

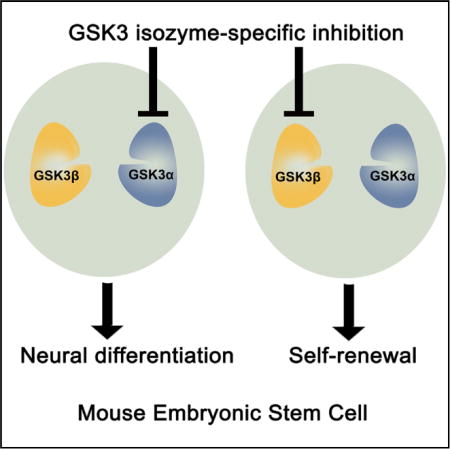
SUMMARY
Glycogen synthase kinase 3 (GSK3) plays a central role in diverse cellular processes. GSK3 has two mammalian isozymes, GSK3α and GSK3β, whose functions remain ill-defined because of a lack of inhibitors that can distinguish between the two highly homologous isozymes. Here, we show that GSK3α and GSK3β can be selectively inhibited in mouse embryonic stem cells (ESCs) using a chemical-genetic approach. Selective inhibition of GSK3β is sufficient to maintain mouse ESC self-renewal, whereas GSK3α inhibition promotes mouse ESC differentiation toward neural lineages. Genome-wide transcriptional analysis reveals that GSK3α and GSK3β have distinct sets of downstream targets. Furthermore, selective inhibition of individual GSK3 isozymes yields distinct phenotypes from gene deletion, highlighting the power of the chemical-genetic approach in dissecting kinase catalytic functions from the protein’s scaffolding functions. Our study opens new avenues for defining GSK3 isozyme-specific functions in various cellular processes.
Graphical abstract
INTRODUCTION
Glycogen synthase kinase 3 (GSK3), a serine/threonine protein kinase, plays a central role in multiple intracellular signaling pathways, including those activated by Wnt/β-catenin, sonic hedgehog, Notch, growth factors/receptor tyrosine kinases, and G-protein-coupled receptors (Beurel et al., 2014; Sutherland, 2011). In mammals, GSK3 is encoded by two paralogous genes, Gsk3α and Gsk3β, which are ~90% identical with respect to the amino acid sequences within their kinase domains. Previous studies have suggested that GSK3α and GSK3β had overlapping functions in regulating the Wnt/β-catenin signaling pathway (Doble et al., 2007). On the other hand, knockout of GSK3α or GSK3β in mice produced distinct phenotypes. While GSK3β knockout resulted in embryonic lethality, GSK3α knockout mice were viable and had only minor defects in hepatic glycogen metabolism (Hoeflich et al., 2000; MacAulay et al., 2007). These results indicate that the two GSK3 isozymes have non-redundant roles during development, the basis of which is not clear, however.
GSK3 is an attractive drug target because dysregulation of GSK3 activity has been implicated in the pathogenesis of diverse diseases, including diabetes, cancer, and neurodegenerative diseases (Cohen and Goedert, 2004; Eldar-Finkelman and Martinez, 2011; Rayasam et al., 2009). However, the role of individual GSK3 isozymes in the pathogenesis of these diseases is poorly defined. Furthermore, no inhibitor is currently available that can well distinguish between the two isozymes (Eldar-Finkelman and Martinez, 2011). These factors might have underlain the limited success in targeting GSK3 for therapeutic purposes. Understanding the mechanism by which GSK3α and GSK3β differentially regulate cellular processes could, in the future, facilitate the development of more specific drugs to target individual GSK3 isozymes for the treatment of various diseases caused by dysregulation of GSK3 activity.
Although gene knockout/knockdown experiments have provided bountiful information regarding the function of individual GSK3 kinases, these approaches not only eliminate catalytic functions but also abolish non-catalytic functions and perturb protein stoichiometry in cells, thereby confounding data analysis and interpretation. Dr. Kevan Shokat and colleagues have developed a chemical-genetic approach that involves genetic modification in the protein of interest and the use of inhibitor analogs that specifically recognize the modified protein to efficiently distinguish between highly homologous proteins (Bishop et al., 2000). This approach is ideal for defining the function of individual GSK3 isozymes due to its acute action, preservation of protein stoichiometry, and ability to distinguish between highly homologous kinases in cells.
Mouse embryonic stem cells (ESCs) can be maintained in culture indefinitely while retaining the capacity to generate any type of cells in the body. Inhibition of GSK3 by pan-GSK3 inhibitor CHIR99021 (CHIR) can activate both self-renewal and differentiation programs, and favors ESC self-renewal only when the mitogen-activated protein kinase kinase (MEK) is simultaneously inhibited (Ying et al., 2008). The roles of individual GSK3 isozymes in ESC self-renewal and differentiation and how GSK3 inhibition produces divergent effects, however, remain largely unknown. The pluripotency of ESCs, and the ease with which they can be genetically manipulated and selected, make these cells a powerful platform for dissecting the functions of individual GSK3 isozymes. Here, we combined the chemical-genetic approach with ESC-based technologies to define the roles of GSK3α and GSK3β in ESC self-renewal and differentiation. Our results reveal unexpectedly that these two GSK3 isozymes play distinct roles in the regulation of ESC fate despite their high homology to each other.
RESULTS
Either GSK3 Isozyme Can Phosphorylate β-Catenin When the Other Is Ablated
To define the roles of individual GSK3 isozymes, we first generated Gsk3α−/−, Gsk3β−/−, and Gsk3α/β double-knockout (DKO) E14TG2a mouse ESC lines via CRISPR/Cas9-mediated gene targeting (Figure 1A). E14TG2a ESCs were derived from the 129 strain of mouse and maintained on gelatin-coated plates in the presence of leukemia-induced factor (LIF) and serum (Smith et al., 1988; Williams et al., 1988). Next, we examined β-catenin activity in these GSK3 mutant ESC lines. β-Catenin is a well-established substrate of GSK3 in the canonical Wnt signaling pathway. GSK3 phosphorylates β-catenin at S33/S37/T41, leading to proteasome-mediated degradation of β-catenin (Kimelman and Xu, 2006; Liu et al., 2002). Inhibition of GSK3 activates canonical Wnt/β-catenin signaling through stabilization and subsequent nuclear translocation of β-catenin, which has been suggested to be the key mechanism underlying ESC self-renewal mediated by GSK3 inhibition (Merrill, 2012). β-Catenin phosphorylation at S33/S37/T41 was absent in Gsk3α/β DKO ESCs (Figure 1A), confirming that GSK3 is essential for phosphorylating β-catenin at these sites. Interestingly, the levels of β-catenin phosphorylation in Gsk3α−/− and Gsk3β−/− ESCs were comparable with that in the wild-type ESCs (Figure 1A), suggesting that either GSK3 isozyme is capable of phosphorylating β-catenin when the other is physically absent. We used the luciferase-based TopFlash reporter assay to measure the Wnt/β-catenin signaling activity (Korinek et al., 1997; Veeman et al., 2003). Addition of CHIR increased TopFlash activity in both Gsk3α−/− and Gsk3β−/− ESCs, but not in Gsk3 DKO ESCs (Figure 1B). Together, these results confirm that either GSK3 isozyme can inhibit canonical Wnt/β-catenin signaling when the other is ablated, which is consistent with a previous report (Doble et al., 2007).
Figure 1. Generation and Characterization of GSK3 Mutant ESC Lines.
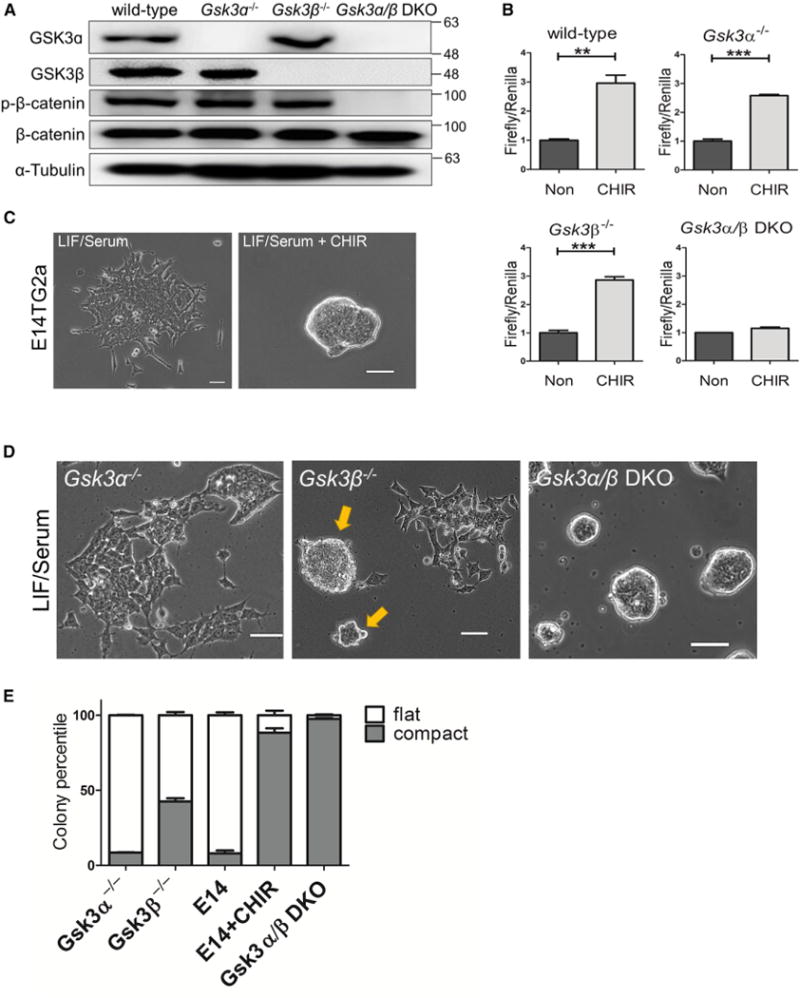
(A) Western blot analysis of Gsk3α−/−, Gsk3β−/−, and Gsk3α/β DKO ESCs. Gsk3α/β DKO ESCs were generated by knocking out Gsk3α in Gsk3β−/− ESCs. β-Catenin phosphorylation at S33/S37/T41 was examined in the indicated ESC lines using a phospho-β-catenin antibody from Cell Signaling Technology (no. 9561, lot 12). The same antibody was used to examine phosphor-β-catenin throughout the study.
(B) TopFlash assay in wild-type and GSK3 mutant ESCs treated with 3 μM CHIR for 8 hr. Non, no treatment control. Data represent means ± SD of three biological replicates. **p < 0.01; ***p < 0.001.
(C) Representative images of E14TG2a ESCs cultured in LIF/serum or LIF/serum + 3 μM CHIR for 5 days. Scale bars, 100 μm.
(D) Representative images of different GSK3 mutant ESC lines cultured in LIF/serum for 5 days. Gsk3β−/− ESCs formed both compact (arrows) and flat colonies. Scale bars, 100 μm.
(E) Quantification of flat and compact colonies formed from the indicated ESC lines. ESCs were plated onto six-well plates at clonal density and cultured in LIF/serum (if not specified) or LIF/serum + 3 μM CHIR (E14 + CHIR) for 7 days. E14, E14TG2a ESCs. Data represent means ± SD of three biological replicates.
Deletion of Either Gsk3α or Gsk3β Is Not Sufficient to Mimic the Effect of CHIR in Promoting ESC Self-Renewal
Next, we examined whether deletion of Gsk3α, Gsk3β, or both can mimic the effect of CHIR in promoting ESC self-renewal. Under LIF/serum condition, E14TG2a ESC colonies displayed a flat morphology, and addition of CHIR resulted in the formation of mainly compact colonies (Figure 1C). Gsk3α−/− ESCs, like the parental E14TG2a ESCs, formed mostly flat colonies in LIF/serum. In contrast, Gsk3β−/− ESCs cultured in LIF/serum displayed a mixture of colonies with both flat and compact morphologies (Figures 1D and 1E). Gsk3α/β DKO ESCs formed uniform compact colonies in LIF/serum, similar to E14TG2a ESCs cultured in LIF/serum plus CHIR (Figures 1C–1E). These results suggest that genetic deletion of neither Gsk3α nor Gsk3β can fully recapitulate the effect of GSK3 inhibition by CHIR. Nonetheless, Gsk3α−/− and Gsk3β−/− ESCs displayed different colony morphologies under the LIF/serum condition, suggesting that GSK3α and GSK3β may function differently in ESCs.
To further explore the roles of individual GSK3 isozymes, we cultured wild-type and GSK3 mutant ESCs in serum-free N2B27 medium supplemented with MEK inhibitor PD0325901 (PD03). Wild-type mouse ESCs cannot be maintained in PD03/N2B27 and addition of CHIR is necessary for ESC self-renewal (Ying et al., 2008). In contrast, Gsk3α/β DKO ESCs could be continuously passaged and remained undifferentiated in PD03/N2B27, and addition of CHIR made no difference, strongly suggesting that CHIR promotes ESC self-renewal through inhibition of GSK3 (Figures 2A and 2B). Gsk3α−/− ESCs differentiated in PD03/N2B27, and CHIR was required in order to maintain their self-renewal (Figure 2C). Gsk3β−/− ESCs cultured in PD03/N2B27 displayed a mixed cell population containing both differentiated and undifferentiated ESCs at the first passage and eventually differentiated after passaging. Interestingly, although all Gsk3β−/− ESCs remained undifferentiated in PD03 + CHIR/N2B27 (2i/N2B27), they proliferated significantly more slowly and formed much smaller colonies compared with E14TG2a and Gsk3α−/− ESCs cultured in the same 2i/N2B27 condition (Figures 2D–2F). These results suggest that, while inhibition of GSK3α is necessary for the self-renewal of Gsk3β−/− ESCs, it might negatively regulate ESC proliferation.
Figure 2. Characterization of GSK3 Mutant ESCs Cultured under Serum-Free N2B27 Condition.
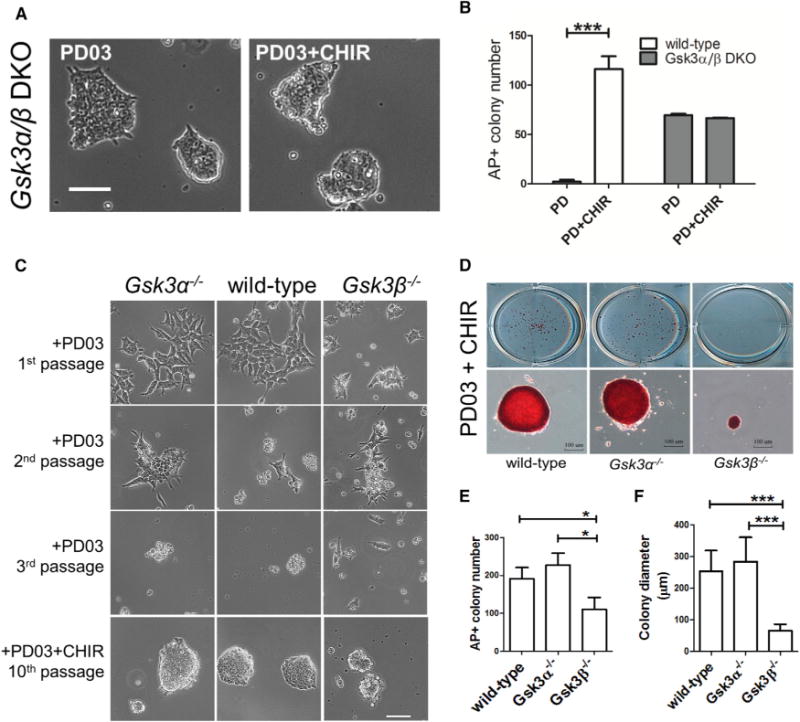
(A) Representative images of Gsk3α/β DKO ESCs cultured in the indicated conditions for five passages. Scale bar, 100 μm.
(B) Quantification of alkaline phosphatase positive (AP+) colonies formed from wild-type and Gsk3α/β DKO ESCs cultured in N2B27 medium supplemented with PD03 or PD03 + CHIR. ESCs were plated onto 12-well plates at a density of 200 cells/well and cultured in the indicated conditions for 7 days before performing AP staining. AP+ is an indicator of undifferentiated ESCs. Data represent means ± SD of three biological replicates. ***p < 0.001.
(C) Representative images of wild-type and GSK3 mutant ESCs cultured in N2B27 medium supplemented with the indicated inhibitors. Scale bar, 100 μm.
(D) Representative images of AP+ colonies formed from the indicated ESC lines cultured in PD03 + CHIR for 7 days. ESCs were plated onto 12-well plates at a density of 500 cells/well and cultured in PD03 + CHIR/N2B27 for 7 days. Scale bars, 100 μm.
(E) Quantification of the numbers of AP+ colonies formed from the indicated ESC lines as shown in (D). For each ESC line, the AP+ colony data represent means ± SD from a total of nine wells in three independent experiments. *p < 0.05.
(F) Quantification of the diameters of AP+ colonies formed from the indicated ESC lines as shown in (D). The diameter of the AP+ colonies was calculated using the image software AxioVision. For each ESC line, the colony diameter data represent means ± SD from a total of 60 randomly selected AP+ colonies. ***p < 0.001.
Selective Chemical Inhibition of GSK3α and GSK3β Is Achieved through a Chemical-Genetic Approach
It is well known that pharmacological inhibition and genetic deletion could result in very different outcomes (Knight and Shokat, 2007). Next, we investigated the effect of chemically inhibiting just one GSK3 isozyme in ESCs when both isozymes are present. Because no existing small molecules can distinguish with high selectivity between GSK3α and GSK3β, we employed a chemical-genetic approach to achieve specific inhibition of individual GSK3 isozymes.
We generated GSK3α-L195G and GSK3β-L132G mutants by substituting a structurally conserved bulky leucine residue at the “gatekeeper” position in the kinase active site with a smaller glycine residue. This mutation creates an expanded pocket within the kinase active site which can then be inhibited by “bumped” inhibitors that contain enlarged groups to occupy the pocket (Bishop et al., 2000). A cell-free kinase activity assay revealed that the kinase activity of GSK3α-L195G and GSK3β-L132G was reduced by 50% ± 6% and 56% ± 9%, respectively, compared with their wild-type counterparts (Figure 3A). This reduction of GSK3 kinase activity upon introduction of the gatekeeper mutations is consistent with previous studies (Chen et al., 2012; Kaasik et al., 2013). The kinase assay also confirmed that bumped inhibitors such as 1NA-PP1, 1NM-PP1, and 3MB-PP1 could specifically inhibit the kinase activity of mutant GSK3α-L195G and GSK3β-L132G but not that of wild-type GSK3α and GSK3β. In contrast, CHIR inhibited both the mutant and wild-type GSK3 kinase activity (Figure 3B). Among the three bumped inhibitors tested, 3MB-PP1 was found to be the most potent and selective inhibitor of GSK3α-L195G and GSK3β-L132G and therefore was used for all the following experiments. The same approach has been previously employed to study GSK3 (Chen et al., 2012; Kaasik et al., 2013), in which a less potent inhibitor 1NA-PP1 was used to selectively inhibit GSK3β.
Figure 3. GSK3α and GSK3β Can Be Selectively Inhibited in ESCs through a Chemical-Genetic Approach.
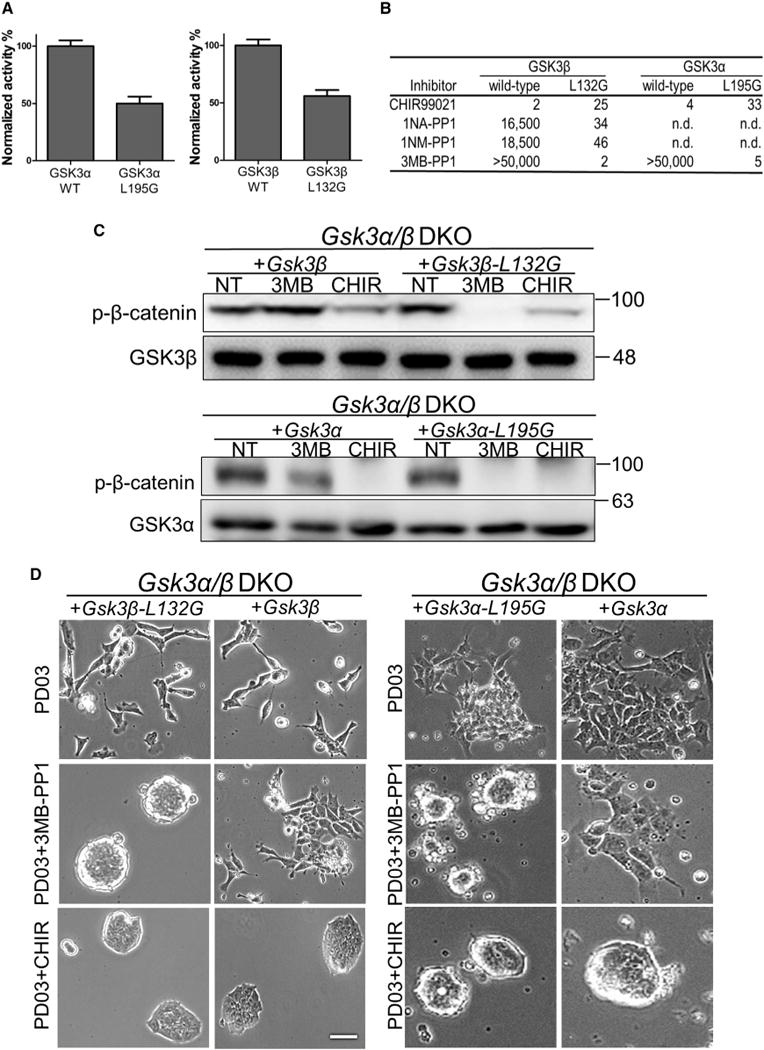
(A) Relative in vitro activity of the GSK3 kinases. Activity of GSK3 kinases (10 nM) was measured using the phosphocellulose disk assay with the resulting counts per minute values normalized to those of the wild-type kinase. Data represent means ± SD of three biological replicates.
(B) Half maximal inhibitory concentration values (nM) of the inhibitors against wild-type and mutant GSK3α/β in vitro. n.d., not determined.
(C) Western blot analysis of β-catenin phosphorylation at S33/S37/T41 in Gsk3α/β DKO ESCs expressing the indicated transgenes. ESCs were treated with 3 μM 3MB-PP1 (3MB) or 3 μM CHIR for 30 min. NT, no treatment control.
(D) Representative images of different GSK3 mutant ESCs cultured in N2B27 supplemented with the indicated inhibitors for 5 days. Scale bar, 100 μm.
Next, we investigated whether the in vivo function of GSK3α-L195G and GSK3β-L132G is compromised by the gatekeeper mutation. To this end, we overexpressed Gsk3α-L195G, Gsk3β-L132G, or their wild-type counterparts in Gsk3α/β DKO ESCs and measured β-catenin phosphorylation at S33/S37/T41. Phosphorylation of β-catenin at these sites was not detectable in Gsk3α/β DKO ESCs (Figure 1A). Introducing Gsk3α-L195G or Gsk3β-L132G restored the phosphorylation of β-catenin to a level similar to that in Gsk3α/β DKO ESCs overexpressing wild-type GSK3 (Figure 3C), suggesting that the mutant GSK3 kinases retain their normal catalytic activity in ESCs. The addition of 3MB-PP1 specifically inhibited the kinase activity of GSK3α-L195G and GSK3β-L132G, but not that of wild-type GSK3, as indicated by the change in β-catenin phosphorylation levels at S33/S37/T41 (Figure 3C). The kinase activity of both mutant and wild-type GSK3 could still be inhibited by CHIR (Figure 3C), in agreement with the results from in vitro kinase assays. Gsk3α/β DKO ESCs expressing GSK3α-L195G or GSK3β-L132G remained undifferentiated in the presence of 3MB-PP1 and PD03 but differentiated in PD03 alone. Gsk3α/β DKO ESCs expressing wild-type GSK3α or GSK3β could be maintained in PD03 + CHIR but not in PD03 + 3MB-PP1 (Figure 3D). Together, these data demonstrate the feasibility of selective chemical inhibition of individual GSK3 isozymes using the chemical-genetic approach.
Chemical Inhibition of GSK3β, but Not GSK3α, Promotes ESC Self-Renewal
Having demonstrated that individual GSK3 isozymes can be specifically inhibited, we next investigated the resulting phenotypes with regard to ESC fate. To this end, we introduced Gsk3α-L195G and Gsk3β-L132G transgenes into Gsk3α−/− and Gsk3β−/− ESCs, respectively. In these cells, the kinase activity of the engineered GSK3, but not the wild-type GSK3, can be specifically inhibited by 3MB-PP1, while CHIR inhibits the activity of both kinases (Figure 4A). Since expression of a transgene beyond physiological levels may affect its cellular function, we chose ESC clones with the transgene expressed at levels similar to that of the endogenous gene for subsequent characterization (Figure S1). Gsk3β−/− + Gsk3β-L132G ESCs could be efficiently expanded while remaining undifferentiated in PD03 plus either CHIR or 3MB-PP1. In contrast, Gsk3α−/− + Gsk3α-L195G ESCs could only be expanded in PD03 + CHIR and rapidly differentiated or died in PD03 + 3MB-PP1. The same results were observed in multiple ESC clones we tested (Figure 4B).
Figure 4. Selective Chemical Inhibition of GSK3β, Not GSK3α, Promotes ESC Self-Renewal.
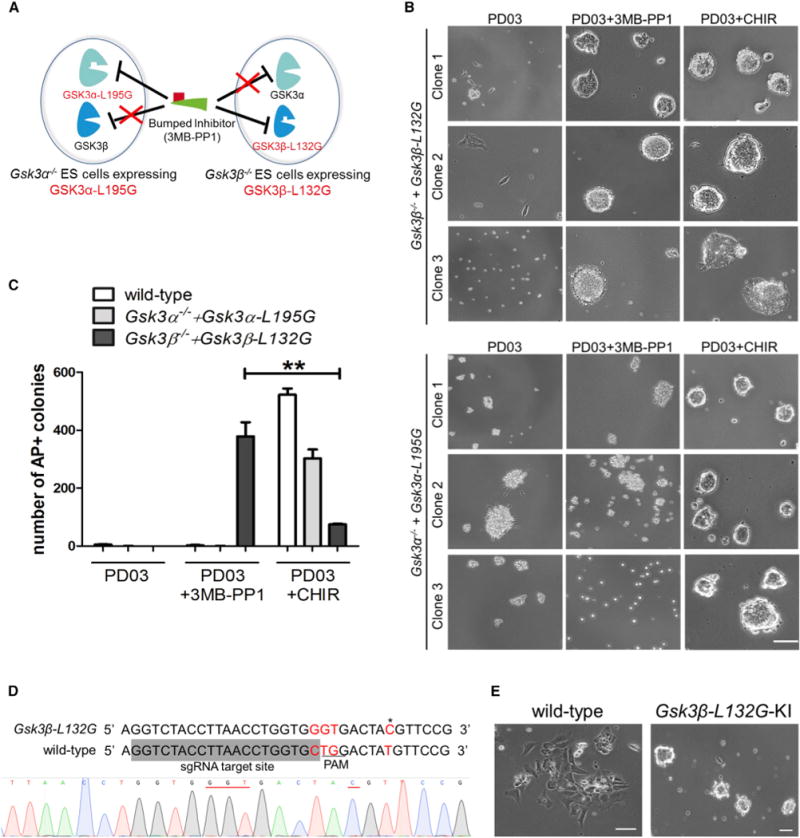
(A) Diagram showing the chemical-genetic strategy to selectively inhibit GSK3α or GSK3β.
(B) Phase-contrast images of Gsk3β−/− + Gsk3β-L132G and Gsk3α−/− + Gsk3α-L195G ESCs plated at a clonal density and cultured in N2B27 medium supplemented with the indicated inhibitors for 7 days. Representative images show results from thee independent clones of mutant ESC lines with GSK3 transgene levels similar to the physiological levels. Scale bar, 100 μm.
(C) Quantification of AP+ colonies of different ESC lines. ESCs were plated onto six-well plates at a density of 1,000 cells/well and cultured for 7 days in N2B27 medium supplemented with the indicated inhibitors. Data represent means ± SD of three biological replicates. **p < 0.01.
(D) Strategy and sequencing result for introducing the L132G point mutation at the Gsk3β locus in mouse ESCs by CRISPR/Cas9. The single guide RNA (sgRNA) target site is shaded in gray. A targeting vector containing 5′ homologous arm-loxP-CAG-PuroR-loxP-3′ homologous arm was used as the template for homologous recombination. The 5′-NGG PAM site (TGG) is underlined in the wild-type locus. The mutated code (CTG→GGT) is highlighted in red. The restriction fragment length polymorphism (RFLP) site is labeled in red and indicated by an asterisk. This RFLP site (AˆCGT) can be digested by HpyCH4IV. Sequencing result showing the knockin of the CTG→GGT (underlined in red) mutation at the Gsk3β-L132G locus in one of the ESC clones.
(E) Representative images of wild-type and Gsk3β-L132G knockin (Gsk3β-L132G-KI) ESCs cultured in N2B27 medium supplemented with PD03 and 3MB-PP1 for 5 days. Gsk3β-L132G-KI ESCs self-renewed continuously in PD03 + 3MB-PP1, whereas wild-type ESCs differentiated rapidly under the same conditions. Scale bars, 100 μm.
Next, we performed a colony-formation assay to quantify the effect of inhibiting individual GSK3 isozymes in promoting ESC self-renewal. Gsk3β−/− + Gsk3β-L132G ESCs formed abundant alkaline phosphatase (AP) positive undifferentiated colonies in the presence of PD03 + 3MB-PP1, whereas Gsk3α−/− + Gsk3α-L195G ESCs differentiated under the same condition, suggesting that selective inhibition of GSK3β, but not GSK3α, recapitulates the self-renewal-promoting effect of CHIR. Notably, Gsk3β−/− + Gsk3β-L132G ESCs formed a significantly greater number of AP positive colonies when GSK3β was selectively inhibited (PD03 + 3MB-PP1) than when both GSK3 isozymes were inhibited (PD03 + CHIR) (Figure 4C), supporting the notion that GSK3α inhibition might have a negative effect on ESC self-renewal (Figures 2C–2E).
To further confirm that selective inhibition of GSK3β promotes ESC self-renewal, we generated Gsk3β-L132G knockin ESCs by introducing a CTG/GGT point mutation at position 394–396 to both endogenous Gsk3β alleles via CRISPR/Cas9-mediated homologous recombination (Figures 4D and S1). A combination of PD03 and 3MB-PP1 was able to maintain long-term self-renewal of the knockin ESCs, while wild-type ESCs rapidly differentiated in the same condition (Figure 4E). Collectively, these results demonstrate that GSK3α and GSK3β play distinct and non-redundant roles in regulating cell fates when both kinases are present in ESCs.
GSK3β Has a Stronger Affinity than GSK3α to Interact with and Phosphorylate β-Catenin
The distinct phenotypes caused by selective inhibition of GSK3α/β in ESCs prompted us to investigate the underlying mechanism. First, we examined β-catenin phosphorylation status upon selective inhibition of individual GSK3 isozymes in ESCs. As expected, CHIR treatment for 30 min inhibited β-catenin phosphorylation at S33/S37/T41 in wild-type, Gsk3α−/− + Gsk3α-L195G, and Gsk3β−/− + Gsk3β-L132G ESCs (Figure 5A). The level of β-catenin phosphorylation in Gsk3β−/− + Gsk3β-L132G ESCs also significantly decreased 30 min after the addition of 3MB-PP1. In contrast, addition of 3MB-PP1 to Gsk3α−/− + Gsk3α-L195G ESCs did not inhibit β-catenin phosphorylation (Figure 5A). Next, we examined the change of cytosolic β-catenin level and β-catenin transcriptional activity after selective inhibition of GSK3α or GSK3β. Treatment with 3MB-PP1 significantly increased both cytosolic β-catenin level and β-catenin transcriptional activity in Gsk3β−/− + Gsk3β-L132G ESCs, but not in wild-type or Gsk3α−/− + Gsk3α-L195G ESCs, while CHIR treatment increased cytosolic β-catenin level and β-catenin transcriptional activity in all three cell lines (Figures 5B and 5C).
Figure 5. GSK3β Has a Stronger Affinity than GSK3α to Interact with and Phosphorylate β-Catenin.
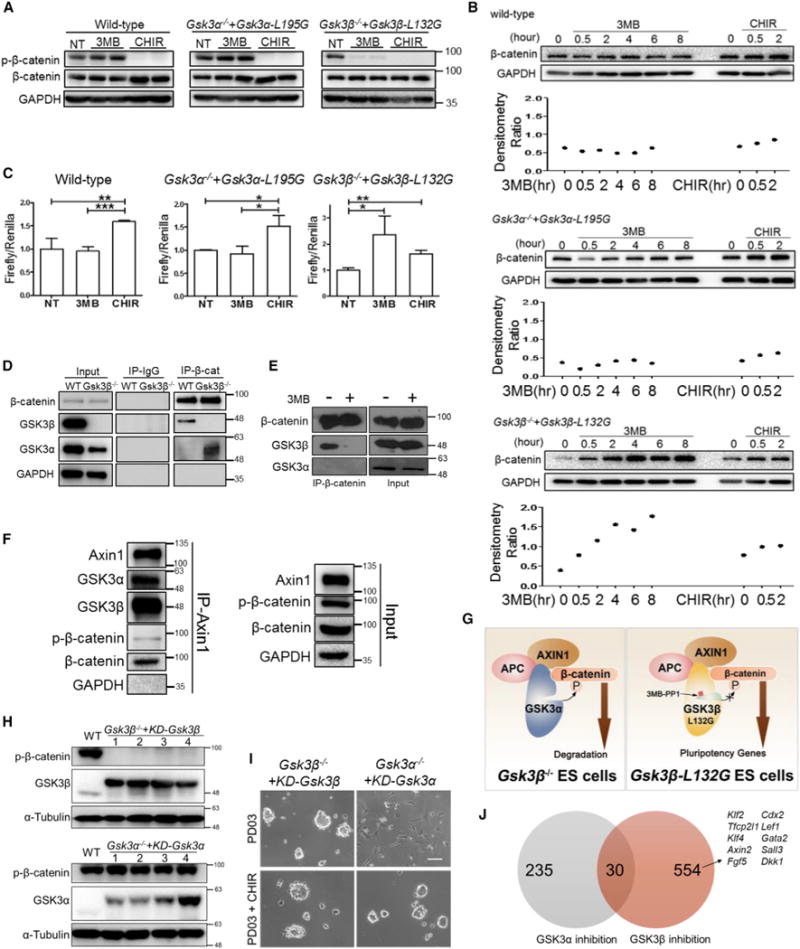
(A) Western blot analysis of the indicated ESC lines treated with 5 μM 3MB-PP1 or CHIR for 30 min (3MB and CHIR-treated samples were duplicated).
(B) Western blot analysis of cytosolic β-catenin levels in ESCs in which GSK3α or GSKβ is selectively inhibited. The indicated ESC lines were treated with 5 μM 3MB-PP1 or CHIR for various times and cytosolic lysates were isolated by 0.015% digitonin and used for western blot analysis. The relative cytosolic β-catenin levels were quantified using ImageJ software and normalized to GAPDH expression.
(C) TopFlash assay in the indicated ESC lines treated with 5 μM 3MB-PP1 or CHIR for 24 hr. NT, no treatment control. Data represent means ± SD of three biological replicates. *p < 0.05; **p < 0.01; ***p < 0.001.
(D) CoIP assay was performed in wild-type ESCs and Gsk3β−/− ESCs using β-catenin antibody. Mouse immunoglobulin G (IgG) antibody was used as a control.
(E) CoIP assay was performed by pulling down β-catenin in Gsk3β−/− + Gsk3β-L132G ESCs treated with or without 3 μM 3MB-PP1 for 24 hr.
(F) CoIP assay was performed by pulling down Axin1 in wild-type ESCs cultured in N2B27 medium for 4 hr, followed by immunoblotting of GSK3α and GSK3β.
(G) A working model to explain the distinct phenotypes between gene deletion and chemical inhibition of GSK3 in ESCs. Upon the deletion of the Gsk3β gene, GSK3α can substitute GSK3β’s function to bind with and phosphorylate β-catenin. In contrast, selective chemical inhibition of GSK3β blocks phosphorylation and degradation of β-catenin, as GSK3α has a lower affinity than GSK3β to bind with β-catenin.
(H) Western blot analysis of β-catenin phosphorylation at S33/S37/T41 in the indicated ESCs maintained in LIF/serum. The numbers 1–4 represent individual Gsk3β−/− ESC clones expressing kinase-dead (KD)-GSK3β or individual Gsk3α−/− ESC clones expressing KD-GSK3α. Both KD-GSK3α and KD-GSK3β contain 2xFLAG tag.
(I) Phase-contrast images of the indicated ESC lines cultured in PD03/N2B27 or PD03 + CHIR/N2B27 for 4 days. Scale bar, 100 μm.
(J) Venn diagram showing the number of genes differentially induced or suppressed by selective inhibition of GSK3α or GSK3β. Arrow indicates the known Wnt/β-catenin downstream targets that are induced by selective inhibition of GSK3β.
See also Table S1.
These results suggest that, when both GSK3 isozymes are present, GSK3β has a stronger ability than GSK3α in phosphorylating β-catenin. In the absence of GSK3β, however, GSK3α is capable of phosphorylating β-catenin, as shown in Figure 3C. To elucidate the underlying mechanism, we performed co-immunoprecipitation (coIP) experiments in wild-type and Gsk3β−/− ESCs. CoIP analysis in wild-type ESCs showed that GSK3β preferentially bound with β-catenin when both isozymes are present. In Gsk3β−/− ESCs, however, GSK3α could substitute for GSK3β in binding with and phosphorylating β-catenin (Figure 5D). Interestingly, the binding between GSK3β and β-catenin was disrupted in Gsk3β−/− + Gsk3β-L132G ESCs when GSK3β was specifically inhibited by 3MB-PP1 (Figure 5E).
GSK3 binds with Axin1 and adenomatosis polyposis coli (APC), forming the so-called destruction complex, which phosphorylates and marks β-catenin for proteasomal degradation (Stamos and Weis, 2013). Since interaction of GSK3 with Axin1 is known to promote GSK3-mediated phosphorylation of β-catenin (Dajani et al., 2003; Ikeda et al., 1998; Thomas et al., 1999), we investigated the potential preference of Axin1 to bind with either GSK3α or GSK3β. CoIP analysis revealed that both GSK3α and GSK3β bound with Axin1 in wild-type ESCs (Figure 5F). Taken together, our results suggest that, while both GSK3 isozymes can bind with Axin1, GSK3β has a stronger affinity than GSK3α to interact with and phosphorylate β-catenin when both isozymes are present. In the absence of GSK3β, however, GSK3α can replace GSK3β to interact with and phosphorylate β-catenin (Figure 5G).
To further validate GSK3’s isozyme-specific interaction with β-catenin, we generated kinase-dead versions of GSK3α and GSK3β (KD-GSK3) and introduced them into Gsk3α−/− and Gsk3β−/− ESCs, respectively. β-Catenin phosphorylation at S33/S37/T41 was barely detectable in Gsk3β−/− + KD-Gsk3β ESCs, whereas β-catenin phosphorylation in Gsk3α−/− + KD-Gsk3α ESCs was comparable with that in wild-type cells (Figure 5H). These results were confirmed in multiple clones with different transgene levels, suggesting that the preferential interaction between β-catenin and GSK3β is real rather than an artifact due to the variation in the GSK3 isozyme levels. Gsk3β−/− ESCs differentiated in PD03 condition, whereas Gsk3β−/− + KD-Gsk3β ESCs could be continuously propagated while remaining undifferentiated in the same condition. In contrast, both Gsk3α−/− and Gsk3α−/− + KD-Gsk3α ESCs required PD03 + CHIR for self-renewal (Figures 2C and 5I). Collectively, these data suggest that GSK3β, not GSK3α, preferentially interacts with and phosphorylates β-catenin when both isozymes are present, and that inhibition of just GSK3β is sufficient to mimic the effect of CHIR in promoting ESC self-renewal.
RNA-Seq Analysis Reveals Distinct Sets of Downstream Targets between GSK3α Inhibition and GSK3β Inhibition
To gain further insight into the mechanism underlying the distinct functions of GSK3α and GSK3β, we performed RNA sequencing (RNA-seq) analysis to examine genome-wide transcriptional profile changes of mouse ESCs upon selective inhibition of GSK3α or GSK3β. We treated Gsk3α−/− + Gsk3α-L195G and Gsk3β−/− + Gsk3β-L132G ESCs with 3MB-PP1 for 2 hr to selectively inhibit just one GSK3 isozyme. RNA-seq analysis on these cells showed that GSK3α inhibition induced a distinct set of downstream targets compared with GSK3β inhibition (Figure 5J and Table S1) (GSE98080). Notably, the known Wnt/β-catenin downstream targets such as Klf2, Tfcp2l1, Klf4, Axin2, Fgf5, Cdx2, Lef1, Gata2, Sall3, and Dkk1 were induced by GSK3β inhibition but not by GSK3α inhibition (Figure 5J and Table S1). These results from transcriptional profiling further confirmed that inhibition of GSK3β has a stronger ability than inhibition of GSK3α in activating the canonical Wnt/β-catenin pathway.
Selective Inhibition of GSK3β Is More Robust in Promoting ESC Self-Renewal than Dual Inhibition of GSK3α and GSK3β
Mouse ESCs cultured in PD03 formed significantly more undifferentiated colonies when GSK3β was selectively inhibited compared with dual inhibition of GSK3α and GSK3β (Figure 4C). To further validate the distinct roles of GSK3α and GSK3β in ESC self-renewal, we cultured Gsk3β−/− + Gsk3β-L132G ESCs in the presence of either 3MB-PP1 or CHIR alone (without PD03). Gsk3β−/− + Gsk3β-L132G ESCs cultured with 3MB-PP1 could be continuously passaged while remaining undifferentiated, whereas the same ESCs cultured with CHIR underwent differentiation and could not be maintained beyond passage 3 (Figures 6A and 6B). Notably, Gsk3β−/− + Gsk3β-L132G ESCs cultured in 3MB-PP1 contained both differentiated and undifferentiated ESCs (Figure 6B), and addition of PD03 resulted in the formation of a uniform undifferentiated ESC population (Figure 4B). These results confirm that, while inhibition of GSK3β promotes ESC self-renewal, this effect seems to be impaired by simultaneous inhibition of GSK3α.
Figure 6. Chemical Inhibition of GSK3β Alone Maintains ES Cell Self-Renewal.
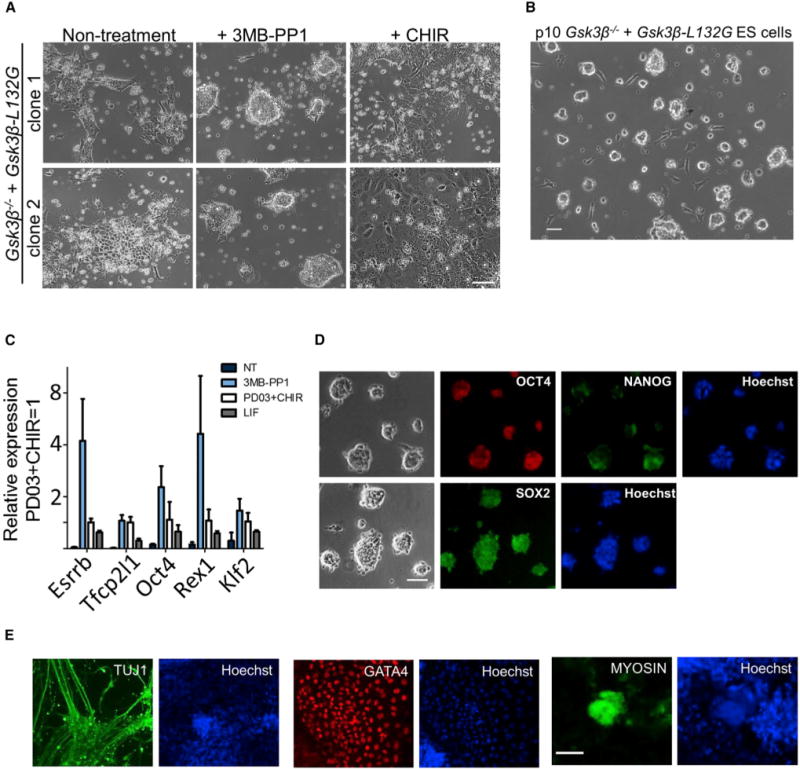
(A) Phase-contrast images of two independent Gsk3β−/− + Gsk3β-L132G ESC clones cultured in N2B27 medium supplemented with CHIR or 3MB-PP1 for 5 days. Scale bar, 100 μm.
(B) A representative image of Gsk3β−/− + Gsk3β-L132G ESCs maintained in 3MB-PP1/N2B27 for ten passages. Scale bar, 100 μm.
(C) qRT-PCR analysis of the expression of naive pluripotency markers. Gsk3β−/− + Gsk3β-L132G ESCs were cultured in the indicated conditions for three passages. Data represent means ± SD of three biological replicates.
(D) Immunofluorescence staining of Gsk3β−/− + Gsk3β-L132GESCs cultured in 3MB-PP1/N2B27 for ten passages. Scale bar, 100 μm.
(E) Embryoid bodies (EBs) were generated from Gsk3β−/− + Gsk3β-L132G ESCs cultured in 3MB-PP1/N2B27 for ten passages. The outgrowths of EBs were immunostained with the indicated antibodies. Scale bar, 100 μm.
Next, we examined whether Gsk3β−/− + Gsk3β-L132G ESCs maintained in 3MB-PP1 remain pluripotent. RT-PCR experiments confirmed that pluripotent genes, including some naive markers, were upregulated after GSK3β inhibition (Figure 6C). Immunohistochemistry staining also showed that Gsk3β−/− + Gsk3β-L132G ESCs maintained with 3MB-PP1 expressed pluripotency markers OCT4, NANOG, and SOX2 and retained the ability to differentiate into all three germ layers (Figures 6D and 6E). Altogether, these findings strongly support that inhibition of GSK3β, not GSK3α, mediates mouse ESC self-renewal.
Inhibition of GSK3α Promotes ESC Differentiation toward Neural Lineages
While characterizing Gsk3α−/− ESCs, we noticed that these ESCs have a strong propensity to differentiate into neural cells even in the presence of PD03 (Figure 7A). This suggests that GSK3α activity might function to block ESC differentiation toward the neural lineage. To facilitate functional characterization of GSK3α in neural differentiation, we engineered 46C mouse ESCs using CRISPR/Cas9 to generate Gsk3α−/− + Gsk3α-L195G and Gsk3β−/− + Gsk3β-L132G ESCs. 46C ESCs carry a Sox1-GFP knockin reporter (Ying et al., 2003). SOX1 is a specific marker of neuroectoderm (Wood and Episkopou, 1999). The neural differentiation potential of these mutant ESC lines was examined using an adherent monolayer differentiation protocol (Ying et al., 2003). Under the N2B27 condition, overexpression of GSK3α-L195G in Gsk3α−/− 46C ESCs dramatically decreased their ability to differentiate into Sox1-GFP positive cells. This effect could be reversed when GSK3α-L195G was specifically inhibited by 3MB-PP1. In contrast, overexpression of GSK3β-L132G in Gsk3β−/− 46C ESCs significantly enhanced differentiation into Sox1-GFP-positive cells and inhibition of GSK3β-L132G by 3MB-PP1 abolished this effect (Figures 7B and 7C). In addition, 3MB-PP1-treated Gsk3α−/− + Gsk3α-L195G 46C ESCs could efficiently differentiate into TUJ1 positive mature neurons (Figure 7D). Taken together, our results suggest that GSK3α and GSK3β play distinct roles in regulating neural differentiation of ESCs.
Figure 7. Chemical Inhibition of GSK3α Promotes Neural Differentiation of ESCs.
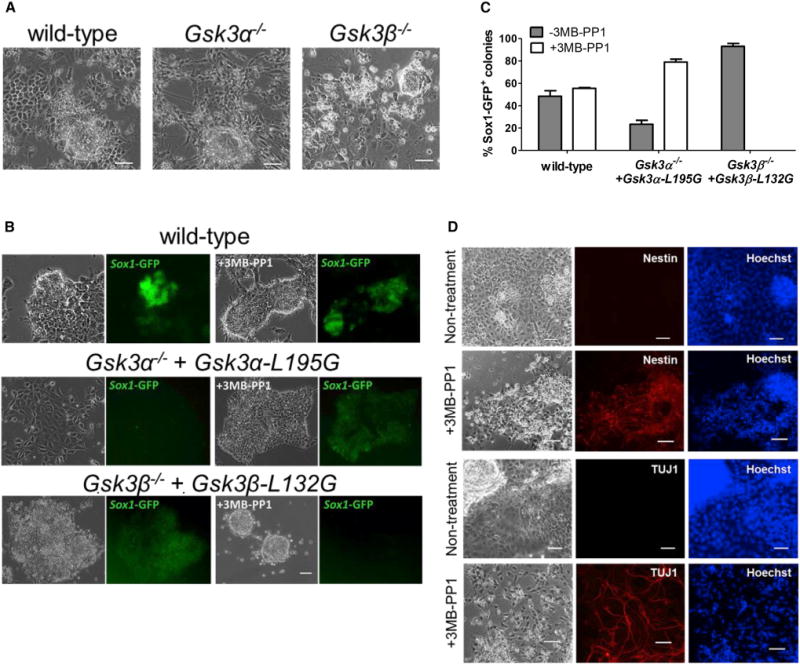
(A) Representative images of the indicated ESCs cultured in PD03/N2B27 for 9 days. Scale bars, 100 μm.
(B) Representative phase-contrast and fluorescent images of the indicated ESCs cultured in N2B27 medium only (the leftmost two columns) or N2B27 medium supplemented with 3 μM 3MB-PP1 (the rightmost two columns). ESCs were plated at clonal density and cultured in LIF/serum for 1 day. The medium was then changed to either N2B27 or 3MB-PP1/N2B27 and cultured for another 6 days before the images were taken. Scale bar, 100 μm.
(C) Quantification of Sox1-GFP positive colonies from experiments described in (B). Data represent means ± SD of three biological replicates.
(D) Representative phase-contrast and immunofluorescence staining images of Gsk3α−/− + Gsk3α-L195G 46C ESCs cultured in N2B27 medium with or without 3 μM 3MB-PP1 for 11 days. Scale bar, 100 μm.
DISCUSSION
In this study, by combining a chemical-genetic approach with ESC-based technologies, we have achieved specific and individual inhibition of GSK3α and GSK3β and demonstrated that selective inhibition of the two isozymes produces distinct phenotypes in ESC self-renewal and neural differentiation. Our study also provides novel mechanistic insights into the functional differences between the two GSK3 isozymes and highlights the phenotypic differences between chemical and genetic perturbations.
In the canonical Wnt/β-catenin pathway, GSK3 forms the destruction complex with Axin and APC and phosphorylates β-catenin at S33/S37/T41. Phosphorylated β-catenin is targeted for ubiquitination and subsequent proteasome-mediated degradation (Kimelman and Xu, 2006). Our study has expanded our understanding of the current model of Wnt/β-catenin signaling by providing the following findings. First, we demonstrated that GSK3β has a stronger ability than GSK3α in interacting with and phosphorylating β-catenin when both GSK3 isozymes are present in ESCs (Figures 5A and 5D). Second, we showed that β-catenin phosphorylation at S33/S37/T41 is barely detectable in mouse ESCs in which the endogenous GSK3β is replaced by a kinase-dead mutant form. In contrast, replacing the endogenous GSK3α with the kinase-dead GSK3α does not inhibit β-catenin phosphorylation (Figure 5H). These results further support the notion that GSK3β, not GSK3α, is the primary kinase for phosphorylating β-catenin under physiological conditions. Finally, we found that, when GSK3β is genetically ablated, GSK3α can replace GSK3β by interacting with and phosphorylating β-catenin (Figures 1A and 5D). We anticipate that GSK3β can also compensate for the function of GSK3α when GSK3α is physically absent. Although our study has clearly revealed the differential binding affinity between GSK3α and GSK3β, whether the two GSK3 isozymes interact with β-catenin in an exclusive manner awaits to be answered in future studies. Importantly, the model that we propose here can explain not only our observations from chemical and genetic perturbations but also why the functions of GSK3α and GSK3β have been previously thought to be redundant to each other in the Wnt/β-catenin pathway using the knockout approach (Doble et al., 2007).
Until now, the study of the function of individual GSK3 isozymes has largely relied on genetic ablation of one isozyme and the characterization of the resulting phenotypes (Banerji et al., 2012; Doble et al., 2007; Grassilli et al., 2014; Guo et al., 2016; Hurtado et al., 2012; Jaworski et al., 2011; Kaidanovich-Beilin et al., 2009; Lee et al., 2007; MacAulay et al., 2007; Patel et al., 2008; Phiel et al., 2003; Urs et al., 2012; Zhang et al., 2014; Zhou et al., 2010, 2013). As revealed in this study, genetic ablation and chemical inhibition of individual GSK3 isozymes yield distinct phenotypes in mouse ESC self-renewal (Figures 2C and 4B). This is likely due to the preservation of the scaffolding function of GSK3 isozymes with chemical perturbation but not in the genetic approach. These results highlight the importance of preserving the scaffolding function of GSK3 isozymes for the understanding of their functions under physiological conditions.
While this study is focused on dissecting the functions of GSK3α and GSK3β in ESC self-renewal and differentiation, the same chemical-genetic approach can be readily applied to dissect the functions of individual GSK3 isozymes and other pairs of highly homologous kinases in different cellular processes. We anticipate that GSK3α and GSK3β are also likely to have different functions in other cellular and developmental processes. One caveat of the chemical-genetic approach is that the activity of the engineered kinase carrying the gatekeeper mutation will likely be reduced, which may affect its physiological function. Hence, it is important to further validate the findings using other biological approaches such as the kinase-dead mutants, as we did in this study. Because ESCs retain the ability to differentiate into nearly every cell type in vitro and in vivo, the GSK3-engineered ESC lines generated in this study can also be readily used to study the functions of individual GSK3 isozymes in different cell types and animal models.
Our finding that GSK3α and GSK3β play distinct roles in ESCs is likely to have broad implications. Elevated GSK3 kinase activity has been linked to the pathogenesis of a variety of diseases, including Alzheimer disease, amyotrophic lateral sclerosis, and diabetes (Cohen and Goedert, 2004; Eldar-Finkelman and Martinez, 2011; Rayasam et al., 2009), but the roles of individual GSK3 isozymes in the disease process have not been well defined. Our results suggest that it is critical to define the roles of individual GSK3 isozymes in order to develop more effective therapies targeting GSK3. While numerous small-molecule inhibitors of GSK3 have been developed, none of them can well distinguish between GSK3α and GSK3β (Eldar-Finkelman and Martinez, 2011). The lack of specific inhibitors for GSK3α and GSK3β has precluded chemical mapping of their precise functions and the development of therapies targeting the specific GSK3 isozyme that causes the pathogenesis of the disease. The combination of CRISPR/Cas9 and ESC-based gene-targeting technologies with the chemical-genetic approach should allow us to create animal disease models in which GSK3α and GSK3β can be specifically and individually inhibited. These animal models will provide a powerful platform for defining the roles of individual GSK3 isozymes and validating the inhibition of individual GSK3 isozymes as drug targets under various disease settings.
The successful derivation of ESCs from multiple strains of mice and rats relies on blockade of GSK3 and MEK kinase activities with CHIR and PD03, respectively (Buehr et al., 2008; Li et al., 2008; Ying et al., 2008). The range of CHIR concentrations that permits ESC derivation and maintenance is rather narrow, especially in rat ESCs (Chen et al., 2013). Because CHIR binds to GSK3α and GSK3β with similar affinity, it is expected to simultaneously inhibit the two GSK3 isozymes, causing opposite effects on ESC fate, based on our findings. There may be a fine window in which inhibition of GSK3β is more pronounced than inhibition of GSK3α to exert maximal promotion of ESC self-renewal. This may explain why ESC self-renewal is so sensitive to CHIR concentration. So far, authentic naive ESCs have only been derived from mice and rats. It would be of interest to test whether inhibition of GSK3β without affecting GSK3α kinase activity would increase the efficiency of ESC derivation in mice and rats, and, more importantly, whether it can facilitate ESC derivation from additional species.
STAR*METHODS
Detailed methods are provided in the online version of this paper and include the following:
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti-alpha-Tubulin (B-5-1-2) | Thermo Fisher Scientific | Cat#32-2500; RRID: AB_2533071 |
| Rabbit monoclonal anti-GSK-3α (D80E6) | Cell Signaling Technology | Cat#4337; RRID: AB_10859910 |
| Mouse monoclonal anti-GSK-3 beta (clone 7) | BD Biosciences | Cat#610201; RRID: AB_397600 |
| Mouse monoclonal anti-β-catenin (clone 14) | BD Biosciences | Cat#610153; RRID: AB_397554 |
| Rabbit polyclonal anti-phospho-β-catenin (Ser33/37/Thr41) | Cell Signaling Technology | Cat#9561; RRID: AB_331729 |
| Rabbit monoclonal anti-GAPDH (D16H11) | Cell Signaling Technology | Cat#5174; RRID: AB_10622025 |
| Rabbit monoclonal anti-Axin1 (C76H11) | Cell Signaling Technology | Cat#2087; RRID: AB_2274550 |
| Mouse monoclonal anti-Oct-3/4 (C-10) | Santa Cruz Biotechnology | Cat#sc-5279; RRID: AB_628051 |
| Goat polyclonal anti-Sox-2 (Y-17) | Santa Cruz Biotechnology | Cat#sc-17320; RRID: AB_2286684 |
| Goat polyclonal anti-mouse Nanog | R&D Systems | Cat#AF2729; RRID: AB_2150103 |
| Mouse monoclonal anti-Neuronal Class III β-Tubulin (TUJ1) | Covance Research Products Inc | Cat#MMS-435P; RRID: AB_2313773 |
| Mouse monoclonal anti-Myosin (MF-20) | Developmental Studies Hybridoma Bank (DSHB), University of Iowa, IA | Cat#MF-20; RRID: AB_2147781 |
| Mouse monoclonal IgG1 Isotype Control | Cell Signaling Technology | Cat#5415S; RRID: AB_10829607 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| CHIR99021 | Division of Signal Transduction Therapy, University of Dundee, UK | CAS: 252917-06-9 |
| PD0325901 | Division of Signal Transduction Therapy, University of Dundee, UK | CAS: 391210-10-9 |
| N-2 Supplement (100X) | Thermo Fisher Scientific | Cat#17502048 |
| B-27™ Supplement (50X), serum free | Thermo Fisher Scientific | Cat#17504044 |
| 3MB-PP1 | Cayman Chemical | Cat#17860; CAS: 956025-83-5 |
| 1NA-PP1 | Cayman Chemical | Cat#10954; CAS: 221243-82-9 |
| 1NM-PP1 | Cayman Chemical | Cat#13330; CAS: 221244-14-0 |
| Leukemia inhibitory factor | Our own lab | N/A |
| ATP, [γ-32P]- 3000Ci/mmol 5mCi/ml EasyTide, 250 μCi | PerkinElmer | Cat#BLU502H500UC |
| Peptide substrate (RRRAAEELDSRAG{pSer}PQL) | GenScript | N/A |
| Critical Commercial Assays | ||
| Leukocyte Alkaline Phosphatase Kit | Sigma-Aldrich | Cat#85L3R |
| Dual-Luciferase Reporter Assay System | Promega | Cat#E1910 |
| iTaq™ Universal SYBR® Green Supermix | Bio-Rad | Cat#1725124 |
| RNeasy Mini Kit | Qiagen | Cat#74104 |
| Deposited Data | ||
| RNA-seq data of gene expression profiles in mouse ES cells upon GSK3α or GSK3β inhibition | This paper | GEO: GSE98080 |
| Full western blots | This paper | Mendeley Data: https://data.mendeley.com/datasets/sncsy3phsy/draft?a=87bb5918-ac94-46e4-a46b-40dac003f5ef |
| Experimental Models: Cell Lines | ||
| Mouse ES-E14 cell line | Ying et al., 2008 | RRID: CVCL_C320 |
| Mouse ES-46C cell line | Ying et al., 2003 | RRID: CVCL_Y482 |
| Gsk3α−/− mouse ES cells | This paper | N/A |
| Gsk3β−/− mouse ES cells | This paper | N/A |
| Gsk3α−/−; Gsk3β−/− mouse ES cells | This paper | N/A |
| Oligonucleotides | ||
| Gapdh: F-tgtgagggagatgctcagtg, R-tgttcctacccccaatgtgt | Qiu et al., 2015 | N/A |
| Esrrb: F-aacagcccctacctgaacct, R-ctcatctggtccccaagtgt | Tai et al., 2014 | N/A |
| Tfcp2l1: F-aggtgctgacctcctgaaga, R-gttttgctccagctcctgac | Qiu et al., 2015 | N/A |
| Oct4: F-gaagcagaagaggatcaccttg, R-ttcttaaggctgagctgcaag | Tai et al., 2014 | N/A |
| Rex1: F-tcactgtgctgcctccaagt, R-gggcactgatccgcaaac | Qiu et al., 2015 | N/A |
| Klf2: F-aggcctgtgggttcgctataaa, R-ggcaaattatggctcaaagtagcag | Qiu et al., 2015 | N/A |
| Gsk3α sgRNA. 5′-TGATTGGTAATGGCTCATT-3′ | This paper | N/A |
| Gsk3β sgRNA. 5′-ATTCCAAAGGAATGGATAT-3′ | This paper | N/A |
| Recombinant DNA | ||
| PiggyBac vector (CAG promoter) | Ye et al., 2016 | N/A |
| px330 | Cong et al., 2013 | Addgene Plasmid #42230 |
| px330-P2A-puro | This paper | N/A |
| pGL2-SuperTop plasmid | Kim et al., 2013 | N/A |
| pRL-TK | Promega | Cat#E2241 |
| pET-41a | EMD Biosciences | Cat#70556-3 |
| Software and Algorithms | ||
| FIJI (ImageJ) | NIH | ImageJ ver2 |
| Excel | Microsoft | 2016 |
| Prism | GraphPad | Ver 5 |
| Partek Flow | Partek Inc. | V6.0.17.0219 |
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Qi-Long Ying (qying@med.usc.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell Culture
Male E14TG2a Mouse ES cells were cultured on 0.1% gelatin-coated dishes, at 37 °C in 5% CO2. Medium used for routine ES cell maintenance was DMEM (Invitrogen) supplemented with 10% FBS (HyClone), 1% MEM non-essential amino acids (Invitrogen), 2 mM GlutaMAX (Invitrogen), 0.1 mM β-mercaptoethanol (Invitrogen), and 100 units/ml LIF (prepared in-house). For serum-free culture, mouse ES cells were maintained in N2B27 medium (Ying et al., 2008) supplemented with 3 μM CHIR and 1 μM PD03 (both synthesized in the Division of Signal Transduction Therapy, University of Dundee, UK).
METHOD DETAILS
Generation of Gsk3 Mutant ES Cell Lines
CRISPR/cas9 technique was applied to generate Gsk3 knockout mouse ES cell lines. px330 plasmid was a gift from Feng Zhang (Addgene plasmid # 42230) (Cong et al., 2013). A P2A-puro cassette was inserted into the FseI and EcoRI sites of px330 and was in-frame with cas9 protein codon. The following guide RNAs used to knock out Gsk3 were designed using the online CRISPR design tool (crispr.mit.edu) (Ran et al., 2013): Gsk3α: 5′-TGATTGGTAATGGCTCATT-3′; Gsk3β: 5′-ATTCCAAAGGAATGGATAT-3′. In brief, px330-P2A-puro vectors carrying Gsk3 guide RNAs were transfected into ES cells, followed by puromycin selection. Individual puromycin-resistant colonies were picked, expanded, and screened for Gsk3 knockout by sequencing and Western blot.
For the generation of GSK3-expressing stable cell lines, the coding sequences of Gsk3α and Gsk3β were cloned from mouse ES cells cDNA by Q5® High-Fidelity DNA Polymerase (NEB) and inserted into the PiggyBac vector. Overlapping PCR was used to generate the Gsk3α-L195G, Gsk3α-K148R (kinase-dead), Gsk3β-L132G, Gsk3β-K85R (kinase-dead) mutations. ES cells were transfected with PiggyBac vector (Ye et al., 2016) carrying various GSK3 mutants using Lipofectamine LTX (Invitrogen) according to the manufacturer’s instructions. Selection began one day after transfection by adding puromycin/hygromycin/blasticidin/zeocin and continued for one week or until the untransfected cells were all killed by the antibiotics in the untransfected control group. Drug-resistant colonies were picked and expanded. Gsk3β-L132G knockin mouse ES cell line was generated by CRISPR/Cas9-mediated homology recombination. A targeting vector was designed and cloned to include 5′-homology arm-loxP-CAG-Puro-loxP-3′-homology arm. In 5′-homology arm, coding sequence of leucine 132 at exon 4 of Gsk3β was mutated to glycine. Gsk3β guide RNA was inserted into the original px330 vector and the plasmid was co-transfected into ES cells with the linearized targeting vector. Individual clones with the correct point mutation were verified by sequencing.
Alkaline Phosphatase Activity Assay
The alkaline phosphatase activity of mouse ES cells was detected using the Alkaline Phosphatase Kit (Sigma). In brief, cells for each treatment were washed with PBS, then fixed with fixative solution (citrate buffered acetone, 60%) for 45 seconds. Cells were then washed with deionized water and incubated with alkaline-dye mixture (one Fast Violet B capsule in 48 ml distilled water) at room temperature in dark for 30 min. After incubation, cells were washed with deionized water twice and kept wet. The number of colonies with alkaline phosphatase staining in each well was manually counted under microscope. Pictures were taken with Olympus CKX41 using Infinity X2 software.
Western Blot and Co-IP
Western blot was performed according to a standard protocol. Cells were lysed in ice-cold RIPA cell buffer (TEKNOVA) supplemented with protease inhibitors (Thermo Scientific) and phosphatase inhibitors (Roche). The proteins were separated with a 4-15% PAGE gel (Bio-Rad) and electrotransferred onto a PVDF membrane. Probing was performed with specific primary antibodies and HRP-conjugated secondary antibodies. For fractionation of cytosolic extracts for free β-catenin detection, lysates were harvested by scraping in PBS+ 0.015% digitonin containing a cocktail of protease inhibitors and phosphatase inhibitors. The mixtures were gently shaken on an orbital shaker at 4 °C for 10 min. Supernatant was then collected as cytosolic extracts. For Co-IP, immune complexes were obtained from cells lysed in mild lysis buffer (50mM Tris, pH7.5, 150mM NaCl, 0.1% NP-40, 5mM EDTA, supplemented at time of lysis with 1× protease inhibitors (Thermo Scientific) and 1× phosphatase inhibitors (Roche)). The supernatant was collected and incubated with anti-β-catenin, anti-Axin1 or anti-IgG antibody overnight at 4 °C following incubation with protein A/G Plus Agarose (Santa Cruz) for 4 hrs. The beads were then washed five times with lysis buffer and resuspended in SDS sample buffer. The primary antibodies used were α-tubulin (#32-2500; Invitrogen, 1:1000), GSK3α (#4337; Cell Signaling, 1:1000), GSK3β (#610201; BD, 1:1000), β-catenin (#610153; BD, 1:1000), phospho-β-catenin (Ser33/37/Thr41) (#9561, lot 12; Cell Signaling, 1:1000), GAPDH (#5174, Cell Signaling, 1:1000), Axin1 (#2087, Cell Signaling, 1:1000).
Of note, in the p-β-catenin blots, an extra band above the expected p-β-catenin band was detected. Our data suggested that this was a non-specific band of a particular lot of the p-β-catenin antibody we used (#9561, lot 12, Cell Signaling technology). To avoid confusion, we have opted not to include this upper band in the Western blot figures shown in the paper. The full Western blots have been deposited in Mendeley Data.
TopFlash Luciferase Assay
For quantifying relative β-catenin/Tcf transcriptional activity, pGL2-SuperTOP plasmid (Kim et al., 2013) was co-transfected with the Renilla vector (pRL-TK, Promega) and assayed accordingly. Dual Luciferase Assay (Promega) was performed according to the manufacturer’s instructions. After co-transfected with SuperTOP and Renilla plasmids, cells were plated onto 24-wells and treated with or without CHIR/3MB in N2B27 medium for 24 hrs. Then growth medium was removed and cells were rinsed with 1× PBS. 1× PLB was directly added into each well and the culture dishes were gently shaken for 15 min at room temperature. Lysates were then transferred to 96-well plate with 20 μl per well. One hundred μl of LAR II was added into each well and firefly luciferase activity was measured. One hundred μl of Stop & Glo® Reagent was then dispensed into the same well and Renilla luciferase activity was measured.
Immunostaining
Cells were fixed in 4% paraformaldehyde for 20 min at room temperature and then incubated in blocking buffer (PBS containing 5%BSA and 0.2% Triton X-100) at 37 °C for 1 hr. Cells were then incubated with primary antibodies diluted in blocking buffer at 4 °C overnight followed by incubation with secondary antibodies diluted in blocking buffer at 37 °C for 1 hr. Nuclei were stained with Hoechst33342 (Invitrogen, 1:10000). Primary antibodies used include the following: Oct4 (C-10, Santa Cruz, 1:200), Sox2 (Y-17, Santa Cruz, 1:200), GATA4 (G-4, Santa Cruz, 1:200), Nanog (AF2729, R&D Systems, 1:200), TUJ1 (Covance, Princeton, NJ, 1:500), Myosin (MF-20, DSHB, 1:50). Alexa Flour fluorescent secondary antibodies (Invitrogen) were used at a 1:2000 dilution.
Quantitative Real-Time PCR (qRT-PCR)
Total RNA was extracted using the RNEasy Mini Kit (QIAGEN) according to manufacturer’s instructions. cDNA was synthesized from 0.5 μg total RNA using the iScript™ Reverse Transcription Supermix (BIO-RAD) according to the manufacturer’s instructions. qRT-PCR was performed using the iTaq™ Universal SYBR® Green Supermix (BIO-RAD) on a Roche lightcycle 480 Real-Time PCR machine (Roche). Gene expression was normalized to Gapdh expression. The qRT-PCR primers used are listed in Key Resources Table (Qiu et al., 2015; Tai et al., 2014).
RNA-Seq Sample Preparation and Analysis
For RNA-seq analysis to compare the downstream targets of GSK3α and GSK3β, E14 Gsk3α−/− + Gsk3α-L195G and Gsk3β−/− + Gsk3β-L132G ES cells were treated with or without 5μM 3MB-PP1 for 2 hrs after serum starvation in N2B27 medium for 6 hrs. Each experiment (control and 3MB-PP1 treated) was performed in duplicate. Total RNA was extracted from the above cells with the Qiagen RNeasy kit. Library preparation and RNA-seq were performed at the USC Epigenome Centre Core Facility. In short, libraries were prepared from 0.5μg total RNA using the Illumina TruSeq Sample Prep kit (with polyA selection), barcoded and the samples multiplexed for sequencing on the Illumina NextSeq500 with read lengths of 75bp (single-end). Pre-alignment QA/QC and base trimming were performed in Partek Flow v6.0.17.0219 using fastqc and quality_trim. The trimmed reads were aligned to mouse mm10 (whole genome) using TopHat2. Quantification and normalization were performed with Partek expectation maximization (EM) algorithm. The Partek Flow software suite was made available through the USC Norris Medical Library Bioinformatics Services (http://norris.usc.libguides.com/nml-bioinfo). Genes whose change in expression was greater than 1.5-fold were counted as differentially expressed. Computation for the work described in this paper was supported by the University of Southern California’s Center for High-Performance Computing (hpc.usc.edu).
In Vitro Differentiation of ES Cells
ES cells were converted to Sox1-GFP positive neural stem cells using a monolayer differentiation protocol as described (Ying et al., 2003). In brief, ES cells were plated onto 6-well plates at a density of 1 × 104 cells/cm2 in N2B27 medium. Medium was changed every other day. Sox1-GFP positive neural stem cells appeared 4 days after plating. Differentiation of ES cells into three lineages were induced by the formation of EBs. In brief, mouse ES cells were plated onto 10-cm non-adherent dishes at a density of 5-7 × 106 cells/10-cm dish to allow EB formation in DMEM/10% FBS medium. Medium was changed every other day. After 10-15 days, mouse ES cells-derived EBs were dissociated and replated onto gelatin-coated dishes and cultured in DMEM/10% FBS medium for another 5-7 days before performing immunostaining to examine the expression of lineage markers.
Protein Expression and Purification
The kinase domain of mouse GSK3α/β was cloned into pET-41a vector and expressed in E. coli BL21 (DE3). Cultures were grown at 37 °C to OD600 between 0.6 - 0.8 units, induced with IPTG (0.5 mM), and then grown at 18 °C overnight. The cells were harvested by centrifugation at 4300 rpm, re-suspended in lysis buffer (50 mM Tris, 150 mM NaCl, pH 8.0), and lysed by sonication with 10 short bursts of 15 sec on ice followed by intervals of 15 sec for cooling (Q500 Sonicator, Qsonica). After the cell lysate was centrifuged at 10200 × g for 1 h at 4 °C, the supernatant was incubated with glutathione agarose (Thermo Fisher Scientific). Then the resins were rinsed with washed buffer (50 mM Tris, 150 mM NaCl at pH 8.0) 4 times before the addition of elution buffer (10 mM glutathione, 50 mM Tris, 150 mM NaCl at pH 8.0). Elution fractions were analyzed by SDS-PAGE gel for purity determination and used for in vitro kinase assay.
In Vitro Kinase Assay
The kinase activity was measured by quantifying the amount of 32P transfer from [γ-32P]–ATP to a peptide substrate. The assay was carried out in 50 mM Tris (pH 8.0), 10 mM MgCl2, 5.3 nM [γ-32P]–ATP (PerkinElmer), 1 mg/mL BSA, 0.1 mM peptide substrate (RRRAAEELDSRAG{pSer}PQL, GenScript), along with different concentrations of the inhibitors at 25 °C for 30 minutes. The reaction was then transferred onto phosphocellulose paper (Whatman P81/GE Healthcare), quenched with 10% aq. acetic acid, washed with 0.5% aq. phosphoric acid 3 times, and rinsed with acetone. After air-drying the phosphocellulose disks, scintillation fluid (BetaMax-ES, MP Biomedicals) was added and samples were counted on a scintillation counter (Beckman LC6500). The data were analyzed with GraphPad Prism 5 to calculate the IC50.
QUANTIFICATION AND STATISTICAL ANALYSIS
The relative cytosolic β-catenin levels shown in Figure 5B were quantified using Image J software. All data are reported as the mean ± SD. A Student’s t test was used to determine the significance of differences in comparisons. Values of p < 0.05 were considered as statistically significant. Sample size and p-values are included in the figure legends.
DATA AND SOFTWARE AVAILABILITY
RNA-seq data have been deposited to the NCBI Gene Expression Omnibus under the accession number GEO: GSE98080. The full western blots have been deposited in Mendeley Data with the DOI URL https://data.mendeley.com/datasets/sncsy3phsy/draft?a=87bb5918-ac94-46e4-a46b-40dac003f5ef.
Supplementary Material
Highlights.
Selective inhibition of GSK3α or GSK3β is achieved via a chemical-genetic approach
Selective inhibition of GSK3β maintains mouse ESC self-renewal
Selective inhibition of GSK3α promotes neural differentiation of mouse ESCs
Gene deletion and chemical inhibition of GSK3 yield distinct phenotypes
Acknowledgments
We thank the USC Libraries Bioinformatics Service for assisting with data analysis. The bioinformatics software and computing resources used in the analysis are funded by the USC Office of Research and the Norris Medical Library. This work was supported by California Institute for Regenerative Medicine (CIRM) New Faculty Award II (RN2-00938), the Chen Yong Foundation of the Zhongmei Group, and in part by the National Science Foundation (CHE-1455306) and American Cancer Society (IRG-58-007-51). X.C. was supported by a predoctoral fellowship from the NICHD/USC Joint T32 Training Program in Developmental Biology, Stem Cells, and Regeneration. A.P. was a CIRM Bridges to Stem Cell Research Fellow (TB1-01181).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information includes one figure and one table and can be found with this article online at https://doi.org/10.1016/j.devcel.2017.11.007.
AUTHOR CONTRIBUTIONS
Conceptualization, X.C., C.Z., and Q.-L.Y.; Methodology, X.C., C.Z., and Q.-L.Y.; Investigation, X.C., R.W., X.L., Y.W., T.Z., Y.Y., A.P., Y.-C.C., L.H., J.P.C., A.A., C.Z., and Q.-L.Y.; Formal Analysis, Writing – Original Draft, X.C., C.Z., and Q.-L.Y.; Writing – Review & Editing, X.C., J.P.C., Q.-L.Y., and C.Z.; Funding Acquisition, Q.-L.Y. and C.Z.
References
- Banerji V, Frumm SM, Ross KN, Li LS, Schinzel AC, Hahn CK, Kakoza RM, Chow KT, Ross L, Alexe G, et al. The intersection of genetic and chemical genomic screens identifies GSK-3α as a target in human acute myeloid leukemia. J Clin Invest. 2012;122:935–947. doi: 10.1172/JCI46465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beurel E, Grieco SF, Jope RS. Glycogen synthase kinase-3 (GSK3): regulation, actions, and diseases. Pharmacol Ther. 2014;148:114–131. doi: 10.1016/j.pharmthera.2014.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bishop AC, Ubersax JA, Petsch DT, Matheos DP, Gray NS, Blethrow J, Shimizu E, Tsien JZ, Schultz PG, Rose MD, et al. A chemical switch for inhibitor-sensitive alleles of any protein kinase. Nature. 2000;407:395–401. doi: 10.1038/35030148. [DOI] [PubMed] [Google Scholar]
- Buehr M, Meek S, Blair K, Yang J, Ure J, Silva J, McLay R, Hall J, Ying QL, Smith A. Capture of authentic embryonic stem cells from rat blastocysts. Cell. 2008;135:1287–1298. doi: 10.1016/j.cell.2008.12.007. [DOI] [PubMed] [Google Scholar]
- Chen M, Maloney JA, Kallop DY, Atwal JK, Tam SJ, Baer K, Kissel H, Kaminker JS, Lewcock JW, Weimer RM, et al. Spatially coordinated kinase signaling regulates local axon degeneration. J Neurosci. 2012;32:13439–13453. doi: 10.1523/JNEUROSCI.2039-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Blair K, Smith A. Robust self-renewal of rat embryonic stem cells requires fine-tuning of glycogen synthase kinase-3 inhibition. Stem Cell Reports. 2013;1:209–217. doi: 10.1016/j.stemcr.2013.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen P, Goedert M. GSK3 inhibitors: development and therapeutic potential. Nat Rev Drug Discov. 2004;3:479–487. doi: 10.1038/nrd1415. [DOI] [PubMed] [Google Scholar]
- Cong L, Ran F, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA, Zhang F. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339:819–823. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dajani R, Fraser E, Roe SM, Yeo M, Good VM, Thompson V, Dale TC, Pearl LH. Structural basis for recruitment of glycogen synthase kinase 3beta to the axin-APC scaffold complex. EMBO J. 2003;22:494–501. doi: 10.1093/emboj/cdg068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doble BW, Patel S, Wood GA, Kockeritz LK, Woodgett JR. Functional redundancy of GSK-3alpha and GSK-3beta in Wnt/beta-catenin signaling shown by using an allelic series of embryonic stem cell lines. Dev Cell. 2007;12:957–971. doi: 10.1016/j.devcel.2007.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eldar-Finkelman H, Martinez A. GSK-3 inhibitors: preclinical and clinical focus on CNS. Front Mol Neurosci. 2011;4:32. doi: 10.3389/fnmol.2011.00032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grassilli E, Ianzano L, Bonomo S, Missaglia C, Cerrito MG, Giovannoni R, Masiero L, Lavitrano M. GSK3A is redundant with GSK3B in modulating drug resistance and chemotherapy-induced necroptosis. PLoS One. 2014;9:1–8. doi: 10.1371/journal.pone.0100947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo X, Snider WD, Chen B. GSK3β regulates AKT-induced central nervous system axon regeneration via an eIF2Bε-dependent, mTORC1-independent pathway. Elife. 2016;5:1–18. doi: 10.7554/eLife.11903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoeflich KP, Luo J, Rubie EA, Tsao MS, Jin O, Woodgett JR. Requirement for glycogen synthase kinase-3beta in cell survival and NF-kappaB activation. Nature. 2000;406:86–90. doi: 10.1038/35017574. [DOI] [PubMed] [Google Scholar]
- Hurtado DE, Molina-Porcel L, Carroll JC, MacDonald C, Aboagye AK, Trojanowski JQ, Lee VMY. Selectively silencing GSK-3 isoforms reduces plaques and tangles in mouse models of Alzheimer’s disease. J Neurosci. 2012;32:7392–7402. doi: 10.1523/JNEUROSCI.0889-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda S, Kishida S, Yamamoto H, Murai H, Koyama S, Kikuchi A. Axin, a negative regulator of the Wnt signaling pathway, forms a complex with GSK-3beta and beta-catenin and promotes GSK-3beta-dependent phosphorylation of beta-catenin. EMBO J. 1998;17:1371–1384. doi: 10.1093/emboj/17.5.1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaworski T, Dewachter I, Lechat B, Gees M, Kremer A, Demedts D, Borghgraef P, Devijver H, Kügler S, Patel S, et al. GSK-3α/β kinases and amyloid production in vivo. Nature. 2011;480:E4–E5. doi: 10.1038/nature10615. [DOI] [PubMed] [Google Scholar]
- Kaasik K, Kivimäe S, Allen JJ, Chalkley RJ, Huang Y, Baer K, Kissel H, Burlingame AL, Shokat KM, Ptáček LJ, et al. Glucose sensor O-GlcNAcylation coordinates with phosphorylation to regulate circadian clock. Cell Metab. 2013;17:291–302. doi: 10.1016/j.cmet.2012.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaidanovich-Beilin O, Lipina TV, Takao K, van Eede M, Hattori S, Laliberté C, Khan M, Okamoto K, Chambers JW, Fletcher PJ, et al. Abnormalities in brain structure and behavior in GSK-3alpha mutant mice. Mol Brain. 2009;2:35. doi: 10.1186/1756-6606-2-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H, Wu J, Ye S, Tai CI, Zhou X, Yan H, Li P, Pera M, Ying QL. Modulation of β-catenin function maintains mouse epiblast stem cell and human embryonic stem cell self-renewal. Nat Commun. 2013;4:2403. doi: 10.1038/ncomms3403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimelman D, Xu W. Beta-catenin destruction complex: insights and questions from a structural perspective. Oncogene. 2006;25:7482–7491. doi: 10.1038/sj.onc.1210055. [DOI] [PubMed] [Google Scholar]
- Knight ZA, Shokat KM. Chemical genetics: where genetics and pharmacology meet. Cell. 2007;128:425–430. doi: 10.1016/j.cell.2007.01.021. [DOI] [PubMed] [Google Scholar]
- Korinek V, Barker N, Morin PJ, van Wichen D, de Weger R, Kinzler KW, Vogelstein B, Clevers H. Constitutive transcriptional activation by a beta-catenin-Tcf complex in APC−/− colon carcinoma. Science. 1997;275:1784–1787. doi: 10.1126/science.275.5307.1784. [DOI] [PubMed] [Google Scholar]
- Lee HC, Tsai JN, Liao PY, Tsai WY, Lin KY, Chuang CC, Sun CK, Chang WC, Tsai HJ. Glycogen synthase kinase 3 alpha and 3 beta have distinct functions during cardiogenesis of zebrafish embryo. BMC Dev Biol. 2007;7:93. doi: 10.1186/1471-213X-7-93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li P, Tong C, Mehrian-Shai R, Jia L, Wu N, Yan Y, Maxson RE, Schulze EN, Song H, Hsieh CL, et al. Germline competent embryonic stem cells derived from rat blastocysts. Cell. 2008;135:1299–1310. doi: 10.1016/j.cell.2008.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C, Li Y, Semenov M, Han C, Baeg GH, Tan Y, Zhang Z, Lin X, He X. Control of beta-catenin phosphorylation/degradation by a dual-kinase mechanism. Cell. 2002;108:837–847. doi: 10.1016/s0092-8674(02)00685-2. [DOI] [PubMed] [Google Scholar]
- MacAulay K, Doble BW, Patel S, Hansotia T, Sinclair EM, Drucker DJ, Nagy A, Woodgett JR. Glycogen synthase kinase 3alpha-specific regulation of murine hepatic glycogen metabolism. Cell Metab. 2007;6:329–337. doi: 10.1016/j.cmet.2007.08.013. [DOI] [PubMed] [Google Scholar]
- Merrill BJ. Wnt pathway regulation of embryonic stem cell self-renewal. Cold Spring Harb Perspect Biol. 2012;4:a007971. doi: 10.1101/cshperspect.a007971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel S, Doble BW, MacAulay K, Sinclair EM, Drucker DJ, Woodgett JR. Tissue-specific role of glycogen synthase kinase 3beta in glucose homeostasis and insulin action. Mol Cell Biol. 2008;28:6314–6328. doi: 10.1128/MCB.00763-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phiel CJ, Wilson CA, Lee VMY, Klein PS. GSK-3alpha regulates production of Alzheimer’s disease amyloid-beta peptides. Nature. 2003;423:435–439. doi: 10.1038/nature01640. [DOI] [PubMed] [Google Scholar]
- Qiu D, Ye S, Ruiz B, Zhou X, Liu D, Zhang Q, Ying QL. Klf2 and Tfcp2l1, two Wnt/β-Catenin targets, act synergistically to induce and maintain naive pluripotency. Stem Cell Reports. 2015;5:314–322. doi: 10.1016/j.stemcr.2015.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F. Genome engineering using the CRISPR-Cas9 system. Nat Protoc. 2013;8:2281–2308. doi: 10.1038/nprot.2013.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rayasam GV, Tulasi VK, Sodhi R, Davis JA, Ray A. Glycogen synthase kinase 3: more than a namesake. Br J Pharmacol. 2009;156:885–898. doi: 10.1111/j.1476-5381.2008.00085.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith AG, Heath JK, Donaldson DD, Wong GG, Moreau J, Stahl M, Rogers D. Inhibition of pluripotential embryonic stem cell differentiation by purified polypeptides. Nature. 1988;336:688–690. doi: 10.1038/336688a0. [DOI] [PubMed] [Google Scholar]
- Stamos JL, Weis WI. The β-catenin destruction complex. Cold Spring Harb Perspect Biol. 2013;5:a007898. doi: 10.1101/cshperspect.a007898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutherland C. What are the bona fide GSK3 substrates? Int J Alzheimers Dis. 2011;2011:505607. doi: 10.4061/2011/505607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tai CI, Schulze EN, Ying QL. Stat3 signaling regulates embryonic stem cell fate in a dose-dependent manner. Biol Open. 2014;3:958–965. doi: 10.1242/bio.20149514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas GM, Frame S, Goedert M, Nathke I, Polakis P, Cohen P. A GSK3-binding peptide from FRAT1 selectively inhibits the GSK3-catalysed phosphorylation of axin and beta-catenin. FEBS Lett. 1999;458:247–251. doi: 10.1016/s0014-5793(99)01161-8. [DOI] [PubMed] [Google Scholar]
- Urs NM, Snyder JC, Jacobsen JPR, Peterson SM, Caron MG. Deletion of GSK3β in D2R-expressing neurons reveals distinct roles for β-arrestin signaling in antipsychotic and lithium action. Proc Natl Acad Sci USA. 2012;109:20732–20737. doi: 10.1073/pnas.1215489109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veeman MT, Slusarski DC, Kaykas A, Louie SH, Moon RT. Zebrafish prickle, a modulator of noncanonical Wnt/Fz signaling, regulates gastrulation movements. Curr Biol. 2003;13:680–685. doi: 10.1016/s0960-9822(03)00240-9. [DOI] [PubMed] [Google Scholar]
- Williams RL, Hilton DJ, Pease S, Willson TA, Stewart CL, Gearing DP, Wagner EF, Metcalf D, Nicola NA, Gough NM. Myeloid leukaemia inhibitory factor maintains the developmental potential of embryonic stem cells. Nature. 1988;336:684–687. doi: 10.1038/336684a0. [DOI] [PubMed] [Google Scholar]
- Wood HB, Episkopou V. Comparative expression of the mouse Sox1, Sox2 and Sox3 genes from pre-gastrulation to early somite stages. Mech Dev. 1999;86:197–201. doi: 10.1016/s0925-4773(99)00116-1. [DOI] [PubMed] [Google Scholar]
- Ye S, Zhang D, Cheng F, Wilson D, Mackay J, He K, Ban Q, Lv F, Huang S, Liu D, et al. Wnt/β-catenin and LIF-Stat3 signaling pathways converge on Sp5 to promote mouse embryonic stem cell self-renewal. J Cell Sci. 2016;129:269–276. doi: 10.1242/jcs.177675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying QL, Stavridis M, Griffiths D, Li M, Smith A. Conversion of embryonic stem cells into neuroectodermal precursors in adherent monoculture. Nat Biotechnol. 2003;21:183–186. doi: 10.1038/nbt780. [DOI] [PubMed] [Google Scholar]
- Ying QL, Wray J, Nichols J, Batlle-Morera L, Doble B, Woodgett J, Cohen P, Smith A. The ground state of embryonic stem cell self-renewal. Nature. 2008;453:519–523. doi: 10.1038/nature06968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang JS, Herreros-Vilanueva M, Koenig A, Deng Z, de Narvajas AAM, Gomez TS, Meng X, Bujanda L, Ellenrieder V, Li XK, et al. Differential activity of GSK-3 isoforms regulates NF-кB and TRAIL- or TNFα induced apoptosis in pancreatic cancer cells. Cell Death Dis. 2014;5:e1142. doi: 10.1038/cddis.2014.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J, Lal H, Chen X, Shang X, Song J, Li Y, Kerkela R, Doble BW, MacAulay K, DeCaul M, et al. GSK-3alpha directly regulates beta-adrenergic signaling and the response of the heart to hemodynamic stress in mice. J Clin Invest. 2010;120:2280–2291. doi: 10.1172/JCI41407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J, Freeman TA, Ahmad F, Shang X, Mangano E, Gao E, Farber J, Wang Y, Ma XL, Woodgett J, et al. GSK-3α is a central regulator of age-related pathologies in mice. J Clin Invest. 2013;123:1821–1832. doi: 10.1172/JCI64398. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.