

Introduction
Microinfarcts are prevalent but tiny ischemic lesions that may contribute to vascular cognitive impairment and dementia (VCID)(Fig. 1A and B).1 They are defined as areas of tissue infarction, often with gliosis and/or cavitation, visible only by examination of the autopsied brain at a microscopic level.2, 3 Numerous autopsy studies have now shown that a greater microinfarct burden is correlated with increased likelihood of cognitive impairment.2, 3 Cerebral microinfarcts are observed post-mortem in the brains of approximately 43% of patients with Alzheimer’s disease, 62% of patients with vascular dementia, and 24% of non-demented elderly subjects.4. However, reported microinfarct numbers are a significant underestimation of total burden, as only a small portion of the brain is examined at routine autopsy.1 Indeed, they can number in the hundreds to thousands within a single brain. Microinfarcts can arise from a variety of etiologies, including cerebral small vessel disease, large vessel disease, cerebral hypoperfusion, and cardiac disease, but their role in the pathogenesis of VCID remains poorly understood.3, 5–7
Figure 1. Human microinfarcts and microhemorrhages in CAA cases.
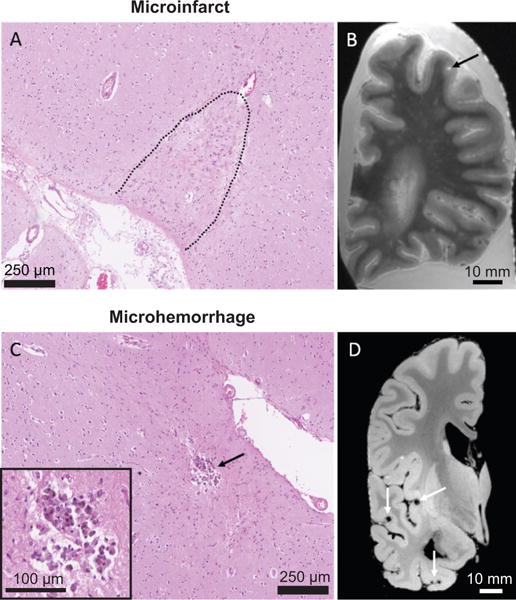
(A) A cortical microinfarct on a Hematoxylin & Eosin-stained section. (B) Microinfarct (black arrow) on a T2-weighted ex vivo MRI scan. (C) A cortical microhemorrhage on a Hematoxylin & Eosin-stained section. (D) Multiple lobar microbleeds (white arrows) on a GRE ex vivo MRI scan.
Microhemorrhages are microscopic bleeds caused by rupture of cerebral microvessels, generating lesions on a similar scale as microinfarcts (Fig. 1C and D).5 Pathologically, old microhemorrhages are defined as focal depositions of iron-positive hemosiderin-containing macrophages. Unlike microinfarcts, microhemorrhages easily escape detection upon neuropathological examination, suggesting that they are not as widespread as microinfarcts. However, they can be detected with high sensitivity using in vivo MRI.5, 6 The two most common etiologies of age-related microhemorrhages are hypertensive arteriopathy and cerebral amyloid angiopathy (CAA). Microhemorrhages are associated with higher likelihood of dementia, and like microinfarcts, their role in VCID remains incompletely understood.7
Clinical studies have emphasized the need to better understand microinfarcts and microhemorrhages (microlesions) because their widespread nature and remote effects may cause broad disruption of brain function. However, it is challenging to measure their functional impact in the human brain because their onset times and locations are unpredictable. Further, microlesions often co-exist with other disease processes, making it difficult to isolate their specific contribution to brain dysfunction. Animal models that allow microlesions to be re-created in a more controlled environment are therefore valuable for understanding their impact on brain function. The purpose of this review is to collate existing rodent models of both microinfarcts and microhemorrhages.
Lesion size criterion
Microinfarcts and microhemorrhages are thought to arise from the occlusion or rupture of small parenchymal arterioles, such as penetrating arterioles and their smaller branches. We define microinfarcts in rodent models as lesions with sizes that could only arise from the occlusion of single penetrating arterioles or their downstream branches. Microinfarcts are typically no larger than 1 mm in diameter in the mouse and rat cortex. We also apply this size criteria to microinfarcts in deeper brain structures, though the relationship between vascular architecture and microinfarcts beyond cortex remains understudied. Microhemorrhages induced in cortex, and occurring spontaneously in rodent models, appear to be ~ 200–300 μm or smaller at histopathology. We have adhered to this size range in our review of the literature. We further note that the term ‘microhemorrhage’ is used for histologically-verified bleeds, and ‘microbleeds’ for the MRI-visible correlate of microhemorrhages. Supplemental information for defining microinfarcts and microhemorrhages in rodent tissues is provided online (Supplemental materials).
1) Models of induced microinfarcts
a. Intra-carotid injection of microemboli
One method of generating cerebral microinfarcts involves the injection of microemboli into the blood circulation, such as occlusive microbeads8, 9 or cholesterol crystals10, 11 (Fig. 2, left side). Injections are typically made through the internal carotid artery. This produces broadly distributed microinfarcts, with cortex, hippocampus, and thalamus being major sites of accrual.8, 10 A spectrum of microinfarct types are seen, including wedge or column-shaped lesions in the cerebral cortex that are continuous with the pial surface (Fig. 3A), as well as smaller circumscribed microinfarcts contained within deeper cortical layers. The choice of microembolus size, type, and/or number injected is important to achieve consistency of microinfarct formation.
Figure 2. Models of induced microinfarct and microhemorrhage.
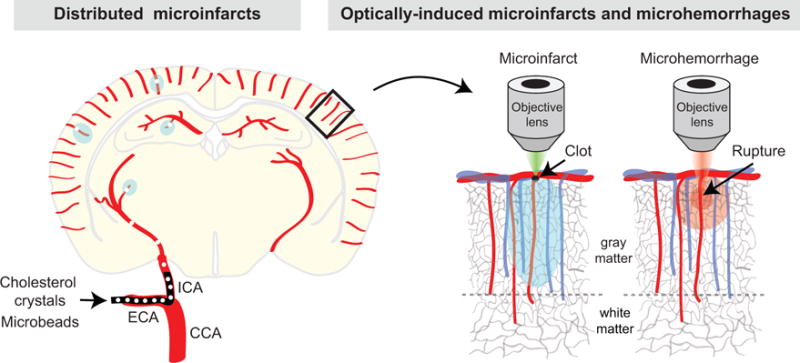
The left hemisphere depicts the production of distributed microinfarcts by injecting microemboli through the internal carotid artery (ICA). CCA = common carotid artery, and ECA = external carotid artery. The right hemisphere shows the selective occlusion of a penetrating arteriole with focal photothrombosis or rupture of a penetrating arteriole with an amplified laser during in vivo optical imaging.
Figure 3. Microinfarcts in rodent models.
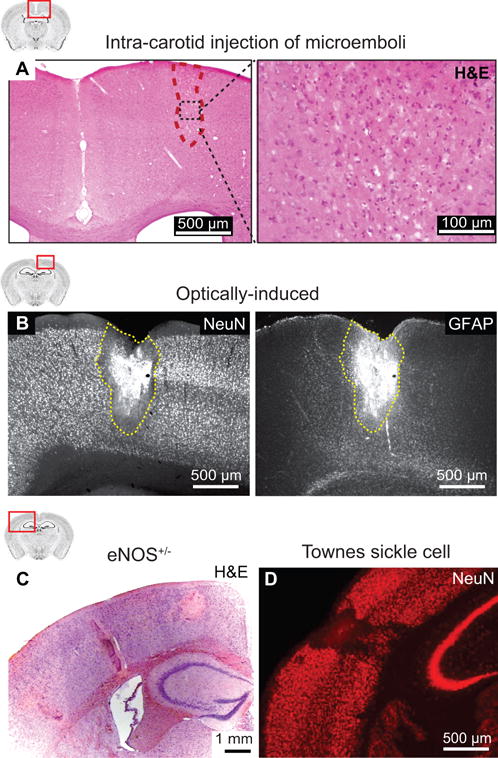
(A) A cortical microinfarct observed in Hematoxylin & Eosin stained mouse brain sections after injection of cholesterol crystals into the internal carotid artery. From Wang et al.10 (B) A cortical microinfarct observed in NeuN and GFAP immunostained rat brain sections after occlusion of a single cortical penetrating arteriole by focal photothrombosis. From Shih et al.12 (C) Spontaneous microinfarcts observed in Hematoxylin & Eosin stained brain sections from an 18 months old eNOS-deficient mouse. From Tan et al.13 (D) Spontaneous microinfarcts observed in NeuN immunostained brain sections from a 13 months old Townes sickle cell mouse. From Hyacinth et al.14
b. Laser-induced occlusion of penetrating arterioles
A second method allows reproducible targeting of microinfarct location and size in rodent cortex (Fig. 2, right side).12 Typically coupled with in vivo two-photon imaging, single penetrating arterioles (or small pial arterioles that support flow to penetrating arterioles) are selectively occluded by inducing clots with precise laser irradiation. This is achieved most commonly by activating circulating photosensitizing agents with focused green lasers, i.e., focal photothrombosis.12, 15, 16 Occlusions can also be made without photosensitizer by using amplified femtosecond laser ablation17, or targeted irradiation with higher laser powers from conventional two-photon imaging lasers18. Targeted penetrating arteriole occlusions primarily generate wedge or column-shaped cortical microinfarcts because clots are made in vessels at or near the pial surface (Fig. 3B).
2) Models with spontaneously occurring microinfarcts
a. Bilateral carotid artery stenosis
Bilateral carotid artery stenosis (BCAS) is a common manipulation to induce chronic cerebral hypoperfusion in rodents. Normal C57Bl/6 mice develop subcortical microinfarcts following long periods of BCAS (6 months)19, but not following shorter periods (2-3 months).20 Two groups recently examined the effects of BCAS in the Tg-SwDI mouse model, which develops early and pronounced CAA.21, 22 Interestingly, only 2-3 months of BCAS was necessary to induce microinfarcts in these mice.23 Microinfarcts were observed in the cerebral cortex and hippocampus, and were related to CAA severity, whereas no microinfarcts were seen in age-matched sham operated Tg-SwDI mice. Further, a recent study showed that atherosclerotic Apoe knockout mice develop microinfarcts within 1.5 months after BCAS.24 Thus, prolonged cerebral hypoperfusion itself can cause microinfarcts, but this effect is exacerbated in transgenic mice with existing cerebrovascular disease.
b. Obesity and diabetes
Spontaneous microinfarcts were observed in a mouse model that crossed the Aβ overexpressing mouse line, APP/PS1, with the db/db mouse model for diabetes.25 The db/db line harbors a mutation of the diabetes (db) gene that leads to a leptin signaling defect, causing severe obesity, hypertension, and type 2 diabetes with hyperglycemia.26 The progeny of the APP/PS1-db/db cross retained features of parental lines, but the combined risk factors led to cortical microinfarcts that were not observed in either parental strain. Microinfarcts appeared as small cystic cavities in various layers of cortex. The authors suspected that aberrant angiogenesis, unique to the crossed mice, led to immature and leaky microvessels that were prone to occlusion.
c. Endothelial NOS deficient mice
Endothelial nitric oxide synthase (eNOS) is critical for regulation of vascular tone and blood pressure. A recent study showed that mice with partial deletion of endothelial nitric oxide synthase (eNOS+/−) develop microinfarcts in cortex, and to a lesser extent, in the hippocampus and thalamus (Fig. 3C).13 Cortical microinfarcts accrued in watershed regions between the perfusion territories of major cerebral arteries.27 They were noticeable by 6 months of age, but were most prevalent at 12 to 18 months. This was accompanied by microvascular pathology, including intravascular clots, diffuse CAA, neuroinflammation and blood-brain barrier disruption. Microinfarcts in eNOS+/− mice was postulated to result from small vessel thrombosis due to endothelial and platelet dysfunction.13
d. Notch3 mutant mice
Microinfarcts (and microhemorrhages) have been reported in a mouse model of cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL).28 CADASIL is a hereditary form of vascular dementia caused by mutations on the gene for Notch3, a transmembrane receptor critical for mural cell-endothelial communication and vascular development. Mice with an Arg170Cys (R170C) mutation knocked into the endogenous Notch3 gene (a prevalent substitution mutation seen in human CADASIL) developed cerebrovascular pathology akin to that seen in human CADASIL. The authors reported microinfarcts in the motor cortex of 20 months old mice, which appeared as small cystic cavities in deeper cortical layers. In contrast, another CADASIL mouse line (PAC-Notch3R169C) carrying rat Notch3 with an Arg169Cys mutation exhibited cerebral hypoperfusion and isolated white matter lesions, but no microinfarcts.29 For reasons still unclear, other mouse lines with various Notch3 mutations develop arteriopathy, but do not exhibit ischemic or hemorrhagic lesions.28, 30
e. Sickle cell mice
Cerebrovascular disease is a well established complication of sickle cell disease (SCD). About 40% of children with SCD develop small “silent” cerebral infarcts, with some falling in the size range of microinfarcts.31 Recently, spontaneous cortical microinfarcts were reported in the Townes model of SCD (Fig. 3D)14, a model that involves replacement of murine β-globin gene with the human sickle β-globin and human γ-globin genes.32 Aged sickle cell mice (13 months old) exhibited faster capillary flow velocities and altered microvascular topology, akin to that described in humans with SCD who were at high risk for stroke.14 Spontaneous cortical microinfarcts in SCD mice were larger and more frequent, and were associated with blood-brain barrier leakage and local tissue hypoxia, suggesting vascular pathology as an origin.
3) Models of induced microhemorrhage
a. Laser-induced rupture of parenchymal vessels
Microhemorrhages can be induced with high spatiotemporal precision in rodent cortex by directly rupturing cortical microvessels using focused lasers (Fig. 4A).33, 34 Much like optically-induced microinfarcts, this model requires implantation of a cranial window and a two-photon microscope to visualize and ablate the desired microvessel. An amplified femtosecond laser is used to damage the wall of target vessels, such as penetrating arterioles34 or capillaries.33 In vivo two-photon imaging of laser-induced microhemorrhages shows rapid extravasation of blood cells forming a lesion core roughly ~100 μm in diameter and broad dissipation of blood plasma over a region ~5-times larger than the core.34
Figure 4. Microhemorrhages in mouse models.
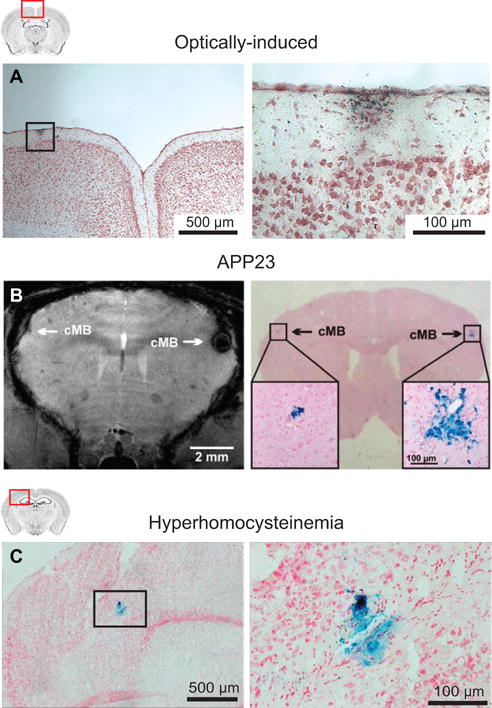
(A) A cortical microhemorrhage observed in Prussian blue stained mouse brain sections after optically-induced rupture of a single cortical penetrating arteriole. From Rosidi et al.34 (B) Microbleeds detected by T2*-weighted MRI (left) and corresponding microhemorrhages detected by Prussian blue (right) in an APP23 mouse. From Reuter et al.35 (C) Microhemorrhages detected by Prussian blue staining in a mouse that received a specialized diet to induce hyperhomocysteinemia. From Sudduth et al.36
4) Models with spontaneous microhemorrhages
a. CAA models
A variety of APP overexpressing mouse lines develop CAA. These models include the PDAPP37, Tg257638, double transgenic APP/PS139, 40, triple transgenic Tg-SwDI41, and APP2342 mouse lines. In these mice, Aβ plaque load as well as CAA increases gradually in an age-dependent fashion, with some model-specific variation. However, reports of spontaneous microhemorrhages (or microinfarcts) have been sparse, and only described in lines that develop severe CAA during old age or after a ‘second hit’.
The most commonly described observations of spontaneous microhemorrhages occur in APP23 mice.43 Cerebral microbleeds could be observed with in vivo T2*-weighted MRI in APP23 mice starting around 16 months of age35, 44–46 Microbleed number and volume increased with animal age. Post-mortem analyses revealed that these MRI-observed microbleeds were true microhemorrhages on corresponding histopathological Prussian blue stained sections (Fig. 4B). Parenchymal microvessels in these mice show severe vascular pathology, including smooth muscle cell degeneration and aneurysm-like vasodilation.43
The APPDutch mouse model bears the mutation (E22Q-mutated Aβ) that causes hereditary cerebral hemorrhage with amyloidosis-Dutch type (HCHWA-D)47, a rare autosomal dominant disorder in humans characterized by early-onset severe CAA and multiple recurrent lobar hemorrhages.48 Interestingly, when APP23 mice are crossed with APPDutch mice, twice as many microhemorrhages arise compared to the APP23 genotype alone.49 Further exacerbation of CAA was observed in crossed mice, which may explain the increased incidence of microhemorrhage.
b. Hypertension models
A widely-used model of severe hypertension is the inbred strain stroke-prone spontaneously hypertensive rat (SHRSP).50 Early characterization studies showed that these rats developed spontaneous ischemic lesions and hemorrhages around 9-12 months of age. Microhemorrhages (and some microinfarcts) co-existed with larger ischemic or hemorrhagic strokes.51, 52 Fibrinoid necrosis and thickening of the vascular walls was regularly observed with cerebral penetrating arterioles, which likely contributes to vascular occlusions and ruptures in these animals.53, 54 Abnormal vascular remodeling may also generate weakened microvessels leading to microhemorrhage.51
Hypertension-induced microhemorrhages in mice require combining transgenic lines with treatments to chronically increase vascular tone. One study used a transgenic mouse line expressing both the human renin and angiotensinogen genes (R+/A+), which develop chronic hypertension but are otherwise normal.55 Challenging these hypertensive mice with a high salt diet and L-NAME (an inhibitor of neuronal and endothelial NOS) led to formation of microhemorrhages in multiple brain regions, including brain stem, cerebellum and basal ganglia. Another study administered chronic Angiotensin II and L-NAME to aged Tg2576 mice, and reported the development of more microhemorrhages compared to mice receiving vehicle.56
c. Hyperhomocysteinemia model
Hyperhomocysteinemia (HHcy) is a risk factor for stroke and Alzheimer’s disease. In diet-induced HHcy, mice are placed on a diet deficient in folate, vitamin B6, and B12 and supplemented with excess methionine.36 Treated mice developed microhemorrhages, visualized by in vivo MRI and Prussian blue-staining of brain sections post-mortem (Fig. 4C).36 When HHcy was induced in APP/PS1 mice, significantly more microhemorrhages where observed in transgenics compared to their wild-type littermates.57 This increase was believed to be mediated by a heightened CAA and activation of matrix metalloproteinase-9 at the cerebrovascular wall.
Model selection: Advantages and disadvantages
We have collated the models discussed in this review in Table 1. Inducible models allow one to ask how a microlesion affects local brain activity or structure, independent of other disease factors. Laser-induced microlesions are limited to cortex, but provide exquisite control over the location and timing of their onset. In a complementary fashion, intra-carotid injection of microemboli provide less control over lesion location, but produced distributed microinfarcts that cause measurable deficits in common tests of cognitive function.
Table 1.
Summary of animal models.
| Microinfarcts | ||||||
|---|---|---|---|---|---|---|
| Model | Model Type | Brain location | Lesion size reported/depicted | Lesion number reported; Consistency | Animal age | Ref. |
| Injected microemboli | Induced | Cortex, hippocampus, striatum, thalamus, corpus callosum | ~100-500 μm
diameter (~0.3 mm2 area) |
Numerous microinfarcts per injection; Very consistent | Any age | 8, 10 |
| Laser-induced | Induced | Cortex | ~500 μm
diameter (~0.2 mm3 volume) |
One microinfarct per microvessel occlusion; Very consistent | Any age | 12, 58 |
| BCAS (C57Bl/6) | Spontaneous | Cortex and thalamus | Wide range of sizes (~0.5 mm3 volume) |
<10 microinfarcts per brain; ~50% of mice examined | 3 months start plus 6 months of BCAS | 19 |
| BCAS (TgSwDI) | Spontaneous | Cortex and hippocampus | ~100-300 μm
diameter (0.01 to 0.03 mm2 area) |
1-10 microinfarcts per brain; ~25-40% of mice examined | 4-8 months start plus 2-3 months of BCAS | 22, 23 |
| BCAS (Apoe−/−) | Spontaneous | Hippocampus | ~200 μm diameter | Multiple microinfarcts per brain; ~75% of mice examined | 3 months start plus 1.5 months of BCAS | 24 |
| APP/PS1 × db | Spontaneous | Cortex | ~500 μm diameter | Multiple microinfarcts per brain; ~75% of mice examined | 7 months | 25 |
| eNOS+/− | Spontaneous | Cortex, hippocampus, thalamus | ~100-500 μm diameter | Multiple microinfarcts per brain; Consistency unspecified | 12, 18 months | 13 |
| Notch3 mutant (R170C) | Spontaneous | Cortex (motor) | ~50-100 μm in diameter | Number per brain not specified; 12% of mice examined | 20-22 months | 28 |
| Townes sickle cell mouse | Spontaneous | Cortex | ~100-500 μm
diameter (~0.05 mm2 area) |
~5 microinfarcts over 20 sections per brain; 100% of mice examined | 13 months | 14 |
| Microhemorrhages | ||||||
| Laser-induced | Induced | Cortex | ~100 μm diameter (hemorrhage core) | 1 microhemorrhage per rupture; Very consistent | Any age | 33, 34 |
| APP23 | Spontaneous | Cortex, thalamus | ~50-300 μm diameter | 15-30 microbleeds per brain; Very consistent | 20-28 months | 35, 43–46 |
| BCAS (TgSwDI) | Spontaneous | Thalamus | Not depicted | Very few microhemorrhages per brain; 30% of mice examined | 4-8 months | 22, 23 |
| BCAS (C57Bl/6) | Spontaneous | Thalamus | ~150 μm diameter, but wide range reported | Multiple microhemorrhages per brain; ~75% of mice examined | 6 months | 19 |
| eNOS+/− | Spontaneous | Cortex | ~25 μm diameter | ~8 microhemorrhages over 9-10 sections per brain; Consistency unspecified | 18 months | 13 |
| Notch3 mutant (R170C) | Spontaneous | Cortex | ~25 μm diameter | Number per brain not specified; 12% of mice examined | 20-22 months | 28 |
| SPSHR | Spontaneous | Cortex, hippocampus, basal ganglia, corpus callosum | ~50-150 μm diameter | Multiple microhemorrhages per brain; ~60% of rats examined | 8-12 months | 52 |
| R+/A+ (high salt and L-NAME) | Spontaneous | Brain stem, cerebellum, basal ganglia | ~50-150 μm diameter | Multiple microhemorrhages per brain; Very consistent | ~11 months start plus 6 month treatment | 55 |
| Tg2576 (AngII and L-NAME) | Spontaneous | Cortex and hippocampus | <100 μm in diameter | Multiple microhemorrhages per brain; Very consistent | ~15 months start plus 1 week treatment | 56 |
| HHcy (C57Bl/6) | Spontaneous | Cortex and hippocampus | ~100 μm diameter | ~2-3 microhemorrhages per section; Consistency unspecified | 7 months start plus 4 months diet | 36 |
| HHcy (APP/PS1) | Spontaneous | Cortex and hippocampus | ~100 μm diameter | ~4 microhemorrhages per section; Consistency unspecified | 12 months start plus 6 months diet | 57 |
Models developing spontaneous microinfarct or microhemorrhage provide an opportunity to understand the vascular deficiencies that lead to lesion formation, and to identify potential targets for prevention. A wide variety of model types develop spontaneous microinfarcts and microhemorrhages, and this supports the idea that diverse disease processes can be contributing factors, including CAA, mural cell or endothelial cell dysfunction, cerebral hypoperfusion, and vascular inflammation.
Physiologic impact of induced microinfarcts and microhemorrhages
In vivo optical imaging studies have revealed that microinfarcts induce lasting neural and hemodynamic deficits in surrounding tissues.59, 60 When microinfarcts were photothrombotically induced in the cortices of APP/PS1 or Tg2576 mice, increase in Aβ plaque formation was seen in surrounding tissues.61, 62 This effect was attributed to impaired drainage of interstitial fluid. Indeed, two recent studies reported that distributed microinfarcts produced bi-hemispheric disruption of the brain’s glymphatic system.63, 64 Further, consistent with having a large effect on brain function, distributed microinfarcts also lead to deficits in cognitive tasks, despite total microinfarct volume being small compared to overall brain volume.10, 11
Surrounding the microinfarct core, neurons are viable but there is atrophy of neuronal dendrites, reduced dendritic spine density,59 axonal damage, and myelin loss.8, 10, 64 This is consistent with recent histopathological findings from human microinfarcts.65 Extensive peri-lesional astrogliosis, mislocalization of aquaporin 4 and blood-brain barrier disruption is also observed, indicating neuroinflammation and altered neuronal-glial signaling.10, 12, 59 Collectively, these findings support the idea that microinfarcts impair the function of tissues well beyond their restricted lesion cores.
The existing data on experimentally-induced microhemorrhages suggest that these lesions also produce neural deficits in surrounding tissues, but very transiently. In vivo calcium imaging revealed impaired neuronal responses up to 150 μm from the lesion core, but tissue function recovered within 1 day.66 However, microhemorrhages induced persistent peri-lesional microgliosis and astrogliosis.34 Penetrating arterioles often remained flowing even after rupture34, which suggests that local ischemia (as seen with microinfarcts) is necessary to induce neural deficits distant to the lesion core.
Summary and future directions
Clinical efforts are now focused on understanding the causes, risk factors, and functional effects of microinfarcts and microhemorrhages in VCID.3, 5 However, these microlesions can be difficult to study in humans due to their small size, unpredictable onset, and co-existence with other disease factors. Preclinical studies can complement these clinical efforts by providing insight into the impact and etiology of microlesions. This review has shown that microlesions similar those seen in humans can be reliably induced through microvascular occlusion and manipulation. Further, microlesions develop spontaneously in a variety of genetic and dietary-induced models of cerebrovascular disease. Future studies could use high-field MR imaging and white matter tractography to understand how microlesion accrual affects white matter integrity and brain connectivity. This addresses the possibility that individually small, but broadly distributed microinfarcts can impair brain function on a global scale. Mechanistic studies can also be performed to understand the cellular/molecular changes occurring beyond the lesion core. Technologies such as in vivo multiphoton imaging allow direct visualization of local neuronal, glial, vascular and glymphatic changes in tissues affected by ischemic injury. Further, longitudinal imaging of models that develop spontaneous microlesions can shed light on disease etiology, by identifying changes to the neurovascular unit that could account for narrowing or weakening brain microvessels. Finally, animal models serve as test beds for therapeutics. A small number of past studies have shown that microinfarct volume can be extensively reduced by neuroprotectants12, 18, suggesting a relatively large penumbra and window for therapeutic intervention10. Thus, neuroprotective strategies previously considered for large ischemic stroke, may be worth re-evaluating as preventative therapies for smaller ischemic events.
Supplementary Material
Acknowledgments
We would like to thank Brian Bacskai for critical reading of the manuscript and helpful discussions.
Funding: A.Y.S. is supported by NIH-NINDS (NS085402, NS096997, NS097775), NIH-NIGMS (P20GM109040), National Science Foundation (1539034), the Dana Foundation, the American Heart Association (14GRNT20480366), Alzheimer’s Association, and the Charleston Conference on Alzheimer’s Disease. H.I.H. is supported by NIH-NHLBI (U01HL117721, R01HL138423) and an Emory University Pilot/HeRO Award. D.A.H. is supported by awards from NIH (T32 GM08716), NIH-NCATS (UL1 TR001450, TL1 TR001451), and NIH-NINDS (F30NS096868). S.J.v.V. is supported by a Rubicon grant from the Netherlands Organization for Scientific Research (019.153LW.014).
Footnotes
Note added in proof. During proofing stages, an article was published describing occlusion of single cortical penetrating arterioles using focal application of FeCl3 from a micropipette (Donmez-Demir et al. Brain Research, 1679:84-90, 2018).
Disclosure/Conflict of Interest. The authors declare no conflicts of interest.
References
- 1.Westover MB, Bianchi MT, Yang C, Schneider JA, Greenberg SM. Estimating cerebral microinfarct burden from autopsy samples. Neurology. 2013;80:1365–1369. doi: 10.1212/WNL.0b013e31828c2f52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Smith EE, Schneider JA, Wardlaw JM, Greenberg SM. Cerebral microinfarcts: The invisible lesions. Lancet Neurology. 2012;11:272–282. doi: 10.1016/S1474-4422(11)70307-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.van Veluw SJ, Shih AY, Smith EE, Chen C, Schneider JA, Wardlaw JM, et al. Detection, risk factors, and functional consequences of cerebral microinfarcts. Lancet Neurology. 2017;16:730–740. doi: 10.1016/S1474-4422(17)30196-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brundel M, de Bresser J, van Dillen JJ, Kappelle LJ, Biessels GJ. Cerebral microinfarcts: A systematic review of neuropathological studies. Journal of Cerebral Blood Flow & Metabolism. 2012;32:425–436. doi: 10.1038/jcbfm.2011.200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Greenberg SM, Vernooij MW, Cordonnier C, Viswanathan A, Al-Shahi Salman R, Warach S, et al. Cerebral microbleeds: A guide to detection and interpretation. Lancet Neurology. 2009;8:165–174. doi: 10.1016/S1474-4422(09)70013-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.van Veluw SJ, Charidimou A, van der Kouwe AJ, Lauer A, Reijmer YD, Costantino I, et al. Microbleed and microinfarct detection in amyloid angiopathy: A high-resolution mri-histopathology study. Brain. 2016;139:3151–3162. doi: 10.1093/brain/aww229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cordonnier C, van der Flier WM, Sluimer JD, Leys D, Barkhof F, Scheltens P. Prevalence and severity of microbleeds in a memory clinic setting. Neurology. 2006;66:1356–1360. doi: 10.1212/01.wnl.0000210535.20297.ae. [DOI] [PubMed] [Google Scholar]
- 8.Silasi G, She J, Boyd JD, Xue S, Murphy TH. A mouse model of small-vessel disease that produces brain-wide-identified microocclusions and regionally selective neuronal injury. Journal of Cerebral Blood Flow and Metabolism. 2015;35:734–738. doi: 10.1038/jcbfm.2015.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lam CK, Yoo T, Hiner B, Liu Z, Grutzendler J. Embolus extravasation is an alternative mechanism for cerebral microvascular recanalization. Nature. 2010;465:478–482. doi: 10.1038/nature09001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang M, Iliff JJ, Liao Y, Chen MJ, Shinseki MS, Venkataraman A, et al. Cognitive deficits and delayed neuronal loss in a mouse model of multiple microinfarcts. Journal of Neuroscience. 2012;32:17948–17960. doi: 10.1523/JNEUROSCI.1860-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rapp JH, Pan XM, Neumann M, Hong M, Hollenbeck K, Liu J. Microemboli composed of cholesterol crystals disrupt the blood-brain barrier and reduce cognition. Stroke. 2008;39:2354–2361. doi: 10.1161/STROKEAHA.107.496737. [DOI] [PubMed] [Google Scholar]
- 12.Shih AY, Blinder P, Tsai PS, Friedman B, Stanley G, Lyden PD, et al. The smallest stroke: Occlusion of one penetrating vessel leads to infarction and a cognitive deficit. Nature Neuroscience. 2013;16:55–63. doi: 10.1038/nn.3278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tan XL, Xue YQ, Ma T, Wang X, Li JJ, Lan L, et al. Partial enos deficiency causes spontaneous thrombotic cerebral infarction, amyloid angiopathy and cognitive impairment. Molecular Neurodegeneration. 2015;10:24. doi: 10.1186/s13024-015-0020-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hyacinth HI, Sugihara CL, Spencer TL, Archer DR, Shih AY. Higher prevalence of spontaneous cerebral vasculopathy and cerebral infarcts in a mouse model of sickle cell disease. Journal of Cerebral Blood Flow & Metabolism. 2017 doi: 10.1177/0271678X17732275. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Blinder P, Shih AY, Rafie CA, Kleinfeld D. Topological basis for the robust distribution of blood to rodent neocortex. Proceedings of the National Academy of Sciences USA. 2010;107:12670–12675. doi: 10.1073/pnas.1007239107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sigler A, Goroshkov A, Murphy TH. Hardware and methodology for targeting single brain arterioles for photothrombotic stroke on an upright microscope. Journal of Neuroscience Methods. 2008;170:35–44. doi: 10.1016/j.jneumeth.2007.12.015. [DOI] [PubMed] [Google Scholar]
- 17.Nishimura N, Rosidi NL, Iadecola C, Schaffer CB. Limitations of collateral flow after occlusion of a single cortical penetrating arteriole. Journal of Cerebral Blood Flow & Metabolism. 2010;30:1914–1927. doi: 10.1038/jcbfm.2010.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang Q, Lan Y, He XF, Luo CM, Wang QM, Liang FY, et al. Allopurinol protects against ischemic insults in a mouse model of cortical microinfarction. Brain research. 2015;1622:361–367. doi: 10.1016/j.brainres.2015.07.010. [DOI] [PubMed] [Google Scholar]
- 19.Holland PR, Searcy JL, Salvadores N, Scullion G, Chen G, Lawson G, et al. Gliovascular disruption and cognitive deficits in a mouse model with features of small vessel disease. Journal of Cerebral Blood Flow & Metabolism. 2015;35:1005–1014. doi: 10.1038/jcbfm.2015.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shibata M, Yamasaki N, Miyakawa T, Kalaria RN, Fujita Y, Ohtani R, et al. Selective impairment of working memory in a mouse model of chronic cerebral hypoperfusion. Stroke. 2007;38:2826–2832. doi: 10.1161/STROKEAHA.107.490151. [DOI] [PubMed] [Google Scholar]
- 21.Davis J, Xu F, Deane R, Romanov G, Previti ML, Zeigler K, et al. Early-onset and robust cerebral microvascular accumulation of amyloid beta-protein in transgenic mice expressing low levels of a vasculotropic dutch/iowa mutant form of amyloid beta-protein precursor. Journal of Biological Chemistry. 2004;279:20296–20306. doi: 10.1074/jbc.M312946200. [DOI] [PubMed] [Google Scholar]
- 22.Salvadores N, Searcy JL, Holland PR, Horsburgh K. Chronic cerebral hypoperfusion alters amyloid-β peptide pools leading to cerebral amyloid angiopathy, microinfarcts and haemorrhages in tg-swdi mice. Clinical science. 2017;131:2109–2123. doi: 10.1042/CS20170962. [DOI] [PubMed] [Google Scholar]
- 23.Okamoto Y, Yamamoto T, Kalaria RN, Senzaki H, Maki T, Hase Y, et al. Cerebral hypoperfusion accelerates cerebral amyloid angiopathy and promotes cortical microinfarcts. Acta Neuropathologica. 2012;123:381–394. doi: 10.1007/s00401-011-0925-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lee ES, Yoon JH, Choi J, Andika FR, Lee T, Jeong Y. A mouse model of subcortical vascular dementia reflecting degeneration of cerebral white matter and microcirculation. Journal of Cerebral Blood Flow & Metabolism. 2017 doi: 10.1177/0271678X17736963. [published online ahead of print October 20, 2017]. http://journals.sagepub.com/doi/pdf/10.1177/0271678X17736963 Accessed December 17, 2017. [DOI] [PMC free article] [PubMed]
- 25.Niedowicz DM, Reeves VL, Platt TL, Kohler K, Beckett TL, Powell DK, et al. Obesity and diabetes cause cognitive dysfunction in the absence of accelerated β-amyloid deposition in a novel murine model of mixed or vascular dementia. Acta Neuropathologica Communications. 2014;2:64. doi: 10.1186/2051-5960-2-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Coleman DL. Obese and diabetes: Two mutant genes causing diabetes-obesity syndromes in mice. Diabetologia. 1978;14:141–148. doi: 10.1007/BF00429772. [DOI] [PubMed] [Google Scholar]
- 27.Suter OC, Sunthorn T, Kraftsik R, Straubel J, Darekar P, Khalili K, et al. Cerebral hypoperfusion generates cortical watershed microinfarcts in alzheimer disease. Stroke. 2002;33:1986–1992. doi: 10.1161/01.str.0000024523.82311.77. [DOI] [PubMed] [Google Scholar]
- 28.Wallays G, Nuyens D, Silasi-Mansat R, Souffreau J, Callaerts-Vegh Z, Van Nuffelen A, et al. Notch3 arg170cys knock-in mice display pathologic and clinical features of the neurovascular disorder cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy. Arteriosclerosis, thrombosis, and vascular biology. 2011;31:2881–2888. doi: 10.1161/ATVBAHA.111.237859. [DOI] [PubMed] [Google Scholar]
- 29.Joutel A, Monet-Leprêtre M, Gosele C, Baron-Menguy C, Hammes A, Schmidt S, et al. Cerebrovascular dysfunction and microcirculation rarefaction precede white matter lesions in a mouse genetic model of cerebral ischemic small vessel disease. Journal of Clinical Investigation. 2010;120:433–445. doi: 10.1172/JCI39733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Joutel A. Pathogenesis of cadasil: Transgenic and knock-out mice to probe function and dysfunction of the mutated gene, notch3, in the cerebrovasculature. Bioessays. 2011;33:73–80. doi: 10.1002/bies.201000093. [DOI] [PubMed] [Google Scholar]
- 31.DeBaun MR, Armstrong FD, McKinstry RC, Ware RE, Vichinsky E, Kirkham FJ. Silent cerebral infarcts: A review on a prevalent and progressive cause of neurologic injury in sickle cell anemia. Blood. 2012;119:4587–4596. doi: 10.1182/blood-2011-02-272682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wu LC, Sun CW, Ryan TM, Pawlik KM, Ren J, Townes TM. Correction of sickle cell disease by homologous recombination in embryonic stem cells. Blood. 2006;108:1183–1188. doi: 10.1182/blood-2006-02-004812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nishimura N, Schaffer CB, Friedman B, Tsai PS, Lyden PD, Kleinfeld D. Targeted insult to individual subsurface cortical blood vessels using ultrashort laser pulses: Three models of stroke. Nature Methods. 2006;3:99–108. doi: 10.1038/nmeth844. [DOI] [PubMed] [Google Scholar]
- 34.Rosidi NL, Zhou J, Pattanaik S, Wang P, Jin W, Brophy M, et al. Cortical microhemorrhages cause local inflammation but do not trigger widespread dendrite degeneration. Public Library of Science ONE. 2011;6:e26612. doi: 10.1371/journal.pone.0026612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Reuter B, Venus A, Heiler P, Schad L, Ebert A, Hennerici MG, et al. Development of cerebral microbleeds in the app23-transgenic mouse model of cerebral amyloid angiopathy-a 9.4 tesla mri study. Frontiers in Aging Neuroscience. 2016;8:170. doi: 10.3389/fnagi.2016.00170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sudduth TL, Powell DK, Smith CD, Greenstein A, Wilcock DM. Induction of hyperhomocysteinemia models vascular dementia by induction of cerebral microhemorrhages and neuroinflammation. Journal of Cerebral Blood Flow & Metabolism. 2013;33:708–715. doi: 10.1038/jcbfm.2013.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Games D, Adams D, Alessandrini R, Barbour R, Berthelette P, Blackwell C, et al. Alzheimer-type neuropathology in transgenic mice overexpressing v717f beta-amyloid precursor protein. Nature. 1995;373:523–527. doi: 10.1038/373523a0. [DOI] [PubMed] [Google Scholar]
- 38.Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, et al. Correlative memory deficits, abeta elevation, and amyloid plaques in transgenic mice. Science. 1996;274:99–102. doi: 10.1126/science.274.5284.99. [DOI] [PubMed] [Google Scholar]
- 39.Savonenko A, Xu GM, Melnikova T, Morton JL, Gonzales V, Wong MP, et al. Episodic-like memory deficits in the appswe/ps1de9 mouse model of alzheimer’s disease: Relationships to beta-amyloid deposition and neurotransmitter abnormalities. Neurobiology of Disease. 2005;18:602–617. doi: 10.1016/j.nbd.2004.10.022. [DOI] [PubMed] [Google Scholar]
- 40.Radde R, Bolmont T, Kaeser SA, Coomaraswamy J, Lindau D, Stoltze L, et al. Abeta42-driven cerebral amyloidosis in transgenic mice reveals early and robust pathology. EMBO Reports. 2006;7:940–946. doi: 10.1038/sj.embor.7400784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Davis J, Xu F, Deane R, Romanov G, Previti ML, Zeigler K, et al. Early-onset and robust cerebral microvascular accumulation of amyloid beta-protein in transgenic mice expressing low levels of a vasculotropic dutch/iowa mutant form of amyloid beta-protein precursor. Journal of Biological Chemistry. 2004;279:20296–20306. doi: 10.1074/jbc.M312946200. [DOI] [PubMed] [Google Scholar]
- 42.Sturchler-Pierrat C, Abramowski D, Duke M, Wiederhold KH, Mistl C, Rothacher S, et al. Two amyloid precursor protein transgenic mouse models with alzheimer disease-like pathology. Proceedings of the National Academy of Sciences. 1997;94:13287–13292. doi: 10.1073/pnas.94.24.13287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Winkler DT, Bondolfi L, Herzig MC, Jann L, Calhoun ME, Wiederhold KH, et al. Spontaneous hemorrhagic stroke in a mouse model of cerebral amyloid angiopathy. Journal of Neuroscience. 2001;21:1619–1627. doi: 10.1523/JNEUROSCI.21-05-01619.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Beckmann N, Doelemeyer A, Zurbruegg S, Bigot K, Theil D, Frieauff W, et al. Longitudinal noninvasive magnetic resonance imaging of brain microhemorrhages in bace inhibitor-treated app transgenic mice. Neurobiology of Aging. 2016;45:50–60. doi: 10.1016/j.neurobiolaging.2016.05.009. [DOI] [PubMed] [Google Scholar]
- 45.Marinescu M, Sun L, Fatar M, Neubauer A, Schad L, van Ryn J, et al. Cerebral microbleeds in murine amyloid angiopathy: Natural course and anticoagulant effects. Stroke. 2017;48:2248–2254. doi: 10.1161/STROKEAHA.117.017994. [DOI] [PubMed] [Google Scholar]
- 46.Dudeffant C, Vandesquille M, Herbert K, Garin CM, Alves S, Blanchard V, et al. Contrast-enhanced mr microscopy of amyloid plaques in five mouse models of amyloidosis and in human alzheimer’s disease brains. Scientific Reports. 2017;7:4955. doi: 10.1038/s41598-017-05285-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Herzig MC, Winkler DT, Burgermeister P, Pfeifer M, Kohler E, Schmidt SD, et al. Abeta is targeted to the vasculature in a mouse model of hereditary cerebral hemorrhage with amyloidosis. Nature Neuroscience. 2004;7:954–960. doi: 10.1038/nn1302. [DOI] [PubMed] [Google Scholar]
- 48.Bornebroek M, Haan J, Maat-Schieman ML, Van Duinen SG, Roos RA. Hereditary cerebral hemorrhage with amyloidosis-dutch type (hchwa-d): I–a review of clinical, radiologic and genetic aspects. Brain Pathology. 1996;6:111–114. doi: 10.1111/j.1750-3639.1996.tb00793.x. [DOI] [PubMed] [Google Scholar]
- 49.Herzig MC, Eisele YS, Staufenbiel M, Jucker M. E22q-mutant abeta peptide (abetadutch) increases vascular but reduces parenchymal abeta deposition. American Journal of Pathology. 2009;174:722–726. doi: 10.2353/ajpath.2009.080790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Okamoto K, Yamori Y, Nagaoka A. Establishment of the stroke prone spontaneously hypertensive rat. Circulation Research. 1974;34:143–153. [Google Scholar]
- 51.Henning EC, Warach S, Spatz M. Hypertension-induced vascular remodeling contributes to reduced cerebral perfusion and the development of spontaneous stroke in aged shrsp rats. Journal of Cerebral Blood Flow & Metabolism. 2010;30:827–836. doi: 10.1038/jcbfm.2009.246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Schreiber S, Bueche CZ, Garz C, Kropf S, Angenstein F, Goldschmidt J, et al. The pathologic cascade of cerebrovascular lesions in shrsp: Is erythrocyte accumulation an early phase? Journal of Cerebral Blood Flow & Metabolism. 2012;32:278–290. doi: 10.1038/jcbfm.2011.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cova L, Gelosa P, Mura E, Mauro A, Stramba-Badiale M, Michailidis G, et al. Vascular and parenchymal lesions along with enhanced neurogenesis characterize the brain of asymptomatic stroke-prone spontaneous hypertensive rats. Journal of Hypertension. 2013;31:1618–1628. doi: 10.1097/HJH.0b013e3283619d7f. [DOI] [PubMed] [Google Scholar]
- 54.Fredriksson K, Nordborg C, Kalimo H, Olsson Y, Johansson BB. Cerebral microangiopathy in stroke-prone spontaneously hypertensive rats. An immunohistochemical and ultrastructural study. Acta Neuropathologica. 1988:241–252. doi: 10.1007/BF00690532. [DOI] [PubMed] [Google Scholar]
- 55.Iida S, Baumbach GL, Lavoie JL, Faraci FM, Sigmund CD, Heistad DD. Spontaneous stroke in a genetic model of hypertension in mice. Stroke. 2005;36:1253–1258. doi: 10.1161/01.str.0000167694.58419.a2. [DOI] [PubMed] [Google Scholar]
- 56.Passos GF, Kilday K, Gillen DL, Cribbs DH, Vasilevko V. Experimental hypertension increases spontaneous intracerebral hemorrhages in a mouse model of cerebral amyloidosis. Journal of Cerebral Blood Flow & Metabolism. 2016;36:399–404. doi: 10.1177/0271678X15606720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sudduth TL, Weekman EM, Brothers HM, Braun K, Wilcock DM. B-amyloid deposition is shifted to the vasculature and memory impairment is exacerbated when hyperhomocysteinemia is induced in app/ps1 transgenic mice. Alzheimers Research and Therapy. 2014;6:32. doi: 10.1186/alzrt262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Taylor ZJ, Hui ES, Watson AN, Nie X, Deardorff RL, Jensen JH, et al. Microvascular basis for growth of small infarcts following occlusion of single penetrating arterioles in mouse cortex. Journal of Cerebral Blood Flow & Metabolism. 2016;36:1357–1373. doi: 10.1177/0271678X15608388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Summers PM, Hartmann DA, Hui ES, Nie X, Deardorff RL, McKinnon ET, et al. Functional deficits induced by cortical microinfarcts. Journal of Cerebral Blood Flow & Metabolism. 2017;37:3599–3614. doi: 10.1177/0271678X16685573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Harrison TC, Silasi G, Boyd JD, Murphy TH. Displacement of sensory maps and disorganization of motor cortex after targeted stroke in mice. Stroke. 2013;44:2300–2306. doi: 10.1161/STROKEAHA.113.001272. [DOI] [PubMed] [Google Scholar]
- 61.Garcia-Alloza M, Gregory J, Kuchibhotla KV, Fine S, Wei Y, Ayata C, et al. Cerebrovascular lesions induce transient β-amyloid deposition. Brain. 2011;134:3697–3707. doi: 10.1093/brain/awr300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Arbel-Ornath M, Hudry E, Eikermann-Haerter K, Hou S, Gregory JL, Zhao L, et al. Interstitial fluid drainage is impaired in ischemic stroke and alzheimer’s disease mouse models. Acta Neuropathologica. 2013;126:353–364. doi: 10.1007/s00401-013-1145-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wang M, Ding F, Deng S, Guo X, Wang W, Iliff JJ, et al. Focal solute trapping and global glymphatic pathway impairment in a murine model of multiple microinfarcts. Journal of Neuroscience. 2017;37:2870–2877. doi: 10.1523/JNEUROSCI.2112-16.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Venkat P, Chopp M, Zacharek A, Cui C, Zhang L, Li Q, et al. White matter damage and glymphatic dysfunction in a model of vascular dementia in rats with no prior vascular pathologies. Neurobiology of Aging. 2017;50:96–106. doi: 10.1016/j.neurobiolaging.2016.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Coban H, Tung S, Yoo B, Vinters HV, Hinman JD. Molecular disorganization of axons adjacent to human cortical microinfarcts. Frontiers in Neurology. 2017;8:405. doi: 10.3389/fneur.2017.00405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Cianchetti FA, Kim DH, Dimiduk S, Nishimura N, Schaffer CB. Stimulus-evoked calcium transients in somatosensory cortex are temporarily inhibited by a nearby microhemorrhage. PLoS One. 2013;8:e65663. doi: 10.1371/journal.pone.0065663. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.