

Abstract
Gene expression varies across cells in a population or a tissue. This heterogeneity has come into sharp focus in recent years through developments in new imaging and sequencing technologies. However, our ability to measure variation has outpaced our ability to interpret it. Much of the variability may arise from random effects occurring in the processes of gene expression (transcription, RNA processing and decay, translation). The molecular basis of these effects is largely unknown. Likewise, a functional role of this variability in growth, differentiation and disease has only been elucidated in a few cases. In this review, we highlight recent experimental and theoretical advances for measuring and analyzing stochastic variation.
Introduction
Gene expression is the link that connects environmental stimuli to the phenotypic responses of an organism. Early experiments aimed at identifying the key cellular factors and genetic elements that regulated expression were in vitro and population based. Recent advances in sequencing and fluorescence microscopy now allow scientists to probe gene expression at the basic unit of information flow — the single cell. From a systems biology viewpoint, the methods of single-cell imaging and single-cell RNA sequencing (scRNA-seq) hold tremendous promise for providing an essential link between stimulus and response with the ability to directly visualize and quantify the production of RNA transcripts.
However, gene expression at the single-cell level is heterogeneous and stochastic — it varies across cells in a population and within a given cell over time. This phenomenon adds a perplexing challenge not only in being able to predict the expression behavior of a gene given known environmental inputs, but also the converse: to infer the state of the environment from a given gene’s behavior. The field of single-cell gene expression is not a straightforward survey of how information gets transmitted from environment to gene product but rather grapples with a fundamentally philosophical question that often goes unappreciated: how do we get from randomness to order? At this point in time, the field appears directed towards the following questions: How does an organism coordinate a response within its body when the expression behavior of its individual cells is so stochastic? How much of gene expression heterogeneity is stable and represents a true biological subpopulation with different phenotypic properties? How does nuclear architecture contribute to gene regulation and variability? How do we generate a quantitative understanding of transcription and gene networks with computational modeling? In this review, we highlight recent literature which is at the forefront of addressing these questions.
Current developments in single-cell imaging and sequencing
The first methods to probe single-cell gene expression at the mRNA level were based on imaging, by labeling RNA transcripts in fixed cells via single molecule RNA FISH [1] (smFISH) and in living cells using the MS2-PP7 system [2–4]. Within a decade, imaging was followed by RNA sequencing (scRNA-seq), which allowed for in-depth gene expression profiling of individual cells[5,6]. For the sake of brevity we refer readers to recent reviews [7,8] for an up-to-date history of the methodologies. In this section (Figure 1) we outline the current advantages and limitations of live-cell imaging, smFISH, and scRNA-seq, and the recent work done to extend the capabilities of each technique.
Figure 1. Single cell gene expression analysis methods.
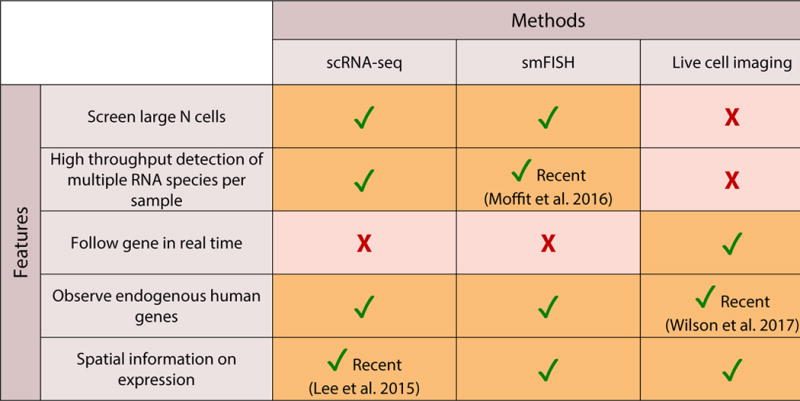
An evaluation of the current features and limitations of single cell gene expression methods, with recent advances noted.
Live-cell imaging with the MS2-PP7 stem loop system remains the most direct method for truly capturing the history of a gene’s expression behavior, as it follows transcription activity of a living cell in real time. One primary limitation of the system is that it is low-throughput: of the three techniques, it is the most laborious and takes months to design and integrate constructs into the desired model system [9]. This time-intensive aspect has hindered the ability to make an extensive survey and classification of real-time transcription across genes. The method is inherently limited by having only two orthogonal stem loop sequences (PP7 and MS2), which allows for at most two elements to be labeled within a single cell. However recent studies have used dual color labeling to their advantage to explore different phenomenon, such as splicing kinetics [10,11], sense and antisense transcription from a single promoter on a yeast gene [12], two genes regulated by a common enhancer [13], and translation of individual RNAs [14].
Due to the difficulty of genetic manipulation in higher order eukaryotes, most studies on transcription kinetics are conducted with exogenous genes. However, homologous recombination has been successfully used to integrate stem loops into the endogenous loci of bacteria and yeast, and was recently used to integrate MS2 loops in mouse embryonic stem cells [15], and to visualize endogenous transcription dynamics of the pluripotency factors Nanog and Oct4 [16]. Within the next few years we expect advances in CRISPR/Cas9 gene editing to greatly aid in making endogenous integrations possible.
The second imaging-based technique, smFISH, has advantages to complement the limitations of live-cell imaging. The fluorescent oligo probes can be designed and commercially synthesized for virtually any endogenous gene. And because the sample is fixed, high throughput imaging can be used to routinely collect data on tens of thousands of cells for a given time point. Initially, smFISH was limited by the number of spectrally separable colors that could be used within a single cell. Earlier methods were able to visualize 10–30 RNA within a single cell through spectral separation [17], sequential labeling [18] or combinatorial labeling [19]. A recent technique (MERFISH) that uses sequential labeling increased the number of mRNA to 100–1000 [20], and a high-throughput version has also been recently published [21]. Progress has also been made in detecting small variations in nucleic acid sequence [22,23]. Although smFISH is only a single time point measurement, advances in labeling and imaging are making the method increasingly high-throughput, allowing a significant number of genes and cells to be analyzed in a single experiment.
The main advantage of the final technique, scRNA-seq, is its breadth and depth: it produces extensive gene expression profiles that quantify the variation in abundance and sequence of all the transcripts in a cell. scRNA-seq has historically dealt with problems of bias in the type and quantity of transcripts it identifies, and several recent protocols (CEL-Seq [24], MARS-Seq [25], Cyto-Seq [26], and Drop-Seq [27]), have sought to overcome these issues. Solutions include in vitro transcription, which uses linear instead of exponential amplification, the use of barcodes to relate each transcript sequence to a unique molecular identifier, and spike-in RNA to normalize the output signal to the relative transcript abundance in each cell, to reduce technical variation. The advantage is that one can quantify the transcriptome in depth, and quantify the variation or heterogeneity of all the expressed genes of a given cell. The primary limitation of traditional scRNA-seq is that it does not inherently contain spatial information, nor does it allow one to follow the transcriptome over time in a single cell. Recently, the protocol ‘FISSEQ’ [28] combined spatial information of transcripts from smFISH with sequencing of the individual transcriptomes of those cells. Sequencing methods that incorporate spatial information of the transcript, or imaging methods that incorporate sequencing, will be essential contributions to ultimately achieving a “4D” transcriptome atlas of gene regulation.
Heterogeneity across cells
Variation can be due to genetic or non-genetic causes. It can be fixed or time-dependent, programmed or random. A significant component of non-genetic heterogeneity is due to the discontinuous nature of transcription. Genes are transcribed in “bursts” and this phenomenon has recently been observed in mouse liver tissue with smFISH [29], Drosophila with live-cell imaging [30], mouse embryonic stem cells with scRNA-seq [31], zebrafish embryos with smFISH [32], and human brain tumors [33] and melanoma [34] with single-cell qPCR.
The prevailing view of considering heterogeneity as ‘extrinsic’ or ‘intrinsic’, was first developed from experimental data using two-color reporter assays in bacteria [35], and later in yeast [36]. Extrinsic factors are those that influence many genes, for example the concentration of RNA polymerase in a cell. Intrinsic noise arises from stochastic fluctuations inherent in biochemical reactions between molecules at low copy number. This dichotomy of heterogeneity continues to be examined. Recently, Fu and Pachter [37] revisited previous data from Elowitz and colleagues [35] and highlighted the importance of experimentally determining whether the two fluorescent reporters have the same distribution of mean and variance in fluorescence intensity, and to normalize them if not, as this is a major assumption of the model. Also recently, Sherman and colleagues [38] proposed that extrinsic and intrinsic variability are not exclusively orthogonal to each other. The authors examined extrinsic variability with the yeast heat shock protein SSA1, and with modeling showed how intrinsic variability can be dependent on external factors. The terms ‘extrinsic’ and ‘intrinsic’ are subjectively defined. Considering that upstream extrinsic factors may also have a timescale of fluctuation (for example due to bursting), one person’s extrinsic noise could be another person’s intrinsic noise.
How stable is heterogeneity, and what are its functional consequences?
Whether the ‘noisiness’ of gene expression has a functional purpose or evolutionary advantage is still an open question. Examinations of ‘bet-hedging’ have largely been confined to bacteria (for a recent review see [39]), but could such transient heterogeneity confer any advantages in eukaryotes? A recent paper by Shaffer and colleagues [40] provides a compelling single-cell viewpoint of how transient switching of phenotype profiles of patient-derived melanoma cells leads to stable populations resistant to the drug vemurafenib. Using a variation on the classic Luria-Delbruck experiment [41], they observed that before application of the drug, cells transition between ‘non-resistant’ and ‘pre-resistant states’, as observed by their transient expression of resistance markers. The pre-resistant state was not heritable, and it was only with addition of vemurafenib that cellular reprogramming and a stable resistance phenotype emerged. This intriguing example of transient heterogeneity in mammalian cells may be seen as a manifestation of “dynamical instability” [42], a model from the field of network theory based on Boolean logic to explain the mechanisms underlying gene expression variability observed in some cancer types. Supporting evidence for the dynamical instability hypothesis has been observed at the population level in ‘anti-profile’ studies, which shows that many cancer subtypes exhibit a high degree of gene expression variability across individuals [43,44]. Hypervariability of gene expression is a reproducible signature of cancer tissue types. It would be interesting to determine whether at the single cell level, transient gene expression heterogeneity is an illustration of dynamical instability of gene networks, and whether this is related to hypervariability in cancer or observed more generally.
Another open question about heterogeneity is whether stochastic transcription ultimately gets transmitted through the nucleus. A few recent studies propose that stochastic expression is in fact buffered, and limits the variation in cytoplasmic mRNA that is ultimately available for translation. One study attributes this buffering to microRNAs [45], and two other studies provide examples of buffering by the nuclear pore complex [46,47]. Halpern and colleagues used whole genome RNA-seq and smFISH in various mouse cell types and found that mRNA was retained in the nuclear pore. The authors found a difference in retention times: immediate early genes tended to have the shortest retention time, and protein coding genes the longest. Their interpretation was that mature processed RNA was retained in the nuclear pore and that fast induction time was due to their release from the pore, not the mRNA synthesis rate itself. The transient nature of heterogeneity and its potential buffering are interesting observations, and it remains to be seen whether they are observed more generally in higher eukaryotes.
Nuclear Architecture and the role of cis elements in gene regulation
Our understanding of nuclear architecture and the role of enhancers has increased profoundly over the past few years. We refer readers to recent reviews on promoter-enhancer interaction [48] and the role of nuclear architecture on gene expression [49], and here focus on new research examining the role of nuclear topology in gene regulation. Hi-C, a population based assay to determine long range chromosome interactions, led to the identification of topologically associated domains (TADs) [50,51] and associations with the proteins CTCF and cohesin to act as insulators of chromosome ‘neighborhoods’, where enhancers interact with the promoters of genes within a neighborhood. Within TADs, transcriptionally active genes are shown to share spatial co-regulation [52,53], and disruption of these topological boundaries have consequences for disease. For example, recent studies looking at the role of nuclear topology and cancer show that gene duplication (a common feature of cancer) is mis-regulated if it occurs at the boundary of a neighborhood rather than within it. A model of ‘enhancer hijacking’ has been proposed [54], which occurs when a boundary is disrupted and an enhancer is able to interact with the promoters of oncogenes and promote their expression. Manipulation of TAD boundaries with CRISPR was recently shown to cause oncogene activation of gliomas [55] and leukemia [56].
Recently, Bartman and colleagues [57] manipulated enhancer-promoter contacts at the locus control region in mouse erythrocytes and human primary erythroid cells. Their observations used smFISH to evaluate how transcription burst features of the beta- and gamma-globin genes were affected when they minimized contacts (via deletion) or increased contacts with forced looping. They saw that enhancer contacts increase burst frequency, supporting the idea that enhancers increase the probability of transcription, similar to what was shown for reporter genes [58]. Importantly, they also found that active transcription of one allele lowered the probability of activity of the other allele, giving evidence to support a model of promoter-enhancer interaction where the enhancer alternates between contacts of the promoters it regulates. Enhancer manipulation has also been carried out with live-cell imaging in Drosophila [13]. In this study, enhancers and insulators were placed between two reporter genes in developing embryos, resulting in modulations of burst frequency. Here, the authors concluded that one enhancer could activate two genes at once, in contrast to the model from the globin locus. More generally, disruption of boundary elements or mutation of CTCF results in increases in gene expression noise [59]. Thus, the interaction between enhancer and promoter as reflected in chromosome topology is a prime determinant of metazoan expression heterogeneity. Single-cell imaging coupled with manipulation of cis nuclear architecture will continue to provide rich insight into the physical factors governing gene regulation.
Modeling the transcription process and gene networks
Finally, one of the goals of studying single-cell gene expression is to reconcile the complexity of biology with the desire to find universal principles that govern living behavior. In pursuit of that understanding, researchers have drawn upon methodology from information theory [60,61]— the study of how information is received, processed, and transmitted within a system. In gene regulation, it is increasingly clear that the gene receives information not just in trans (i.e. chemical modifications, binding of activating and inhibitory transcription factors) but in cis as well, as noted in the previous section. Thus, the question we now ask from a theoretical standpoint is, how can a cell decode the complex collection of incoming signals to produce an effective response? The earliest ‘Telegraph’ model for describing how information is processed through gene expression dynamics [62] was based on a single active and inactive state. The model proved to fit expression data in some instances [36,63,64], but there are increasing examples which illustrate that two states are insufficient to represent the data [65–68]. Recently, Rieckh and colleagues [69] identified instances in which a multi-state promoter model performs better than a simple two-state model; however, they advocate the two-state model as the simplest theoretical baseline to start from, as it is possible to overfit the data with too many states. Many cis and trans factors have the potential to affect promoter states on different timescales (Figure 2), and theoretical models coupled with experimental data will continue to help elucidate these key factors.
Figure 2. Possible factors contributing to multiple promoter states.
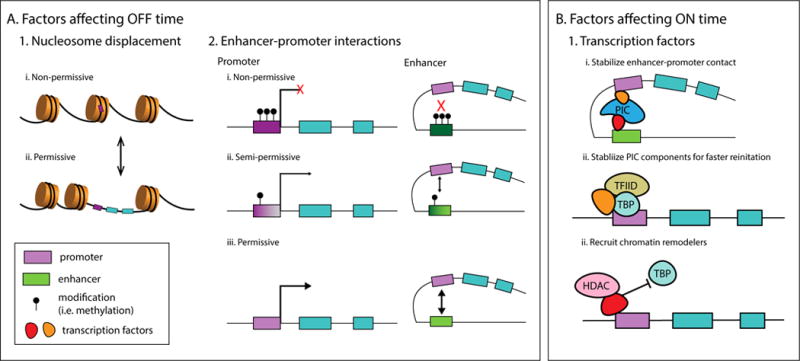
Many factors may affect the OFF and ON time of a gene at different timescales. Factors affecting the OFF time could include chemical modifications of the promoter and enhancer, or the displacement of the +1 nucleosome from the transcription start site. Transcription factors may prolong the ON time of a gene by stabilizing basal transcription machinery at the promoter, or enhancer-promoter contacts, for more successful re-initiation. On the other hand, a repressive transcription factor might recruit factors such as chromatin remodelers that could result in a shortening of the active period of the gene. Abbreviations: PIC, pre-initiation complex; TFIID, general transcription factor II-D; TBP, TATA binding protein; HDAC, histone deacetylase.
Information theory has also recently been used in studies that manipulate trans factors of gene regulation. Hansen and O’Shea [70] controlled the frequency and duration of the yeast transcription factor Msn’s nuclear localization. The authors observed the effect of modulating Msn2 nuclear localization on the burst frequency and amplitude of two target genes and determined that transcriptional bursts from natural Msn2 target promoters encode 1.0–1.3 bits of information about the signal identity and intensity. Another group recently used opto-genetic stimulation to manipulate Ras in NIH3T3 cells and determined its effect on the transcriptome profile of immediate early genes [71]. Along with manipulating cis elements in the nucleus, as described in the previous section, experimental systems that allow researchers to directly control and manipulate the localization of trans elements, such as transcription factors, will be important for achieving a systems biology viewpoint of precisely how information is transmitted to a gene.
A new development in scRNA-seq methodology is the use of principal component analysis (PCA) for stem cell lineage tracing. Several groups [72–74] use scRNA-seq expression data from a population of stem cells that have been induced to differentiate, and use PCA for ‘pseudo-temporal ordering’-a timeline of gene expression changes gathered from the single-timepoint gene expression profiles of many cells. In sequencing, an assumption of pseudo-temporal ordering is that every cell represents a time-point along the same continuum of differentiation. We can see a similar assumption in imaging, where it is assumed that the observed distribution of bursting comes from the same underlying kinetics present in every cell. Both methods assume that modeling will obtain parameters that are reflective of a ‘mean’ process. But what if it is not? Llamosi and colleagues [75] propose that the idea of fitting parameters to a ‘mean cell’ is faulty, and instead suggest that the goal should be to arrive at a distribution of models. This study highlights a major assumption of all three single-cell methods that is gene expression is ergodic—observing a single cell over many timepoints yields the same information as measuring the population of cells at a single timepoint. Recent work has shown the fallacies of the ergodic assumption for dynamical models in which time averages are commonly replaced by population averages [76]. As a result, we believe the assumption of ergodicity in gene expression should be studied carefully.
Another aspect of gene expression to which mathematical modeling has begun to contribute is the elucidation of gene networks from single-cell expression data. Datasets that obtain measurements of gene expression profiles from single cells are becoming increasingly prevalent, and there is potentially much information to be gained from pairwise correlations of genes. But what can co-expression tell us about connectivity? Simply looking for correlation in mRNA as a sign of connectivity proves challenging at the single-cell level because transcription is stochastic and dynamic—timescales of the birth and decay rates of mRNA affects how much will be present at any given point in time (Figure 3). This difficulty has been seen in single-cell imaging and scRNA-seq, where a clear functional response at the population level yields poor or no correlation when examining the RNA of pairs of involved genes within single cells [77,78]. These observations necessitate the following questions for the field: Under what conditions can we expect to see correlations in expression between two interacting genes? And when is co-expression more reflective of a direct interaction rather than an indirect one?
Figure 3. Capturing dynamic transcription regulation at a single timepoint.
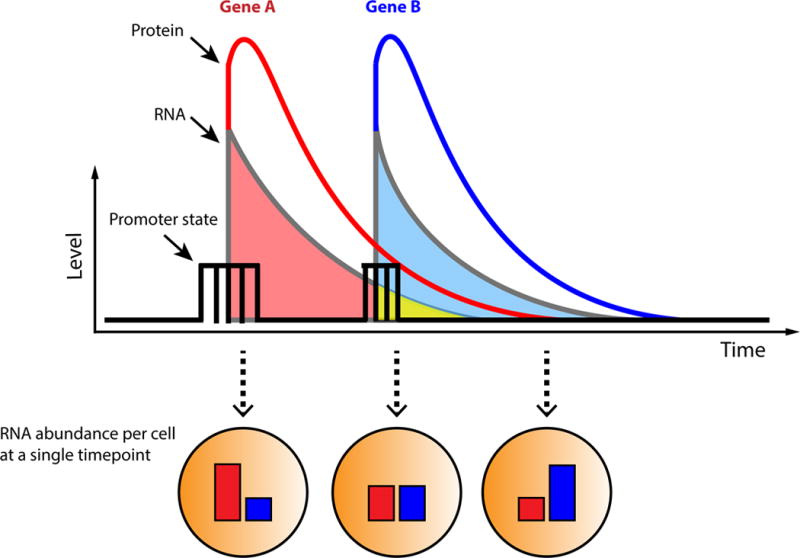
The plot shows a hypothetical gene regulatory interaction Gene A → Gene B, where the protein product of Gene A promotes the expression of Gene B (i.e. the interaction between a transcription factor and its gene target). This interaction may be present in every cell of a population; however, each cell may be at different stages of the regulatory process at any given point in time. As such, fixing the population at a single time point (as is done for smFISH and scRNA-seq) may yield different combinations of Gene A and Gene B mRNA with no apparent correlation at the single cell level. As illustrated in the figure, the half-lives of both mRNA species may affect the degree to which they overlap in time (highlighted in yellow) within a single cell.
These questions have previously been examined using principles from information theory. Ku and colleagues [79] used microarray datasets to determine whether co-expression of gene pairs was indicative of transcriptional regulatory interactions (TRIs, when Gene A codes for a transcription factor that regulates Gene B) or co-regulation (when two genes A and B are regulated by a common upstream factor). They found that co-expression was indicative of co-regulation in E. coli, but more indicative of TRIs in S. cerevisiae. Additional such studies would be useful to the field of single-cell gene expression, as they provide a framework for us to ask what co-expression indicates in higher eukaryotes, and whether these conclusions are observable in single cells. Will population-based network models need to be modified to describe the observed stochastic nature of expression at the single-cell level? Do gene networks change with transient heterogeneity, or when cis nuclear architecture is altered? Development of single cell assays and their applications have outpaced theoretical work to examine the data for its underlying principles. We see a need for more studies to be done in this area in the future.
Conclusions and Future Outlook
The field of single-cell gene expression has the potential to generate a comprehensive and quantitative view of gene regulation. Developments in gene editing, advances in high-throughput/multiplexed assays for single cells, and increased understanding of nuclear architecture are making significant contributions towards our ability to manipulate and understand the dynamic aspects of gene regulation. In this review, we have highlighted recent literature that advances the capabilities of three frequently-used techniques: scRNA-seq, smFISH, and live-cell imaging. Heterogeneity is still a phenomenon to grapple with – both experimentally and theoretically – but as it continues to be identified in more systems, observations of its dynamic properties are leading to hypotheses about its functionality and consequences. Models of heterogeneity continue to be revisited, and studies that manipulate nuclear architecture reveal the magnitude of its role in regulating gene expression. Population-based assays hold great utility for elucidating gene networks, and we hope single-cell data will begin to provide useful insight in this area as well. Current work and future developments in the field will contribute to a more thorough temporal and spatial understanding of gene regulation at the single-cell level, and ultimately to our ability to relate an environmental stimulus to the response of an organism.
Highlights.
New advances in single-cell gene expression techniques achieve high-throughput multiplexed labeling of RNA.
Time-resolved measurements of gene expression lead to new hypotheses about the functionality and consequences of expression heterogeneity.
Principles from information theory can guide models of transcriptional regulation and gene network connectivity.
Acknowledgments
We thank Murali Palangat for helpful discussions on the figures. This work is supported by NSF (Award No. DGE1632976) and the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Femino AM, Fay FS, Fogarty K, Singer RH. Visualization of Single RNA Transcripts in Situ. Science. 1998;280:585–590. doi: 10.1126/science.280.5363.585. [DOI] [PubMed] [Google Scholar]
- 2.Bertrand E, Chartrand P, Schaefer M, Shenoy SM, Singer RH, Long RM. Localization of ASH1 mRNA particles in living yeast. Mol Cell. 1998;2:437–45. doi: 10.1016/s1097-2765(00)80143-4. [DOI] [PubMed] [Google Scholar]
- 3.Larson DR, Zenklusen D, Wu B, Chao JA, Singer RH. Real-Time Observation of Transcription Initiation and Elongation on an Endogenous Yeast Gene. Science. 2011;332:475–478. doi: 10.1126/science.1202142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4•.Janicki SM, Tsukamoto T, Salghetti SE, Tansey WP, Sachidanandam R, Prasanth KV, Ried T, Shav-Tal Y, Bertrand E, Singer RH, et al. From silencing to gene expression: real-time analysis in single cells. Cell. 2004;116:683–98. doi: 10.1016/s0092-8674(04)00171-0. The first study to use RNA imaging as a method for directly observing gene regulation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tang F, Barbacioru C, Wang Y, Nordman E, Lee C, Xu N, Wang X, Bodeau J, Tuch BB, Siddiqui A, et al. mRNA-Seq whole-transcriptome analysis of a single cell. Nat Methods. 2009;6:377–382. doi: 10.1038/nmeth.1315. [DOI] [PubMed] [Google Scholar]
- 6.Wang Z, Gerstein M, Snyder M. RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet. 2009;10:57–63. doi: 10.1038/nrg2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vera M, Biswas J, Senecal A, Singer RH, Park HY. Single-Cell and Single-Molecule Analysis of Gene Expression Regulation. Annu Rev Genet. 2016;50:267–291. doi: 10.1146/annurev-genet-120215-034854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kolodziejczyk AA, Kim JK, Svensson V, Marioni JC, Teichmann SA, Juréus A, Marques S, Munguba H, He L, Betsholtz C, et al. The Technology and Biology of Single-Cell RNA Sequencing. Mol Cell. 2015;58:610–620. doi: 10.1016/j.molcel.2015.04.005. [DOI] [PubMed] [Google Scholar]
- 9.Coulon A, Larson DR. Fluctuation Analysis. Methods in enzymology. 2016:159–191. doi: 10.1016/bs.mie.2016.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10•.Coulon A, Ferguson ML, de Turris V, Palangat M, Chow CC, Larson DR. Kinetic competition during the transcription cycle results in stochastic RNA processing. Elife. 2014;3 doi: 10.7554/eLife.03939. A useful book chapter outlining the basic considerations in experimental design, acquisition, and interpretation of live-cell transcription data from single genes using the MS2–PP7 stem loop system. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Martin RM, Rino J, Carvalho C, Kirchhausen T, Carmo-Fonseca M. Live-cell visualization of pre-mRNA splicing with single-molecule sensitivity. Cell Rep. 2013;4:1144–55. doi: 10.1016/j.celrep.2013.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lenstra TL, Coulon A, Chow CC, Larson DR. Single-Molecule Imaging Reveals a Switch between Spurious and Functional ncRNA Transcription. Mol Cell. 2015;60:597–610. doi: 10.1016/j.molcel.2015.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13•.Fukaya T, Lim B, Levine M. Enhancer Control of Transcriptional Bursting. Cell. 2016;166:358–368. doi: 10.1016/j.cell.2016.05.025. A study looking at the effect of enhancers on transcriptional bursting of reporter genes in live Drosophila embryos. The authors observe that enhancers primarily regulate burst frequency and show that genes with a shared enhancer exhibit simultaneous bursting. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14••.Halstead JM, Lionnet T, Wilbertz JH, Wippich F, Ephrussi A, Singer RH, Chao JA. An RNA biosensor for imaging the first round of translation from single cells to living animals. Science. 2015;347:1367–671. doi: 10.1126/science.aaa3380. The authors utilize the PP7 stem loop system with a newly developed method (TRICK) to observe translation of mRNA transcripts with live-cell imaging. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lionnet T, Czaplinski K, Darzacq X, Shav-Tal Y, Wells AL, Chao JA, Park HY, de Turris V, Lopez-Jones M, Singer RH. A transgenic mouse for in vivo detection of endogenous labeled mRNA. Nat Methods. 2011;8:165–170. doi: 10.1038/nmeth.1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16•.Ochiai H, Sugawara T, Yamamoto T. Simultaneous live imaging of the transcription and nuclear position of specific genes. Nucleic Acids Res. 2015;43:e127. doi: 10.1093/nar/gkv624. A live-cell imaging method (ROLEX) that uses homologous recombination to label endogenous loci of mouse embryonic stem cells. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17•.Lubeck E, Cai L. Single-cell systems biology by super-resolution imaging and combinatorial labeling. Nat Methods. 2012;9:743–8. doi: 10.1038/nmeth.2069. One of the first groups to utilize combinatorial labeling of smFISH probes to measure up to 32 genes simultaneously. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lubeck E, Coskun AF, Zhiyentayev T, Ahmad M, Cai L. Single-cell in situ RNA profiling by sequential hybridization. Nat Methods. 2014;11:360–1. doi: 10.1038/nmeth.2892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Levsky JM, Shenoy SM, Pezo RC, Singer RH. Single-Cell Gene Expression Profiling. Science. 2002;297:836–840. doi: 10.1126/science.1072241. [DOI] [PubMed] [Google Scholar]
- 20•.Chen KH, Boettiger AN, Moffitt JR, Wang S, Zhuang X. RNA imaging. Spatially resolved, highly multiplexed RNA profiling in single cells. Science. 2015;348:aaa6090. doi: 10.1126/science.aaa6090. Report of a multiplexed system (MERFISH) to label many RNA species within a single cell sample. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Moffitt JR, Hao J, Wang G, Chen KH, Babcock HP, Zhuang X. High-throughput single-cell gene-expression profiling with multiplexed error-robust fluorescence in situ hybridization. Proc Natl Acad Sci U S A. 2016;113:11046–51. doi: 10.1073/pnas.1612826113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mellis IA, Gupte R, Raj A, Rouhanifard SH. Visualizing adenosine-to-inosine RNA editing in single mammalian cells. Nat Methods. 2017;14:801–804. doi: 10.1038/nmeth.4332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Levesque MJ, Ginart P, Wei Y, Raj A. Visualizing SNVs to quantify allele-specific expression in single cells. Nat Methods. 2013;10:865–7. doi: 10.1038/nmeth.2589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hashimshony T, Senderovich N, Avital G, Klochendler A, de Leeuw Y, Anavy L, Gennert D, Li S, Livak KJ, Rozenblatt-Rosen O, et al. CEL-Seq2: sensitive highly-multiplexed single-cell RNA-Seq. Genome Biol. 2016;17:77. doi: 10.1186/s13059-016-0938-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jaitin DA, Kenigsberg E, Keren-Shaul H, Elefant N, Paul F, Zaretsky I, Mildner A, Cohen N, Jung S, Tanay A, et al. Massively parallel single-cell RNA-seq for marker-free decomposition of tissues into cell types. Science. 2014;343:776–9. doi: 10.1126/science.1247651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fan HC, Fu GK, Fodor SPA. Combinatorial labeling of single cells for gene expression cytometry. Science. 2015;347:1258367–1258367. doi: 10.1126/science.1258367. [DOI] [PubMed] [Google Scholar]
- 27.Macosko EZ, Basu A, Satija R, Nemesh J, Shekhar K, Goldman M, Tirosh I, Bialas AR, Kamitaki N, Martersteck EM, et al. Highly Parallel Genome-wide Expression Profiling of Individual Cells Using Nanoliter Droplets. Cell. 2015;161:1202–1214. doi: 10.1016/j.cell.2015.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28•.Lee JH, Daugharthy ER, Scheiman J, Kalhor R, Ferrante TC, Terry R, Turczyk BM, Yang JL, Lee HS, Aach J, et al. Fluorescent in situ sequencing (FISSEQ) of RNA for gene expression profiling in intact cells and tissues. Nat Protoc. 2015;10:442–58. doi: 10.1038/nprot.2014.191. A protocols paper describing a variation of scRNA-seq that preserves spatial information about the location of each RNA species within a given cell or tissue. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bahar Halpern K, Tanami S, Landen S, Chapal M, Szlak L, Hutzler A, Nizhberg A, Itzkovitz S. Bursty gene expression in the intact mammalian liver. Mol Cell. 2015;58:147–56. doi: 10.1016/j.molcel.2015.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gregor T, Garcia HG, Little SC. The embryo as a laboratory: quantifying transcription in Drosophila. Trends Genet. 2014;30:364–75. doi: 10.1016/j.tig.2014.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mantsoki A, Devailly G, Joshi A. Gene expression variability in mammalian embryonic stem cells using single cell RNA-seq data. Comput Biol Chem. 2016;63:52–61. doi: 10.1016/j.compbiolchem.2016.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Oka Y, Sato TN. Whole-mount single molecule FISH method for zebrafish embryo. Sci Rep. 2015;5:8571. doi: 10.1038/srep08571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Möllerström E, Rydenhag B, Andersson D, Lebkuechner I, Puschmann TB, Chen M, Wilhelmsson U, Ståhlberg A, Malmgren K, Pekny M. Classification of Subpopulations of Cells Within Human Primary Brain Tumors by Single Cell Gene Expression Profiling. Neurochem Res. 2015;40:336–352. doi: 10.1007/s11064-014-1431-y. [DOI] [PubMed] [Google Scholar]
- 34.Ennen M, Keime C, Kobi D, Mengus G, Lipsker D, Thibault-Carpentier C, Davidson I. Single-cell gene expression signatures reveal melanoma cell heterogeneity. Oncogene. 2015;34:3251–3263. doi: 10.1038/onc.2014.262. [DOI] [PubMed] [Google Scholar]
- 35.Elowitz MB, Levine AJ, Siggia ED, Swain PS. Stochastic Gene Expression in a Single Cell. Science. 2002;297 doi: 10.1126/science.1070919. [DOI] [PubMed] [Google Scholar]
- 36.Raser JM, O’Shea EK. Control of stochasticity in eukaryotic gene expression. Science. 2004;304:1811–4. doi: 10.1126/science.1098641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fu AQ, Pachter L. Estimating intrinsic and extrinsic noise from single-cell gene expression measurements. Stat Appl Genet Mol Biol. 2016;15:447–471. doi: 10.1515/sagmb-2016-0002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sherman MS, Lorenz K, Lanier MH, Cohen BA. Cell-to-cell variability in the propensity to transcribe explains correlated fluctuations in gene expression. Cell Syst. 2015;1:315–325. doi: 10.1016/j.cels.2015.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dueck H, Eberwine J, Kim J. Variation is function: Are single cell differences functionally important? BioEssays. 2016;38:172–180. doi: 10.1002/bies.201500124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40••.Shaffer SM, Dunagin MC, Torborg SR, Torre EA, Emert B, Krepler C, Beqiri M, Sproesser K, Brafford PA, Xiao M, et al. Rare cell variability and drug-induced reprogramming as a mode of cancer drug resistance. Nature. 2017;546:431–435. doi: 10.1038/nature22794. The authors report on how cancer drug resistance is a transient phenotype within a population of patient-derived melanoma cells but becomes a stable phenotype after the drug is administered. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Luria SE, Delbrück M. Mutations of Bacteria from Virus Sensitivity to Virus Resistance. Genetics. 1943;28:491–511. doi: 10.1093/genetics/28.6.491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42•.Pomerance A, Ott E, Girvan M, Losert W. The effect of network topology on the stability of discrete state models of genetic control. Proc Natl Acad Sci U S A. 2009;106:8209–14. doi: 10.1073/pnas.0900142106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43••.Bravo HC, Pihur V, McCall M, Irizarry RA, Leek JT. Gene expression anti-profiles as a basis for accurate universal cancer signatures. BMC Bioinformatics. 2012;13:272. doi: 10.1186/1471-2105-13-272. A study of gene expression profiles across various cancer subtypes in patients. This work shows that although tumors are highly heterogeneous-often deemed a problem for screening based on particular expression markers-the deviation in expression is itself a highly reproducible signature among cancers. This observation was found not only in tissue biopsies, but also in the blood of patients, possibly opening the door for less invasive types of cancer screening. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44•.Dinalankara W, Bravo HC. Gene Expression Signatures Based on Variability can Robustly Predict Tumor Progression and Prognosis. Cancer Inform. 2015;14:71–81. doi: 10.4137/CIN.S23862. The authors expand on previous work from [43] to show that hypervariability of expression predicts the probability of relapse and survival. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schmiedel JM, Klemm SL, Zheng Y, Sahay A, Bluthgen N, Marks DS, van Oudenaarden A. MicroRNA control of protein expression noise. Science. 2015;348:128–132. doi: 10.1126/science.aaa1738. [DOI] [PubMed] [Google Scholar]
- 46•.Bahar Halpern K, Caspi I, Lemze D, Levy M, Landen S, Elinav E, Ulitsky I, Itzkovitz S. Nuclear Retention of mRNA in Mammalian Tissues. Cell Rep. 2015;13:2653–62. doi: 10.1016/j.celrep.2015.11.036. One of two reports (along with [45]) on mRNA transcript retention at the nuclear pore buffering stochastic variability. The authors used whole genome RNA-seq and smFISH in various mouse cell types and discovered differences in retention times of different mRNA species at the nuclear pore. They conclude that the quickly induced genes behave so because of their short retention time at the nuclear pore compared to other genes, and not due to the transcription rate itself. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Battich N, Stoeger T, Pelkmans L. Control of Transcript Variability in Single Mammalian Cells. Cell. 2015;163:1596–1610. doi: 10.1016/j.cell.2015.11.018. [DOI] [PubMed] [Google Scholar]
- 48.Vernimmen D, Bickmore WA. The Hierarchy of Transcriptional Activation: From Enhancer to Promoter. Trends Genet. 2015;31:696–708. doi: 10.1016/j.tig.2015.10.004. [DOI] [PubMed] [Google Scholar]
- 49.Gibcus JH, Dekker J. The hierarchy of the 3D genome. Mol Cell. 2013;49:773–82. doi: 10.1016/j.molcel.2013.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dixon JR, Selvaraj S, Yue F, Kim A, Li Y, Shen Y, Hu M, Liu JS, Ren B. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature. 2012;485:376–80. doi: 10.1038/nature11082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nora EP, Lajoie BR, Schulz EG, Giorgetti L, Okamoto I, Servant N, Piolot T. Spatial partitioning of the regulatory landscape of the X-inactivation center. Nature. 2012;485:381. doi: 10.1038/nature11049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Schoenfelder S, Sexton T, Chakalova L, Cope NF, Horton A, Andrews S, Kurukuti S, Mitchell JA, Umlauf D, Dimitrova DS, et al. Preferential associations between co-regulated genes reveal a transcriptional interactome in erythroid cells. Nat Genet. 2010;42:53–61. doi: 10.1038/ng.496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Karathia H, Kingsford C, Girvan M, Hannenhalli S. A pathway-centric view of spatial proximity in the 3D nucleome across cell lines. Sci Rep. 2016;6:39279. doi: 10.1038/srep39279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Weischenfeldt J, Dubash T, Drainas AP, Mardin BR, Chen Y, Stütz AM, Waszak SM, Bosco G, Halvorsen AR, Raeder B, et al. Pan-cancer analysis of somatic copy-number alterations implicates IRS4 and IGF2 in enhancer hijacking. Nat Genet. 2016;49:65–74. doi: 10.1038/ng.3722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hnisz D, Weintraub AS, Day DS, Valton A-L, Bak RO, Li CH, Goldmann J, Lajoie BR, Fan ZP, Sigova AA, et al. Activation of proto-oncogenes by disruption of chromosome neighborhoods. Science. 2016;351:1454–1458. doi: 10.1126/science.aad9024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Flavahan WA, Drier Y, Liau BB, Gillespie SM, Venteicher AS, Stemmer-Rachamimov AO, Suvà ML, Bernstein BE. Insulator dysfunction and oncogene activation in IDH mutant gliomas. Nature. 2016;529:110–4. doi: 10.1038/nature16490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57•.Bartman CR, Hsu SC, Hsiung CC-S, Raj A, Blobel GA. Enhancer Regulation of Transcriptional Bursting Parameters Revealed by Forced Chromatin Looping. Mol Cell. 2016;62:237–247. doi: 10.1016/j.molcel.2016.03.007. A study using smFISH to evaluate how transcription burst features of the beta and gamma globin genes are affected when enhancer contacts at the locus control region are increased or minimized. Their observations that enhancers increase burst frequency are in agreement with results from Fukaya and colleagues 13, but in contrast they observe alternating and not simulatenous bursting between the target genes, suggesting a different model of enhancer-promoter interaction than what was described for Drosophila in [13] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Larson DR, Fritzsch C, Sun L, Meng X, Lawrence DS, Singer RH. Direct observation of frequency modulated transcription in single cells using light activation. Elife. 2013;2:e00750. doi: 10.7554/eLife.00750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ren G, Jin W, Cui K, Rodrigez J, Hu G, Zhang Z, Larson DR, Zhao K. CTCF-Mediated Enhancer-Promoter Interaction Is a Critical Regulator of Cell-to-Cell Variation of Gene Expression. Mol Cell. 2017;67:1049–1058.e6. doi: 10.1016/j.molcel.2017.08.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Shannon CE. A Mathematical Theory of Communication. Bell Syst Tech J. 1948;27:379–423. [Google Scholar]
- 61.Tkačik G, Bialek W. Information processing in living systems. 2014;7:89–117. [Google Scholar]
- 62.Peccoud J, Ycart B. Markovian Modeling of Gene-Product Synthesis. Theor Popul Biol. 1995;48:222–234. [Google Scholar]
- 63.So L-H, Ghosh A, Zong C, Sepúlveda LA, Segev R, Golding I. General properties of transcriptional time series in Escherichia coli. Nat Genet. 2011;43:554–60. doi: 10.1038/ng.821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Raj A, Peskin CS, Tranchina D, Vargas DY, Tyagi S. Stochastic mRNA synthesis in mammalian cells. PLoS Biol. 2006;4:1707–1719. doi: 10.1371/journal.pbio.0040309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sanchez A, Choubey S, Kondev J. Stochastic models of transcription: From single molecules to single cells. Methods. 2013;62:13–25. doi: 10.1016/j.ymeth.2013.03.026. [DOI] [PubMed] [Google Scholar]
- 66.Harper CV, Finkenstädt B, Woodcock DJ, Friedrichsen S, Semprini S, Ashall L, Spiller DG, Mullins JJ, Rand DA, Davis JRE, et al. Dynamic analysis of stochastic transcription cycles. PLoS Biol. 2011;9:e1000607. doi: 10.1371/journal.pbio.1000607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Suter DM, Molina N, Gatfield D, Schneider K, Schibler U, Naef F. Mammalian Genes Are Transcribed with Widely Different Bursting Kinetics. Science. 2011;332:472–474. doi: 10.1126/science.1198817. [DOI] [PubMed] [Google Scholar]
- 68.Bothma JP, Garcia HG, Esposito E, Schlissel G, Gregor T, Levine M. Dynamic regulation of eve stripe 2 expression reveals transcriptional bursts in living Drosophila embryos. Proc Natl Acad Sci U S A. 2014;111:10598–603. doi: 10.1073/pnas.1410022111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69•.Rieckh G, Tkačik G. Noise and information transmission in promoters with multiple internal States. Biophys J. 2014;106:1194–204. doi: 10.1016/j.bpj.2014.01.014. A theoretical paper that uses the two-state Telegraph model as the basis for examining multiple promoter states and their functional consequences. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hansen AS, O’Shea EK. Limits on information transduction through amplitude and frequency regulation of transcription factor activity. Elife. 2015;4 doi: 10.7554/eLife.06559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wilson MZ, Ravindran PT, Lim WA, Toettcher JE. Tracing Information Flow from Erk to Target Gene Induction Reveals Mechanisms of Dynamic and Combinatorial Control. Mol Cell. 2017;67:757–769.e5. doi: 10.1016/j.molcel.2017.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.DeLaughter DM, Bick AG, Wakimoto H, McKean D, Gorham JM, Kathiriya IS, Hinson JT, Homsy J, Gray J, Pu W, et al. Single-Cell Resolution of Temporal Gene Expression during Heart Development. Dev Cell. 2016;39:480–490. doi: 10.1016/j.devcel.2016.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Fletcher RB, Das D, Gadye L, Street KN, Baudhuin A, Wagner A, Cole MB, Flores Q, Choi YG, Yosef N, et al. Deconstructing Olfactory Stem Cell Trajectories at Single-Cell Resolution. Cell Stem Cell. 2017;20:817–830 e8. doi: 10.1016/j.stem.2017.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Trapnell C, Cacchiarelli D, Grimsby J, Pokharel P, Li S, Morse M, Lennon NJ, Livak KJ, Mikkelsen TS, Rinn JL. The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat Biotechnol. 2014;32:381–6. doi: 10.1038/nbt.2859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75•.Llamosi A, Gonzalez-Vargas AM, Versari C, Cinquemani E, Ferrari-Trecate G, Hersen P, Batt G. What Population Reveals about Individual Cell Identity: Single-Cell Parameter Estimation of Models of Gene Expression in Yeast. PLOS Comput Biol. 2016;12:e1004706. doi: 10.1371/journal.pcbi.1004706. In this paper the authors argue agains the idea of a ‘mean’ cell, and instead propose the best representation of single cell gene expression is with a distribution of models. In single yeast cell experiments, they show that the inferred distribution of cell-specific parameters were passed from mother to daughter cells as they divide. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Peters O, Klein W. Ergodicity Breaking in Geometric Brownian Motion. Phys Rev Lett. 2013;110:100603. doi: 10.1103/PhysRevLett.110.100603. [DOI] [PubMed] [Google Scholar]
- 77.Cote AJ, McLeod CM, Farrell MJ, McClanahan PD, Dunagin MC, Raj A, Mauck RL. Single-cell differences in matrix gene expression do not predict matrix deposition. Nat Commun. 2016;7:10865. doi: 10.1038/ncomms10865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Hsu SC, Gilgenast TG, Bartman CR, Edwards CR, Stonestrom AJ, Huang P, Emerson DJ, Evans P, Werner MT, Keller CA, et al. The BET Protein BRD2 Cooperates with CTCF to Enforce Transcriptional and Architectural Boundaries. Mol Cell. 2017;66:102–116.e7. doi: 10.1016/j.molcel.2017.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ku WL, Duggal G, Li Y, Girvan M, Ott E. Interpreting Patterns of Gene Expression: Signatures of Coregulation, the Data Processing Inequality, and Triplet Motifs. PLoS One. 2012;7:e31969. doi: 10.1371/journal.pone.0031969. [DOI] [PMC free article] [PubMed] [Google Scholar]