6 Recently, IMiD compounds were shown to bind to cereblon (CRBN), the substrate recognition component of cullin-dependent ubiquitin ligases. Treatment of zebrafish with thalidomide results in fin defects, suggesting that IMiD compounds act by stabilizing CRBN substrates.7-9 In MM, LEN treatment leads to the selective ubiquitination and degradation of 2 lymphoid transcription factors, IKZF1 and IKZF3, by the CRBN-CRL4 ubiquitin ligase.10-12 IKZF1, also known as Ikaros, is a zinc finger-containing DNA-binding protein that plays a pivotal role in immune homeostasis via transcriptional regulation of the earliest stages of lymphocyte ontogeny and differentiation.13 Functional deficiency of IKZF1 has been implicated in the pathogenesis of acute lymphoblastic leukemia, the most common form of childhood cancer.13 This finding raises a concern because long-term follow-up data suggest that IMiD compounds are associated with an increased risk for secondary hematologic malignancies. In particular, patients receiving melphalan plus LEN regimens had a significantly greater risk of developing a second primary malignancy than those who did not receive LEN (hazard ratio, 4.41; 95% confidence interval, 2.4-8.1; P < .0001).14
Interestingly, previous studies have shown that in hematopoietic progenitor cells (HPCs), IMiD compounds do not exhibit direct stem cell toxicity; instead, these compounds affect lineage commitment and induce cell expansion.15,16 Treatment with pomalidomide during the development of primary human erythroid cells induces the suppression of several known repressors of fetal globin gene expression.17 We have previously shown that IMiD compounds shift hematopoietic lineage commitment to myeloid colony formation at the expense of erythroid cell colony formation by downregulation of GATA1.18,19 Other major adverse effects of IMiD compounds are thrombocytopenia (grade 3/4, 14.7%) and neutropenia (grade 3/4, 41.5%), which often compromise optimal treatment with IMiD compounds.20,21 Our previous studies have shown that IMiD compounds downregulate PU.1, a critical transcription factor for myeloid maturation, leading to the maturational arrest of granulocytes, the accumulation of immature myeloid precursors, and subsequent neutropenia.16 However, the precise mechanism involved has not yet been determined.
Here, we report that CRBN is expressed in human CD34+ cells and that POM promotes the CRBN-dependent degradation of the IKZF1 protein in CD34+ cells. Knockdown of CRBN in CD34+ cells induces POM resistance, indicating that CRBN is required for the IMiD compound-induced effects on lineage commitment. Our in vitro findings showing that IMiD compounds affect lineage commitment were confirmed in vivo in a humanized NOD/SCID/γ-c KO (NSG) mouse model.
Methods
Isolation and culture of human CD34+ cells
Primary CD34+ cells were isolated from peripheral blood leukapheresis products generated for autologous stem cell transplantation, according to Institutional Review Board guidelines. The cells were separated over a Ficoll-Paque PLUS gradient (GE Healthcare), and CD34+ cells were purified via immunomagnetic-positive selection using CD34+ beads (Miltenyi Biotec) according to the manufacturer’s instructions. Purified CD34+ cells were grown in serum-free hematopoietic growth medium (HPGM; Lonza) supplemented with 10 ng/mL recombinant human interleukin 3 (rhIL-3), 10 ng/mL rhIL-6, and 50 ng/mL recombinant human stem cell factor (rhSCF) (PeproTech). MG132, PS341, MLN4924 (Sigma Aldrich), LEN, and POM (Celgene) in dimethyl sulfoxide (DMSO) were diluted in culture medium just before use; 0.01% DMSO was used as a control.
Western blot analysis
Cells were harvested, washed 3 times with 1× phosphate-buffered saline (PBS), and lysed with 1× RIPA lysis buffer containing Halt Protease and Phosphatase Inhibitor Cocktail (Thermo Fisher Scientific). The proteins were then separated on 4% to 15% Criterion XT Bis-Tris Gels (Bio-Rad Laboratories) and subsequently transferred to polyvinyl difluoride membranes (Bio-Rad Laboratories). The following antibodies were used for immunoblotting: anti-IKZF1, anti-IKZF3 (Abcam), anti-CRBN, and anti-PU.1 (Cell Signaling Technology). Immunoblotting was performed according to the manufacturer’s instructions. Immunoreactive bands were visualized using SuperSignal West Pico Chemiluminescent Substrate (Thermo Fisher Scientific). We used β-actin (Sigma-Aldrich) to normalize the protein quantity.
Immunolabeling for fluorescence microscopy
Immunofluorescence staining was performed as previously described.22 CD34+ cells treated with or without LEN or POM were attached to poly-l-lysine-coated cover slips via centrifugation at 1000g for 10 minutes, and were subsequently fixed using 4% paraformaldehyde for 10 minutes at room temperature. For whole-cell staining, the cells were washed 3 times with PBS, permeabilized with 0.1% Triton X-100, and blocked with 3% bovine serum albumin in PBST (0.1% Triton X-100 in PBS) for 1 hour. CD34+ cells were stained with anti-IKZF1 (Abcam) and anti-green fluorescent protein (GFP) antibodies (Sigma Aldrich) and counterstained with 4′,6-diamidino-2-phenylindole (DAPI; Cell Signaling Technology) for nuclear visualization. Images were captured using a Leica microscope.
Real-time PCR analysis
Total RNA was isolated using TRIZOL reagent (Invitrogen), and cDNA was generated using SuperScript III reverse transcriptase (Invitrogen), followed by SYBR Green (Invitrogen)-based real-time polymerase chain reaction assays performed as previously described.23 The following primer pairs were used: human CRBN, forward (5′-TCCCGAATGTGTGTTGCCTT-3′) and reverse (5′-CCAGCGAGGCCATGAAGTTA-3′); human PU.1, forward (5′-CAAGCTTCGTGGAACTCTCC-3′) and reverse (5′-ACTGACAATCAGGCGCTTCT-3′); and human β-actin, forward (5′-CGAGCACAGAGCCTCGCCTTTG-3′) and reverse (5′-CGACGAGCGCGGCGATATCAT-3′). Human IKZF1 mRNA was analyzed via TaqMan real-time RT-PCR (Life Technologies).
Thalidomide analog bead pull-down assay
Thalidomide-immobilized beads (structure shown in Figure 1) were synthesized as previously described.7 To measure the binding of IMiD compounds to endogenous CRBN in CD34+ cells, human CD34+ cell extracts were prepared as previously described.8 Aliquots of 0.5 mL (5 mg protein) of the resulting extracts were preincubated (15 minutes, room temperature) with 5 μL DMSO (control) or 5 μL solutions containing LEN or POM in DMSO. Thalidomide analog-coupled beads (0.5 mg) were then added to the protein extracts, and the samples were rotated (2 hours, 4°C). Next, the beads were washed 3 times with 0.5 mL NP40 lysis buffer, and the bound proteins were eluted with sodium dodecyl sulfate–polyacrylamide gel electrophoresis sample buffer. Finally, immunoblot analysis was performed using an anti-CRBN Ab for all experiments.
Figure 1.

IMiD compounds downregulate the IKZF1 protein in human CD34+ cells. (A) The protein expression of CRBN, IKZF1, and IKZF3 was analyzed in human myeloma cell lines (RPMI-8226, H929, and MM.1S) and human CD34+ cells (Patients 1 and 2) by western blotting. (B) CD34+ cells were cultured with either LEN or POM at the indicated concentrations for 24 hours (upper) or for a short period (1-24 hours at 1 μM; bottom) or (C) a long period (1-9 days at 1 μM). Drugs were added every 2 days. The cell lysates were analyzed for IKZF1 and CRBN expression by western blotting. β-Actin was used as a loading control. (D) CD34+ cells were treated with DMSO (0.01%), LEN, or POM (1 μM) for 6 hours. The cells were then fixed and stained for IKZF1 (red color), and nuclear counterstaining was performed with DAPI (blue color). The localization of IKZF1 was observed using a Leica microscope (40×). (E) CD34+ cells were treated with DMSO (0.01%), LEN, or POM (1 μM) for the indicated times. IKZF1 mRNA levels were compared using qRT-PCR. (F) For thalidomide analog bead pull-down assays, we used a thalidomide analog coupled with FG beads (high-performance magnetic nanoparticles) to pull down the bound protein, followed by immunoblot detection of CRBN. Input, 5% of total protein from CD34+ cells before affinity bead binding; DMSO, LEN, or POM, pull-down complex from CD34+ cell extracts preincubated with DMSO (0.01%), lenalidomide, or pomalidomide (10 μM) for 15 minutes at room temperature.
Flow cytometry
For flow cytometry analysis, cultured CD34+ cells were harvested, washed once with fluorescence-activated cell sorter buffer (Hanks balanced salt solution buffer supplemented with 0.5% bovine serum albumin and 0.1% NaN3), and incubated with fluorochrome-labeled monoclonal antibodies, such as anti-human CD45, anti-human CD34, anti-human CD33, anti-human CD45RA, and anti-human CD38, as well as isotype control monoclonal antibodies (BD Pharmingen). Flow cytometry analysis was performed with LSRII (BD Pharmingen) using FlowJo software (FlowJo, LLC). Dead cells were excluded from the analysis by gating out low forward scatter and high DAPI-retaining cells.
Colony-formation assay
Colony-formation assays were performed as previously described.24 In brief, CD34+ cells were seeded in serum-free HPGM hematopoietic growth medium (Lonza) and plated in MethoCult H4434 media (STEMCELL Technologies) for 14 days (with DMSO, LEN, or POM). The resulting colonies were counted using a Leica microscope (25×).
Lentiviral shRNA silencing of CRBN and IKZF1
To silence CRBN and IKZF1 expression, CD34+ cells were transduced using pGreenpuro (System Biosciences) lentiviral particles containing CRBN- or IKZF1-targeting shRNAs expressing GFP as a selection marker. Stable shRNA transfectants were obtained via cell sorting based on GFP expression. The following target sequences were used: CRBN shRNA #1 (5′-GGAAAGGGAAGCACAGTTT-3′), CRBN shRNA #3 (5′-GAAGACCAATGTTCATATAAA-3′), IKZF1 shRNA #1 (5′-CCGCTTCCACATGAGCTAAAG-3′), and IKZF1 shRNA #2 (5′- GCATTTGGAAACGGGAATAAA-3′). CRBN and IKZF1 protein and mRNA levels were determined in control and silenced cells by western blotting and quantitative reverse transcription polymerase chain reaction (qRT-PCR), respectively. IKZF1 and CRBN protein levels were determined in control and silenced cells by western blotting.
Expression constructs and site-directed mutagenesis
The full-length human IKZF1 cDNA clone with an N-terminal FLAG tag was purchased from Genecopoeia (Rockville, MD) and subsequently subcloned into the pCDH-CMV-MCS-EF1-copGFP lentiviral vector (System Biosciences). The IKZF1 Q146H point mutation construct was generated by mutagenesis PCR, using the Q5 Site-Directed Mutagenesis Kit. The following primers were employed for mutagenesis: forward (5′-CCATTGCAATCAGTGCGGGGCC-3′) and reverse (5′-AACGGCCGTTCTCCAGTGTGGCTTC-3′). The constructs were verified by sequencing, and protein expression was subsequently confirmed by western blotting.
Lentiviral production and gene transduction
The viral product was concentrated 100 times, using PEG-it TM Virus Precipitation Solution (System Biosciences), and the titers were subsequently determined. Approximately 4 × 106 viral particles were preloaded onto RetroNectin-coated plates (Takara Shuzo), followed by centrifugation for 2 hours at 32°C. Before infection, the viral supernatant was discarded. Approximately 4 × 105 purified CD34+ cells were grown in serum-free HPGM supplemented with 10 ng/mL rhIL-3, 10 ng/mL rhIL-6, and 50 ng/mL rhSCF for 2 days and then added to the virus-preloaded plate, followed by culture for 3 days. Subsequently, gene-transduced CD34+ cells were reseeded onto HPGM containing 10 ng/mL rhTPO, 10 ng/mL rhIL-3, 10 ng/mL rhIL-6, and 50 ng/mL rhSCF. IMiD compounds (or the control vehicle) were added daily, as previously described. The cells were finally harvested for colony assays and western blot analysis.
Chromatin immunoprecipitation assay
Approximately 2 × 107 CD34+ cells were treated with 1 µM POM or the control vehicle for 24 hours. The cells were then collected, and chromatin immunoprecipitation assays were performed using the Pierce Agarose ChIP kit (Thermo Fisher Scientific) according to the manufacturer’s instructions. The following antibodies were used for IP: anti-IgG and anti-IKZF1 (Santa Cruz Biotechnology, Inc.). EBF1B was employed as positive control for the qRT-PCR analysis.25 The specific pairs of primers used for quantitative chromatin immunoprecipitation were as follows: PU.1 promoter #1, forward (5′-AGTGAATGCATGGTGGGCTT-3′) and reverse (5′-ACCTTTCTTGCCTATCCACCATT-3′); PU.1 promoter #2, forward (5′-CATTGCCAAGGGATGGGGAA-3′) and reverse (5′-TTATACCCTCCCCTCCCAGC-3′); PU.1 promoter #3, forward (5′-GTGTGGCAGAGCTACACGTT-3′) and reverse (5′-CCCCAACCCGTTTGCATAAA-3′); and EBF1 B promoter, forward (5′-GGGTTAGTGTGCCTGTGTTTAG-3′) and reverse (5′-CCCTGCTGGATGGAGATTCTG-3′).
In vivo expansion of human CD34+ cells
All animal procedures were approved by the Institutional Animal Care and Use Committee of Columbia University, New York, NY. Humanized mice were generated by transplanting human CD34+ fetal liver cells pretreated with DMSO (0.1%) or POM 1 µM for 16 hours into NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NSG) mice (The Jackson Laboratory). All recipients received total body irradiation (2 Gy) 4 to 8 hours before intravenous injection of 1 × 105 CD34+ cells. One day after the injection of human CD34+ cells, the mice were IV injected with DMSO (0.1%, 100 µL) or POM (300 µg/kg) every 2 days. Three weeks later, bone marrow cells were subjected to flow cytometry for the detection of human progenitor cells. Cells were gated for human CD45+CD34+ and analyzed using anti-human CD45RA, CD38, CD10 antibodies. Murine erythroid cells and nucleated cells were excluded by gating out mouse Ter119+ and Ly5+ cells. Partial bone marrow cells were sorted using human CD34+ MACS beads. The sorted CD34+ cells were used for colony-formation assays. After 14 days, the colonies were counted using a Leica microscope (25×).
Statistical analyses
Quantitative data are presented as the means ± SDs or SEMs, as indicated. Statistical significance was assessed using Student t test or Wilcoxon’s rank sum test, as appropriate for comparisons between the 2 groups.
Results
IMiD compounds downregulate the IKZF1 protein in human CD34+ cells
To identify the roles of CRBN, IKZF1, and IKZF3 in IMiD compound-treated HPCs, we analyzed their expression in human CD34+ cells isolated from peripheral blood. CRBN and IKZF1 protein and mRNA were expressed in CD34+ cells (Figure 1A; supplemental Figure 1). In contrast, no IKZF3 expression could be detected in human CD34+ cells, consistent with a previous report that IKZF3 is highly expressed in human B cells but not in CD34+ progenitors.26 Consistent with the finding that IKZF1 is a downstream target of CRBN in MM cells, we found that in CD34+ cells, both POM and LEN treatment decreased IKZF1 protein levels in a dose- and time-dependent manner in vitro (Figure 1B). Importantly, we observed sustained downregulation of IKZF1 over the course of at least 9 days with LEN and POM treatment every 2 days in serum-free hematopoietic growth medium supplemented with rhIL-3, rhIL-6, and rhSCF (Figure 1C). Immunofluorescence staining of CD34+ cells confirmed that both POM and LEN decreased the cellular IKZF1 protein level (Figure 1D). However, quantitative RT-PCR revealed that the IKZF1 mRNA level was not affected by IMiD compounds (Figure 1E), suggesting that in human CD34+ cells, IMiD compounds promote IKZF1 protein degradation but do not affect its transcript levels. Interestingly, 1-time treatment of CD34+ cells with IMiD compounds led to irreversible loss of the IKZF1 protein, without recovery within the observation period of 9 days (supplemental Figure 2). To address the question of whether the IMiD-induced loss of IKZF1 in the early phases of HPC development is critical in further lineage commitment, CD34+ cells were pretreated with DMSO or POM for 4, 8, or 24 hours and subjected to colony-formation assays without POM. After IMiD compounds were pulse treated for as little as 4 hours and washed, followed by 14 days of colony formation in culture, the number of BFU-E colonies was significantly decreased (P < .05), whereas that of colony-forming unit granulocyte (CFU-G) colonies was significantly increased (P < .05; supplemental Figure 3), demonstrating the potent effect of IMiD compounds on the induction and expansion of myeloid lineages. Using pull-down assays with thalidomide analog-coupled beads, we next confirmed that CRBN is a direct protein target of IMiD compounds in CD34+ cells. LEN and POM competitively inhibited the binding of thalidomide analog-coupled beads to CRBN, suggesting that CRBN directly binds to IMiD compounds in CD34+ cells (Figure 1F).
IMiD compounds promote the CRBN-dependent degradation of the IKZF1 protein in human CD34+ cells
We next evaluated whether IMiD compounds decrease IKZF1 protein levels by promoting IKZF1 protein ubiquitination and degradation. Pulse-chase assays performed in the presence of cycloheximide demonstrated that POM accelerated IKZF1 protein degradation (Figure 2A). Treatment with the proteasome inhibitors MG132 and PS341 or the cullin-dependent ubiquitin ligase inhibitor MLN4924 blocked LEN- and POM-induced IKZF1 degradation (Figure 2B-D). Ubiquitination assays confirmed that both LEN and POM increased IKZF1 protein poly-ubiquitination, resulting in proteasomal-dependent degradation (Figure 2E). Taken together, these data indicate that IMiD compounds decrease IKZF1 protein levels in human CD34+ cells by promoting protein degradation without altering IKZF1 mRNA expression.
Figure 2.

IMiD compounds induce the degradation of the IKZF1 protein in human CD34+ cells. Immunoblot analysis of IKZF1 or β-actin in CD34+ cells was performed in (A) CD34+ cells treated with DMSO (0.01%) or POM (1 μM) for 1 hour before the addition of cycloheximide (CHX; 100 μg/mL) for the indicated times; CD34+ cells treated with POM at the indicated concentrations with or without (B) MG132 (10 μM) and (C) MLN4924 (1 μM) for 24 hours; (D) CD34+ cells treated with DMSO (0.01%), LEN or POM (1 μM), with or without PS341 (10 nM) for 24 hours. (E) For the analysis of the ubiquitination of IKZF1, CD34+ cells were treated with MG132 (10 μM) and DMSO (0.02%), LEN, or POM (2 μM) for 2 hours. Immunoprecipitation was performed using an IKZF1 antibody, followed by western blotting with an ubiquitin antibody, indicating the protein pulled down by the IKZF1 antibody (right). (Left) Five percent total protein input.
CRBN mediates the IMiD-induced degradation of IKZF1, resulting in induction of myelopoiesis
To confirm that POM-induced IKZF1 protein degradation is mediated by CRBN in human CD34+ cells, CRBN expression was silenced in CD34+ cells via shRNA lentivirus transduction (supplemental Figure 4). The knockdown of CRBN in CD34+ cells decreased POM-induced IKZF1 protein degradation (Figure 3A) without affecting IKZF1 mRNA levels (Figure 3B). Immunofluorescence assays also showed that the IKZF1 protein level was stable in CRBN-knockdown CD34+ cells (shCRBN CD34+) after POM treatment (Figure 3C). In prior studies, we demonstrated that IMiD compounds affect stem cell fate by induction of myeloid progenitors.16 Hence, we investigated the role of CRBN in the POM-induced differentiation of HSCs. Flow cytometric analysis showed that in contrast to shCNTL-CD34+ cells, POM failed to induce myeloid precursor formation in shCRBN-CD34+ cells, as measured on the basis of CD33+ expression (Figure 3D). In colony formation assays, POM-induced CD34+ cell lineage commitment to CFU-G at the expense of BFU-E. This effect was blocked in CRBN knockdown cells (Figure 3E). These data suggest that CRBN is critical for mediating the effects of IMiD compounds, which promote the degradation of IKZF1 and a subsequent shift in lineage commitment.
Figure 3.

CRBN mediates IMiD-induced IKZF1 degradation, resulting in a CD34+ cell lineage shift. CD34+ cells were transduced using a lentivirus with control shRNA (shCNTL), CRBN-shRNA#1 (shCRBN-1), or CRBN-shRNA#3 (shCRBN-3). The transfected cells were then sorted for GFP positivity 3 days after transduction. (A) GFP-sorted cells were treated with DMSO (0.01%) or POM (1 μM) for 6 hours. Cell lysates were analyzed by western blotting to compare the levels of CRBN and IKZF1. β-actin was used as a control. (B) The CRBN and IKZF1 mRNA levels in GFP-sorted CD34+ cells were analyzed by qRT-PCR. (C) shCNTL and shCRBN-3 CD34+ cells were treated with POM (1 μM) or DMSO (0.01%) for 6 hours. The treated cells were subsequently fixed with 4% formaldehyde and stained for IKZF1 (red color) or GFP (green color), using DAPI for nuclear counterstaining (blue color). The localization of IKZF1 was observed using a Leica microscope (200×). (D) To analyze myeloid differentiation, for GFP+, CD33+, and CD45+ were analyzed in shCNTL and shCRBN-3 CD34+ cells using flow cytometry. (E) Control (CNTL), shCRBN-1, and shCRBN-3 CD34+ cells were treated with DMSO (0.01%) or POM (1 μM) and used for the colony-formation assays to evaluate BFU-E and CFU-G colony formation. After 14 day, the colonies were counted using a Leica microscope (25×). *P ≤ .05.
IKZF1 mediates the IMiD-induced CD34+ cell lineage shift
Next, we examined the role of IKZF1 in POM-induced cell lineage commitment. First, the nuclear localization of IKZF1 in CD34+ cells was verified by western blotting (supplemental Figure 5). To address the role of IKZF1 in the IMiD-induced CD34+ cell lineage shift, IKZF1 expression was knocked down in CD34+ cells via lentiviral shRNA infection (Figure 4A-B). In colony-formation assays, IKZF1 silencing induced the formation of CFU-G colonies, which was similar to the effects of POM (Figure 4C), suggesting that IKZF1 downregulation is critical for the POM-induced shift of CD34+ cells to the myeloid lineage. Next, we generated an IKZF1 mutant by substituting Q146 with histidine, which abrogates the CRBN-induced ubiquitination of IKZF1. CD34+ cells were transduced with lentiviral constructs for the overexpression of IKZF1-WT or IKZF1-Q146H. POM failed to induce complete IKZF1 degradation in IKZF1-Q146H CD34+ cells, indicating that IMiD-induced IKZF1 degradation is mediated by CRBN binding and the subsequent ubiquitination of IKZF1 (Figure 4D). Functional assays further confirmed that IKZF1-Q146H CD34+ cells were resistant to the POM-induced shift in lineage commitment from BFU-E to CFU-G (Figure 4E) and myeloid expansion, measured according to CD33+ expression by flow cytometry (Figure 4F).
Figure 4.

The IMiD-induced myeloid lineage shift of CD34+ cells is mediated by IKZF1. CD34+ cells were transduced using a lentivirus carrying the control shRNA (shCNTL), IKZF1-shRNA #1 (shIKZF1-1), or IKZF1-shRNA #2 (shIKZF1-2) sequence. At 3 days after transduction, the cells were sorted, and (A) the lysates were analyzed by western blotting to compare the levels of IKZF1. (B) The sorted cells were treated with POM (1 μM) or DMSO (0.01%) as a control for 6 hours, then fixed with 4% formaldehyde and stained for IKZF1 (red color) and GFP (green color) and with DAPI for nuclear counterstaining (blue color). The localization of IKZF1 was observed using a Leica microscope (200×). (C) shCNTL, shIKZF1-1 and shIKZF1-3 CD34+ cells were treated with DMSO (0.01%) or POM (1 μM) and subjected to colony-formation assays. After 14 days, the BFU-E and CFU-G colony numbers were counted. (D-E) CD34+ cells were transduced with empty vector (EV), PCDH-IKZF1-WT (IKZF1WT) or PCDH-IKZF1-Q146H (IKZF1Q146H) and sorted based on GFP expression after 3 days. (D) Sorted cells were treated with POM (1 μM) or DMSO (0.01%) for 6 hours. Cell lysates were analyzed by western blotting to compare levels of IKZF1. (E) Sorted cells were analyzed for GFP+, CD33+, and CD45+ by flow cytometry for the evaluation of myeloid differentiation. (F) Sorted cells were treated with DMSO (0.01%) or POM (1 μM) and subjected to colony-formation assays. After 14 days, the colonies were counted using a Leica microscope (25×). *P ≤ .05; **P ≤ .01.
PU.1 is downregulated by IMiD compounds treatment in CD34+ cells
Previously, we showed that IMiD compounds induce a myeloid shift in which PU.1 plays a pivotal role in mediating the effects of IMiD compounds on hematopoietic differentiation.16,18 PU.1 is downregulated by POM at both the protein and mRNA levels (Figure 5A-B). To understand the underlying mechanism, PU.1 promoter sequences were analyzed, and several potential IKZF1 binding sites identified (Figure 5C, top). Potential IKZF1 binding sites were confirmed by CHIP assays. Treatment of CD34+ cells with POM resulted in IKZF1 degradation, thereby blocking the binding of IKZF1 to PU.1 sequences (Figure 5C, bottom). We further examined whether IKZF1 regulates PU.1 expression in CD34+ cells by knocking down IKZF1. Indeed, IKZF1 knockdown decreased the protein levels of PU.1, but not those of CRBN, suggesting that PU.1 expression is regulated by IKZF1 (Figure 5D). Consistent with this hypothesis, the POM-induced downregulation of PU.1 was blocked by CRBN knockdown (Figure 5E), further confirming that the ubiquitination of CRBN by IMiD compounds induces the degradation of IKZF1 and that PU.1 is not under CRBN control. We examined whether CRBN knock-down restores IKZF1 binding to PU.1 promoter after treatment with POM. CHIP assays analysis showed that in contrast to shCNTL-CD34+ cells, POM failed to block the binding of IKZF1 to PU.1 promoter in shCRBN-CD34+ cells (Figure 5F).
Figure 5.

PU.1 is regulated by IKZF1 in CD34+ cells. (A) CD34+ cells were cultured with DMSO (0.01%) or POM (1 μM) for the indicated times, and compounds were added every 2 days. The cells were subsequently lysed and analyzed for PU.1 expression by western blotting. (B) CD34+ cells were treated with DMSO (0.01%), LEN, or POM (1 μM). IKZF1 and PU.1 mRNA levels were compared using qRT-PCR. (C) Chromatin immunoprecipitation analysis of DMSO (0.01%) or POM (1 μM)-treated CD34+ cells using an IKZF1 antibody or rabbit IgG as a negative control. The precipitated DNA fragments were subjected to qRT-PCR analysis with primers targeting the PU.1 promoter. (D) shCNTL, shIKZF1-1, and shIKZF1-3 CD34+ cell lysates were analyzed by western blotting to compare the levels of IKZF1, CRBN, and PU.1 (E) shCNTL and shCRBN- CD34+ cells were treated with DMSO (0.01%) or POM (1 μM) for 3 days. The cell lysates were then analyzed by western blotting to compare the levels of CRBN and PU.1. β-Actin was probed as a loading control. (F) shCNTL and shCRBN- CD34+ cells were treated with DMSO (0.01%) or POM (1 μM) for 24 hours and analyzed by chromatin immunoprecipitation assay using an IKZF1 antibody, or rabbit IgG as a negative control. The precipitated DNA fragments were subjected to qRT-PCR analysis with primers targeting the PU.1 promoter. **P ≤ .01.
POM affects lineage commitment in a human immune system mouse model
Reflecting the difference between the mouse and human CRBN gene sequences, IMiD compounds do not bind to mouse CRBN; hence, POM treatment of mice had no effect on IKZF1 levels in mice hematopoietic progenitor cells (supplemental Figure 6), and IMiD compounds cannot be studied in currently established mouse models.27 To overcome this hurdle, we generated a humanized NSG mouse model by establishing a functional human hematopoietic system in mice. This model offers a robust in vivo platform to study stem cell engraftment, to observe the effects of IMiD compounds on the proliferation that allows improved analysis of the shift of lineage commitment of human hematopoietic progenitors. NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NSG) mice received 105 human CD34+ fetal liver cells (Figure 6A) and were treated with DMSO or POM every 2 days for 3 weeks, and bone marrow mononuclear cells were subsequently analyzed using flow cytometry. POM treatment significantly decreased the number of common lymphoid progenitor Lin-CD34+CD10+ cells (DMSO 78.8% vs POM 44.2%), but significantly increased the number of common myeloid progenitor Lin-CD34+CD38+CD45RA− cells (DMSO 19.7% vs POM 53.7%). The common lymphoid progenitor/common myeloid progenitor ratio decreased from 2 to 0.4 (P < .05; Figure 6B).This is consistent with the in vitro finding that IMiD compounds induce myeloid lineage commitment at the expense of erythroid progenitors such as BFU-E.16,18 To further analyze the effects of IMiD compounds on hematopoietic progenitors in vivo, CD34+ MACS bead-selected mouse bone marrow cells were used for colony-formation assays. The CD34+ cells of POM-treated mice exhibited reduced erythroid cell colony formation (BFU-E: DMSO, 74 vs POM, 50; P < .05) and an increased frequency of myeloid colonies (CFU-G, 84 vs POM, 146; P < .05), confirming in vivo that IMiD compounds shift the lineage commitment from BFU-E to CFU-G (Figure 6C). Taken together, these data indicate that IMiD compounds directly bind to CRBN in human CD34+ cells, which leads to IKZF1 ubiquitination and subsequent degradation. The loss of IKZF1 decreases PU.1 expression in human CD34+ cells and is associated with a myeloid lineage commitment shift concomitant with the inhibition of myeloid maturation (Figure 6D).
Figure 6.

Pomalidomide affects lineage commitment in a NSG mouse model. (A) NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NSG) mice received fetal CD34+ cells IV for engrafting with human hematopoiesis. (B) NSG mice exhibiting human hematopoiesis were injected IV with DMSO (0.1%, 100 µL) or POM (300 µg/kg) every 2 days. Three weeks later, bone marrow cells were subjected to flow cytometry for the detection of human progenitor cells, gated with human CD45+CD34+, and analyzed using antihuman CD45RA, CD38, CD10. Murine erythroid Ter119+ and Ly5+ cells and nucleated cells were excluded. (C) The partial bone marrow cells were sorted using human CD34+ MACS beads. The sorted cells were subsequently used for colony-formation assays. After 14 days, colony numbers were counted. (D) IMiD compounds directly bind to CRBN in human CD34+ cells, which leads to IKZF1 ubiquitination and promotes protein degradation. The loss of IKZF1 decreases PU.1 expression in human CD34+ cells and leads to a shift to myeloid lineage commitment with inhibition of myeloid maturation. The inhibition of myeloid maturation might lead to neutropenia and an increased risk for secondary malignancies. *P ≤ .05.
Discussion
HPCs have the ability to undergo self-renewal and differentiate into various lineages of hematopoietic cells. The balance between protein synthesis and protein degradation is likely vital for both processes. Here, we showed that the E3 ligase CRBN is an important regulator of the development and lineage commitment of CD34+ cells. These findings indicated that IMiD compounds promote the CRBN-dependent degradation of the IKZF1 protein in CD34+ cells, and consequently decrease the levels of transcription factors such as PU.1, which is critical for the development and maturation of neutrophils.
Several E3 ubiquitin ligases have been demonstrated to be critical for HSCs differentiation. Deficiency of the E3 ubiquitin ligase Itch in HSCs induces accelerated proliferation and progenitor cell expansion, reflecting the accumulation of activated Notch1.28 Other E3 ubiquitin ligases, such as the RING finger ligase c-Cbl, have been shown to control the development and function of HSCs,29 highlighting the important role of E3 ubiquitin ligases and their potential function in the self-renewal and differentiation of HSCs. The E3 ligase protein CRBN is a direct molecular target that mediates the toxicity of thalidomide.7 Human CRBN was originally identified as a novel adenosine triphosphate-dependent Lon protease that is highly conserved from plants to humans.30 However, little is known about the expression and function of CRBN in human CD34+ cells. Here, we showed that CRBN is expressed in CD34+ cells at a similar level to that in MM cell lines (Figure 1A). Moreover, the E3 ligase CRBN plays a critical role in the control of HPC development, as demonstrated by the fact that CRBN-knockdown cells become resistant to POM-induced IKZF1 degradation and the subsequent induction of myelopoiesis (Figure 3), further suggesting that CRBN is required for POM-induced cell lineage commitment. Previous study also showed ubiquitinated IKZF1 is required for proteasome-mediated degradation.31
IKZF1 plays a pivotal role in immune homeostasis via transcriptional regulation of the earliest stages of lymphocyte ontogeny and differentiation.13 IKZF1 is widely expressed in the hematopoietic system, including in multipotent, self-renewing hematopoietic stem cells and their committed progenitors. Functional deficiency of IKZF1 has been implicated in the pathogenesis of acute lymphoblastic leukemia, the most common form of childhood cancer.32 Previous studies have shown that IKZF1−/−mice exhibit severe defects in lymphocyte function, including an absence of B and T cells.33 In addition, these mice display defects in stem cells and in formation of BFU-E, a measure of early erythroid activity.34 These findings are consistent with the results showing that IKZF1 downregulation contributes to the POM-induced cell lineage shift toward myelopoiesis (Figure 4). Therefore, inhibition of IKZF1 expression by IMiD compounds might also promote leukemogenesis in pre-B cells and subsequently contribute to second primary malignancy. Indeed, there is a slightly higher risk for leukemias and B-cell malignancies in patients treated with IMiD compounds, particularly in combination with melphalan.20
PU.1 is a critical transcription factor that regulates granulopoiesis. PU.1-deficient hematopoietic progenitors show a perturbation in neutrophil maturation. Anderson et al verified that PU.1−/− mice develop neutrophils, but these neutrophils fail to achieve functional competence.35 Dakic et al showed that in contrast to wild-type controls, PU.1−/− granulocytes maintained c-kit expression and displayed some (but not complete) maturation, according to morphological criteria.36 The results of the present study demonstrated that IMiD compounds promote the CRBN-dependent degradation of the IKZF1 protein in human CD34+ cells and the subsequent decrease in the levels of the transcription factor PU.1 (Figure 5), which is critical for the development and maturation of neutrophils. Chromatin immune precipitation analysis showed that IKZF1 can directly repress or activate Sfpi1 transcription via different PU.1 cis-elements.37 Considering that PU.1 is critically involved in the IMiD-induced lineage shift in CD34+ cells, we verified that IKZF1 binds to the promoter regions of PU.1, using chromatin immunoprecipitation assays, which suggested that PU.1 is a direct downstream target of IKZF1 in CD34+ cells.
To investigate the in vivo effects of IMiD compounds, we developed a humanized mouse model by establishing a functional human immune system in NSG mice.38 Bone marrow analysis revealed that POM treatment significantly induced the formation of granulocyte/macrophage progenitor cells (CD34+CD38+CD45RA+ cells) at the expense of common lymphoid progenitors (CD34+CD10+ cells; Figure 6). The shift to myelopoiesis observed in vivo is consistent with the in vitro finding that IMiD compounds affect lineage commitment. In mice, the most salient phenotype associated with the loss of IKZF1 is early and complete blockade of B-cell development.39,40 A decrease in or loss of IKZF1 expression leads to T-cell hyperproliferation and lymphomagenesis, indicating that IKZF1 functions as a tumor suppressor41 and increases the production of HSCs or myeloid progenitors.42
Examining the roles of CRBN and IKZF1 in IMiD-induced effects on CD34+ cells provides the first evidence of the pathomechanism underlying the effects of LEN and POM on hematopoiesis and their induction of neutropenia, thrombocytopenia, and anemia, which will lead to the development of novel therapeutic strategies.
Supplementary Material
The full-text version of this article contains a data supplement.
Acknowledgments
The authors thank the patients and donors for providing samples. The authors are grateful to Hal E. Broxmeyer for his comments on this manuscript.
Research reported in this publication was performed in the CCTI Flow Cytometry Core, supported in part by the Office of the Director of the National Institutes of Health under awards S10RR027050 and S10OD020056. S. Li, J.F., and S. Lentzsch were supported by the National Institutes of Health, National Cancer Institute grant R01CA175313 and the US Food and Drug Administration grant R01FD005110, and M.Y.M. was supported by the National Institutes of Health, National Heart, Lung, and Blood Institute grant R01HL93716.
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Authorship
Contribution: S. Li and J.F. conducted experiments and analyzed data; H.W. and H.M. contributed mouse experiments; X.X. and S.D. helped with thalidomide analog bead pull-down assay; Y.G.Y. and M.Y.M. contributed vital reagents and critically revised the manuscript; and S. Li, J.F., and S. Lentzsch designed the research studies, analyzed data, and wrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Suzanne Lentzsch, Division of Hematology/Oncology, Department of Medicine, College of Physicians and Surgeons, Columbia University, Herbert Irving Pavilion, R 953, 161 Ft. Washington Ave, New York, NY 10032; e-mail: sl3440@columbia.edu.
References
- 1.Dimopoulos MA, Richardson PG, Moreau P, Anderson KC. Current treatment landscape for relapsed and/or refractory multiple myeloma. Nat Rev Clin Oncol. 2015;12(1):42-54. [DOI] [PubMed] [Google Scholar]
- 2.Li S, Gill N, Lentzsch S. Recent advances of IMiDs in cancer therapy. Curr Opin Oncol. 2010;22(6):579-585. [DOI] [PubMed] [Google Scholar]
- 3.Pan B, Lentzsch S. The application and biology of immunomodulatory drugs (IMiDs) in cancer. Pharmacol Ther. 2012;136(1):56-68. [DOI] [PubMed] [Google Scholar]
- 4.Quach H, Ritchie D, Stewart AK, et al. Mechanism of action of immunomodulatory drugs (IMiDS) in multiple myeloma. Leukemia. 2010;24(1):22-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Guirguis AA, Ebert BL. Lenalidomide: deciphering mechanisms of action in myeloma, myelodysplastic syndrome and beyond. Curr Opin Cell Biol. 2015;37:61-67. [DOI] [PubMed] [Google Scholar]
- 6.Bartlett JB, Dredge K, Dalgleish AG. The evolution of thalidomide and its IMiD derivatives as anticancer agents. Nat Rev Cancer. 2004;4(4):314-322. [DOI] [PubMed] [Google Scholar]
- 7.Ito T, Ando H, Suzuki T, et al. Identification of a primary target of thalidomide teratogenicity. Science. 2010;327(5971):1345-1350. [DOI] [PubMed] [Google Scholar]
- 8.Lopez-Girona A, Mendy D, Ito T, et al. Cereblon is a direct protein target for immunomodulatory and antiproliferative activities of lenalidomide and pomalidomide. Leukemia. 2012;26(11):2326-2335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhu YX, Braggio E, Shi CX, et al. Cereblon expression is required for the antimyeloma activity of lenalidomide and pomalidomide. Blood. 2011;118(18):4771-4779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lu G, Middleton RE, Sun HH, et al. The myeloma drug lenalidomide promotes the cereblon-dependent destruction of ikaros proteins. Science. 2014;343(6168):305-309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kronke J, Udeshi ND, Narla A, et al. Lenalidomide causes selective degradation of IKZF1 and IKZF3 in multiple myeloma cells. Science. 2014;343(6168):301-305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stewart AK. How thalidomide works against cancer. Science. 2014;343(6168):256-257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schwickert TA, Tagoh H, Gultekin S, et al. Stage-specific control of early B cell development by the transcription factor Ikaros. Nat Immunol. 2014;15(3):283-293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Palumbo A, Bringhen S, Kumar SK, et al. Second primary malignancies with lenalidomide therapy for newly diagnosed myeloma: a meta-analysis of individual patient data. Lancet Oncol. 2014;15(3):333-342. [DOI] [PubMed] [Google Scholar]
- 15.Verhelle D, Corral LG, Wong K, et al. Lenalidomide and CC-4047 inhibit the proliferation of malignant B cells while expanding normal CD34+ progenitor cells. Cancer Res. 2007;67(2):746-755. [DOI] [PubMed] [Google Scholar]
- 16.Pal R, Monaghan SA, Hassett AC, et al. Immunomodulatory derivatives induce PU.1 down-regulation, myeloid maturation arrest, and neutropenia. Blood. 2010;115(3):605-614. [DOI] [PubMed] [Google Scholar]
- 17.Dulmovits BM, Appiah-Kubi AO, Papoin J, et al. Pomalidomide reverses γ-globin silencing through the transcriptional reprogramming of adult hematopoietic progenitors. Blood. 2016;127(11):1481-1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Koh KR, Janz M, Mapara MY, et al. Immunomodulatory derivative of thalidomide (IMiD CC-4047) induces a shift in lineage commitment by suppressing erythropoiesis and promoting myelopoiesis. Blood. 2005;105(10):3833-3840. [DOI] [PubMed] [Google Scholar]
- 19.Moutouh-de Parseval LA, Verhelle D, Glezer E, et al. Pomalidomide and lenalidomide regulate erythropoiesis and fetal hemoglobin production in human CD34+ cells. J Clin Invest. 2008;118(1):248-258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Palumbo A, Hajek R, Delforge M, et al. Continuous lenalidomide treatment for newly diagnosed multiple myeloma. N Engl J Med. 2012;366(19):1759-1769. [DOI] [PubMed] [Google Scholar]
- 21.Weber DM, Chen C, Niesvizky R, et al. Lenalidomide plus dexamethasone for relapsed multiple myeloma in North America. N Engl J Med. 2007;357(21):2133-2142. [DOI] [PubMed] [Google Scholar]
- 22.Fu J, Yan P, Li S, Qu Z, Xiao G. Molecular determinants of PDLIM2 in suppressing HTLV-I Tax-mediated tumorigenesis. Oncogene. 2010;29(49):6499-6507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li S, Pal R, Monaghan SA, et al. IMiD immunomodulatory compounds block C/EBP{beta} translation through eIF4E down-regulation resulting in inhibition of MM. Blood. 2011;117(19):5157-5165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li Z, Godinho FJ, Klusmann JH, Garriga-Canut M, Yu C, Orkin SH. Developmental stage-selective effect of somatically mutated leukemogenic transcription factor GATA1. Nat Genet. 2005;37(6):613-619. [DOI] [PubMed] [Google Scholar]
- 25.Iacobucci I, Iraci N, Messina M, et al. IKAROS deletions dictate a unique gene expression signature in patients with adult B-cell acute lymphoblastic leukemia. PLoS One. 2012;7(7):e40934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Billot K, Parizot C, Arrouss I, et al. Differential aiolos expression in human hematopoietic subpopulations. Leuk Res. 2010;34(3):289-293. [DOI] [PubMed] [Google Scholar]
- 27.Krönke J, Fink EC, Hollenbach PW, et al. Lenalidomide induces ubiquitination and degradation of CK1alpha in del(5q) MDS. Nature. 2015;523(7559):183-188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rathinam C, Matesic LE, Flavell RA. The E3 ligase Itch is a negative regulator of the homeostasis and function of hematopoietic stem cells. Nat Immunol. 2011;12(5):399-407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rathinam C, Thien CB, Langdon WY, Gu H, Flavell RA. The E3 ubiquitin ligase c-Cbl restricts development and functions of hematopoietic stem cells. Genes Dev. 2008;22(8):992-997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Higgins JJ, Pucilowska J, Lombardi RQ, Rooney JP. A mutation in a novel ATP-dependent Lon protease gene in a kindred with mild mental retardation. Neurology. 2004;63(10):1927-1931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Popescu M, Gurel Z, Ronni T, et al. Ikaros stability and pericentromeric localization are regulated by protein phosphatase 1. J Biol Chem. 2009;284(20):13869-13880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Somasundaram R, Prasad MA, Ungerback J, Sigvardsson M. Transcription factor networks in B-cell differentiation link development to acute lymphoid leukemia. Blood. 2015;126(2):144-152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang JH, Nichogiannopoulou A, Wu L, et al. Selective defects in the development of the fetal and adult lymphoid system in mice with an Ikaros null mutation. Immunity. 1996;5(6):537-549. [DOI] [PubMed] [Google Scholar]
- 34.Nichogiannopoulou A, Trevisan M, Neben S, Friedrich C, Georgopoulos K. Defects in hemopoietic stem cell activity in Ikaros mutant mice. J Exp Med. 1999;190(9):1201-1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Anderson KL, Smith KA, Pio F, Torbett BE, Maki RA. Neutrophils deficient in PU.1 do not terminally differentiate or become functionally competent. Blood. 1998;92(5):1576-1585. [PubMed] [Google Scholar]
- 36.Dakic A, Metcalf D, Di Rago L, Mifsud S, Wu L, Nutt SLPU. 1 regulates the commitment of adult hematopoietic progenitors and restricts granulopoiesis. J Exp Med. 2005;201(9):1487-1502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zarnegar MA, Rothenberg EV. Ikaros represses and activates PU.1 cell-type-specifically through the multifunctional Sfpi1 URE and a myeloid specific enhancer. Oncogene. 2012;31(43):4647-4654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kalscheuer H, Danzl N, Onoe T, et al. A model for personalized in vivo analysis of human immune responsiveness. Sci Transl Med. 2012;4(125):125ra30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Meffre E, Casellas R, Nussenzweig MC. Antibody regulation of B cell development. Nat Immunol. 2000;1(5):379-385. [DOI] [PubMed] [Google Scholar]
- 40.Domen J, Weissman IL. Hematopoietic stem cells need two signals to prevent apoptosis; BCL-2 can provide one of these, Kitl/c-Kit signaling the other. J Exp Med. 2000;192(12):1707-1718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Busslinger M. Transcriptional control of early B cell development. Annu Rev Immunol. 2004;22(1):55-79. [DOI] [PubMed] [Google Scholar]
- 42.Georgopoulos K, Bigby M, Wang JH, et al. The ikaros gene is required for the development of all lymphoid lineages. Cell. 1994;79(1):143-156. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.