

Abstract
We propose a docking method that mimics the way proteins bind. The method accounts for the dominant driving forces at the different length scales of the protein binding process, allowing for an efficient selection of a downhill path on the evolving receptor-ligand-free energy landscape. Starting from encounter complexes with as much as 10 Å rms deviation from the native conformation, the method locally samples the six dimensional space of rigid-body receptor-ligand structures subject to a van der Waals constraint. The sampling is initially biased only by the desolvation and electrostatic components of the free energy, which capture the partial affinity of unbound structures that are more than 4 Å away from the native state. Below this threshold, improved discrimination is attained by adding an increasing fraction of the van der Waals energy to the force field. The method, with no free parameters, was tested in eight different sets of independently crystallized receptor-ligand structures consistently predicting bound conformations with the lowest free energies and appropriate stability gap around 2 Å from the native complex. This multistage approach is consistent with the underlying kinetics and internal structure of the free energy funnel to the bound state. Implications for the nature of the protein binding pathways are also discussed.
The goal of protein docking is to obtain a model for the bound complex from the coordinates of the unbound component molecules. Current docking methods evaluate a vast number of docked conformations by simple functions that measure surface complementarity. However, in addition to near-native states, these methods produce many false positives, i.e., structures with good surface complementarity but high root mean square deviations (rmsd). Substantial efforts have been devoted to the development of methods to eliminate the false positives. Approaches involve the reranking of the complex structures by using scoring functions that account for the chemical affinity between the individual molecules, and the refinement of interacting surfaces (1–7). Although these procedures improve the discrimination such that conformations with less than 5 Å rmsd are generally found within the top ten to hundred structures, for most complexes the highest ranked structures are still far from the native.
Based on the mapping of the interaction free energy between a receptor and its ligand (8), we have previously concluded that a reasonable approach to successfully predicting docked conformations was to divide the problem into two steps (4). The first step entails the identification of the binding region (within 10 Å rmsd), emulating the diffusional search of the ligand for its target on the receptor surface. Before establishing substantial surface contacts, receptor-ligand association is governed by electrostatic and desolvation interactions, and hence the approximate binding region can be found by mapping these smooth components of the free energy in the conformational space of encounter complexes (8). Another option is the use of the low resolution docking method (7), which removes the details of interacting proteins to match the resulting smooth geometric forms. Both methods predict the broadly defined binding mode of the two proteins, but are unable to describe the specific interactions at the atomic level. The second step consists on the refinement of this broad binding region to atomic scale. At this stage, surface complementarity (9), led by short-range van der Waals (vdW) forces, plays a crucial role on the stability and specificity of the high affinity complex.
In this paper, we present a docking algorithm that, inspired by the general principles governing protein binding, docks or refines complex structures with as much as 10 Å rmsd from the bound state to 2 Å rmsd. In particular, the method embodies the changes in protein–protein interactions as the process moves along the association pathways. Fig. 1 illustrates the stages in the free energy as the receptor and ligand approach the bound state along some association pathway. At separation between the proteins on the order of 10 Å, the interactions are purely electrostatic and partial desolvation effects, resulting in a free energy surface that is relatively smooth along some arbitrary configurational coordinate measuring the rmsd from the bound or native conformation (8). As the proteins get closer, the occurrence of vdW interactions yield favorable contributions in several states, including the native state, leading to mostly steric energy barriers (Fig. 1B). Finally, once the molecules are fully desolvated, the free energy surface becomes very rugged because of the high sensitivity of the vdW interactions to structural perturbations (Fig. 1C).
Figure 1.

Shapes of the binding free energy landscape as a function of some arbitrary coordinate measuring the rmsd from the native conformation. (A) Free energy corresponding to the smooth component ΔGs, which dominates the interactions of partially desolvated encounter complexes (8) near the binding region (λ = 0). (B) Intermediate free energy mimicking the transition between the “smooth” and the “rugged” free energy (λ ≃ 0.5). (C) Free energy of a fully desolvated interface of a receptor-ligand system (λ = 1). Square symbols portray structures that, driven by the tunable landscape, are funneled into a single minimum of the free energy.
From the point of view of kinetics, Fig. 1 suggests that protein binding should entail distinct kinetic regimes where different driving forces govern the binding process at different times. This scenario is summarized in Fig. 2 (8, 10, 11), where a sketch of the free energy funnel corresponding to Fig. 1 is plotted against a reaction coordinate. The funnel distinguishes three kinetic regimes. First, nonspecific diffusion (regime I) brings the molecules to close proximity. Second, in the recognition stage (regime II), the chemical affinity steers the molecules into relatively well oriented encounter complexes (≈5 Å), overcoming the mostly entropic barrier to binding. Brownian dynamics simulations of this regime (10) were also found to be consistent with a significant narrowing of the binding pathway to the final bound conformation, as suggested by Fig. 1B. Finally, regime III corresponds to the docking stage where short-range forces mold the high affinity interface of the complex structure.
Figure 2.

Binding free energy funnel. The first barrier to binding is for the most part entropic, accounting for the loss in rotational and translational degrees of freedom. The height of this barrier depends on the interplay between the long-range electrostatic steering and the intermediate range desolvation forces. The former can, in some circumstances, eliminate this barrier (23, 24). As suggested by Fig. 1, the steric barrier associated with structural rearrangements is deeper in the binding pocket. For diffusion limited processes, the maximum height of this barrier must be lower than the barrier for the ligand to escape back to solution, implying that the recognition process is the rate-limiting step. Finally, kinetic estimates suggest that the steric activation barrier is significantly higher than the entropic one, suggesting a relatively slow transition, on the order of milliseconds, to the bound structure.
As already mentioned, earlier attempts to address the problem of predictive protein docking have been based on the thermodynamic hypothesis, which reduces the search of the complex structure to the minimization of a potential approximating the free energy. However, as sketched in Fig. 1C, states that are separated by few angstroms in the configurational space may be separated by large steric barriers in energy space. Thus, the straightforward minimization on such a landscape results in the well known multiple minima problem. Significant steps toward the resolution of this problem have been achieved by novel methods proposed by Scheraga and collaborators (12), and others (13, 14). These new algorithms avoid the multiple minima problem by smoothing the landscape in Fig. 1C. The above notwithstanding, the large number of possibilities in which a protein can bind to a substrate, together with the ruggedness of the free energy surface, has rendered it almost impossible for theoreticians to consistently use the thermodynamic principle to predict bound structures from separately crystallized proteins.
The tunable docking method proposed here is based on a kinetic approach that builds on the aforementioned multistage protein binding process, and the inherently smooth free energy governing the initial affinity between a receptor and its ligand. The method slowly tunes in the contribution of the vdW energy in the ranking free energy (see Fig. 1), from the smooth components of the free energy (λ = 0), to the full free energy dominated by vdW interactions (λ = 1). Starting from complexes within the basin of attraction, around 10 Å rmsd from the native complex structure, the method successfully predicts bound conformations within 2 Å rmsd. Interestingly, productive binding pathways also display some of the expected internal structure of the free energy funnel shown in Fig. 2.
Methods
Crystal Structures.
We present docking applications to eight different sets of independently crystallized (unbound) protein pairs, with known complex structure (see Table 1). The protein structures are denoted by their four letter code in the Protein Data Bank (PDB). We study representative systems from four major classes of protein–protein complexes: the barnase-barstar (PDB code 1brs) system from the RNase-inhibitor family; five systems from the protease-inhibitor family, including the trypsin-bovine pancreatic trypsin inhibitor (2ptc) and subtilisin-streptomyces (2sic) and -chymotrypsin (2sni) inhibitor; the serine esterase complex acetylcholinesterase-fasciculin II (1fss); and hen egg white lysozyme bound to antibody Fab D44.1 (1mlc) from the antigen-antibody family. Some of these structures have been analyzed in numerous docking studies (4–7), and most of them are generally considered to be challenging systems when the separately determined protein structures are used for docking. To test the generality of the method, we also make sure that these systems include different binding specificities. For instance, binding of barnase and barstar (1brs) is driven by electrostatic forces, whereas, for the protease inhibitor complexes 1cho and 1ppf, desolvation is the dominant driving force. The complexes 2ptc and 1mlc have basic residues at key positions in the binding pocket, and their flexibility is a further challenge.
Table 1.
Protein complexes studied
| Complex PDB | Receptor | Ligand | Unbound
|
|
|---|---|---|---|---|
| PDB | PDB | |||
| 1ppf | Human leukocyte elastase | OMTKY | 1ppg | 2ovo |
| 1cho | α-chymotrypsin | OMTKY | 5cha | 2ovo |
| 1fss | Acetylcholinesterase | Fasciculin-II | 2ace | 1fsc |
| 1brs | Barnase | Barstar | 1a2p | 1a19 |
| 2sic | Subtilisin BPN′ | Streptomyces inhibitor | 2st1 | 3ssi |
| 2ptc | Trypsin | BPTI | 2ptn | 6pti |
| 2sni | Subtilisin novo | Chymotrypsin inh.2 | 2sbc | 2ci2 |
| 1mlc | Fab D44.1 | Hen egg lysozyme | 1mlb | 1lza |
Initial Conformations.
In this paper, we do not directly address the question of how the initial set of docked structures is generated. The only restriction or assumption is that these structures are within the basin of attraction of the binding region which, based on previous computations, has been established to be around 10 Å rmsd from the crystal complex structure (4, 8). The initial set of conformations, {A}, for 1ppg/2ovo, 5cha/2ovo, and 1mlb/1lza correspond to a cluster of encounter complexes around the lowest binding free energy pocket between receptor and ligand found in a previous discrimination analysis (4). The starting conformations for the other five protein pairs were extracted from a set of 10,000 structures generated by using the program dot (15). The conformations were chosen such that their rmsd from the crystal structure were between 7 and 14 Å. It is interesting to note that in several cases the output from dot resulted in few or no structures below 5 Å rmsd.
Both of these methods produce a relatively large number of conformations around the binding region. Because the docking method depends on the set of initial structures, for this study we selected clusters composed of structures that would differ among each other by 8–12 Å rmsd. We note that in general the average structure would be less that 10 Å rmsd from the complex—typically, around 7 Å rmsd. We also checked that the deviations from the native structure were both translational and rotational. Indeed, superimposing the centers of the initial structures and the target we find that the typical rmsd arising from the difference in rotational states was between 4 and 7 Å.
Free Energy Decomposition.
The binding free energy is computed by the expression (4)
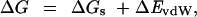 |
1 |
where ΔGs will be referred to as the smooth component of the free energy,
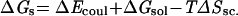 |
2 |
The term ΔEcoul corresponds to the direct electrostatic energy; ΔGsol is the desolvation free-energy change due to transferring the atoms, buried in the interface, from the solvent to a protein environment; ΔSsc is the side-chain entropy loss; and T is the temperature. The van der Waals energy is denoted by ΔEvdW. We note that the binding free energy expression generally also includes the change in internal energy and a further term, TΔS, associated with the loss of rotational, translational, and vibrational entropy. However, in the present analysis, the protein backbones are held rigid at all times, and the relatively small changes in the internal energy are neglected. The TΔS term is omitted because, for a given pair of molecules, it depends weakly on the structure.
The electrostatic term ΔEcoul is calculated by using a distance-dependent dielectric (16) equal to 4r, enforcing a minimum atom-to-atom distance separation equal to the sum of their corresponding vdW radii to avoid artificial overlaps. We estimate the full contribution of the desolvation forces by using Zhang et al. atomic contact potential (ACP) (17). This method is an adaptation of a method first introduced by Miyazawa and Jernigan (18). The ACP potential includes the self-energy change on desolvating charge or polar atom groups and side-chain entropy loss, i.e., ΔGACP = ΔGsol − TΔSsc. The van der Waals energy, ΔEvdW, is computed by using the standard Lennard-Jones potential as implemented in charmm (19). All structural minimizations have been done by using ABNR (adopted basis Newton-Raphson) steps and the charmm-19 potential with polar hydrogens only, distance-dependent dielectrics ɛ = 4r, and fixed backbone.
Method: Tunable Docking.
The algorithm refines a cluster {A} of ten rigid body receptor-ligand structures, constrained to around 10 to 13 Å rmsd from the complex. To specify the geometry of a complex structure, the receptor is centered at the origin of the coordinate system, and the position of the ligand is described in terms of 12 variables. Three of the these, the center-to-center distance dcm between the two molecules, and two Euler angles θcm and φcm, are used to specify the ligand's geometrical center. The relative orientation of the ligand is specified by the remaining 9 variables, corresponding to the Cartesian coordinates of the three unitary vectors, (xi, yi, zi) with i = 1,2,3. The three vectors define a coordinate system fixed on the ligand's center. Because the vectors are orthonormal to each other, only three degrees of freedom are independent. However, the use of this redundant Cartesian system facilitates the sampling of the conformational space. We also define the vector σ⃗, where σk is the standard deviation of the kth variable in the set {A}, constrained to a minimum and maximum of 1o and 6o for θcm and φcm, 0.5 Å and 3 Å for dcm, and 0.08 Å and 0.5 Å for (xi, yi, and zi), respectively.
The docking method consists of the following six steps.
(i) Preprocessing.
The overlaps in the initial set of conformations {A} are removed by rigidly pulling apart the molecules along their center-to-center axis. A second set, {B}, is initialized with the adjusted set {A} ranked according to ΔGs, and contains the top ten best ranked complexes that are sampled during the docking procedure.
(ii) Sampling.
The goal of this step is to generate new structures by a modified version of the nonlinear simplex algorithm (20) using the structures in {A} as templates. We randomly select two structures α and β from {A} to obtain a new point x⃗ with the coordinates d , θ
, θ , φ
, φ , and (x
, and (x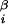 , y
, y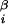 , z
, z ), and define the centroid of the simplex c⃗ as the average of the coordinates in {A} not including x⃗. The basic operations in the simplex method are reflection, expansion, and contraction (20). We perform randomized versions of these operations to sample along the vector r⃗ from x⃗ to c⃗. First, reflection of x⃗ is used to generate the structure γ = c⃗ + r⃗ + ɛ⃗, where ɛ⃗ is a vector of uniformly distributed random real numbers between ±2σ⃗. If γ does not improve the set {B} (see step v of the algorithm), then we perform a contraction to generate a new γ = c⃗ + 0.5r⃗ + ɛ⃗; or, if γ improves the top ranked structure in {B}, then we perform an expansion and generate γ = c⃗ + 2r⃗ + ɛ⃗; otherwise, a new point x⃗(α,β) is selected.
), and define the centroid of the simplex c⃗ as the average of the coordinates in {A} not including x⃗. The basic operations in the simplex method are reflection, expansion, and contraction (20). We perform randomized versions of these operations to sample along the vector r⃗ from x⃗ to c⃗. First, reflection of x⃗ is used to generate the structure γ = c⃗ + r⃗ + ɛ⃗, where ɛ⃗ is a vector of uniformly distributed random real numbers between ±2σ⃗. If γ does not improve the set {B} (see step v of the algorithm), then we perform a contraction to generate a new γ = c⃗ + 0.5r⃗ + ɛ⃗; or, if γ improves the top ranked structure in {B}, then we perform an expansion and generate γ = c⃗ + 2r⃗ + ɛ⃗; otherwise, a new point x⃗(α,β) is selected.
(iii) Constrained vdW optimization.
The energy of the receptor-ligand structure γ from ii is minimized by using 30 adopted basis Newton-Raphson (ABNR) steps in charmm, and the vdW energy of the resulting complex is evaluated. The compactness of the docked conformation is further improved by using the following steps: (a) the ligand is brought closer to the receptor by translating it by an amount equal to −0.02 × dcm along the axis joining the centers of the molecules; (b) 30 ABNR steps are applied to minimize the charmm energy of the structure, and its vdW energy is evaluated; (c) if the new vdW energy is larger than its previous value by more than 10 kcal/mol, then the lower energy structure is minimized by 300 steps. Otherwise, we return to step a.
(iv) Ranking by the free energy.
We compute the electrostatic and desolvation free energy of the minimized complex structure γ. Structures with large overlaps, i.e., with vdW energies larger than 60% of the lowest observed vdW energy, are discarded. If γ is the ith structure sampled during the search, then the target function is evaluated as ΔG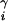 = ΔGs + λiΔEvdw, [3]
= ΔGs + λiΔEvdw, [3]
where λi = i/1,000 for i < 1,000, and λi = 1 otherwise. If ΔG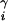 is lower than ΔGi for any of the structures in {B}, then γ is added to {B}, while the highest energy structure is dropped.
is lower than ΔGi for any of the structures in {B}, then γ is added to {B}, while the highest energy structure is dropped.
(v) Updating {A}.
As described, set {B} always contains the 10 lowest free energy structures sampled. These structures are used to periodically update set {A}, but with some delay to avoid shifting the entire cluster because of single outliers. The first update is at i = 140. For i > 140, {A} is updated every time when the best ranked structure in {B} improves, and also at periods of 140 sampled structures.
(vi) Convergence.
Once all the σk are less than their corresponding minima, the method has converged to a solution where the docked structures in the set {B} are within 0.25 Å of each other. Otherwise, the search resumes in step ii.
Results
Our results are summarized in Fig. 3 where the average rmsd of the top 10 best ranked structures in {B} are plotted as a function of the number of structures sampled. The rmsd is computed with respect to the unbound “native-like” ligand—i.e., aligned as in the native complex structure. As indicated by the standard deviation of the average rmsds, the method converges toward a unique binding site (within 0.5 Å rmsd) after ≈500 sampled structures, or about a day of CPU time on a RISC 10000 Silicon Graphics computer. For two cases, 2sbc/2ci2 and 1mlb/1lza, we observed a slower rate of convergence.
Figure 3.

rmsd as a function of the number of sampled structures. The rmsd is computed with respect to the optimal alignment of the unbound ligand in the bound native structure. The + symbols correspond to the average rmsd of the top 10 structures stored in set {B}. Solid lines indicate the standard deviation of the average rmsd.
To appreciate how the refinement proceeds from the initial structures to the final prediction, Fig. 4 shows the locus of the initial set of conformations, the unbound “native-like” ligand, and the predicted docked structure for four complexes, as well as the centers of the 400 initial structures sampled for two systems. In all cases, we find that the method flows nicely to the correct region of the phase space. The reason is that, as shown in Fig. 5, the total free energy of the sampled structures as a function of rmsd for all complexes is a good discriminator, with no false positives. It is also interesting to note that we find an energy gap of around 10 kcal/mol between our predictions and structures with rmsds larger than 4 Å.
Figure 4.

Loci of the initial set of structures and of the predicted structures with respect to the native state. The 10 initial structures have their z axis drawn in green. The predicted structure has its z axis drawn in red, and the axes of the “native-like” structure are in black. For clarity, we show only the position of the center of mass of the first 400 structures sampled in two systems (magenta dots). All distance units are in Å. The center of the receptors are at (0,0,0).
Figure 5.

Free energy as a function of rmsd. The + symbols correspond to the average free energy of the best 10 ranked structures. Dots denote the free energy of the sampled structures.
The breakdown of the free energy to its two main components, ΔGs and ΔEvdW, is shown for four cases in Fig. 6. Most cases behave as for 2sbc/2ci2, where the smooth component of the free energy ΔGs is a good discriminator between 4 to 10 Å rmsd away from the complex, whereas ΔEvdW is typically better below 4 Å rmsd. The only exceptions were found for 2ptn/6pti and 1mlb/1lza, where the level of discrimination of ΔGs is overall weak. These two complexes show a sharp decrease of ΔGs in the final stage of the minimization. However, this feature does not discriminate between high and low rmsd structures; instead, it reflects only the close proximity of some charge groups. Another interesting case is 1a2p/1a19, which shows that ΔGs is a good discriminator between 4 to 10 Å rmsd, but below this range it has a negative correlation. These observations are consistent with the fact that these three complexes show very little chemical affinity if the bound protein conformations are replaced by the unbound structures. Indeed, it has been shown that, before association, these complexes undergo side chain rearrangements that significantly improve their affinity (21). Because our method does not include any preprocessing of the crystal structures, it is not surprising that these cases resulted in docked conformations with the highest rmsd.
Figure 6.

Free energy components as a function of rmsd. Symbols are as in Fig. 5. The vdW energy is represented by the Lennard-Jones potential. The Top four panels correspond to the decomposition of the free energy for the same data shown in Fig. 5. The Bottom panel corresponds to a rerun of the docking algorithm for 1mlb/1lza using the full free energy (λ = 1) as the target function.
We have also checked that a straightforward minimization of the full free energy does not necessarily improve the predictions. A good example of this is shown at the bottom of Fig. 6, where fixing λ = 1 for 1mlb/1lza leads to a different minimum with an rmsd of 4 Å, as well as a very different behavior of ΔGs and ΔEvdW. The two minimizations, varying λ or fixing it, highlight how the improvement of ΔGs (or ΔEvdW) can sometimes come at the expense of ΔEvdW (or ΔGs). In all likelihood, this behavior is artificially (21) enhanced by the poor positioning of side chains at the binding interface.
The thermodynamic analysis provided by Figs. 3, 5, and 6 might be somewhat misleading when analyzed independently of the dynamic process in which λ varies between 0 and 1. For example, for 2ptn/6pti, ΔGs does not seem to be a good discriminator as a function of rmsd. However, as a function of the number of sampled structures (or “time” in a sense analogous to Monte Carlo time steps), ΔGs works very well driving the system to lower free energies and lower rmsd structures (see Fig. 7). On the other hand, a full free energy minimization might trap the docking process in the high rmsd minimum apparent in the plot of ΔEvdW.
Figure 7.

Free energy components as a function of the number of sampled structures for 2ptn/6pti. Symbols are as in Fig. 5.
For all eight of the test cases, we predict a complex structure with around 2 Å rmsd from the unbound “native-like” ligand. The worst prediction is the antibody-antigen 1mlb/1lza system, which predicts a 3 Å rmsd structure. Most predictions fare somewhat better when compared directly with the C-α chain of the native structure located within 10 Å of the interface of the cocrystallized complex structure. Table 2 shows, for the top ranked predictions, the rmsd with respect to the “native-like” unbound ligand, and the rmsd of both the predicted and the “native-like” unbound ligands with respect to the relevant C-α atoms of the native structure near the interface. Because the backbone of the unbound structures is kept rigid, even the best alignment of the unbound ligand to the bound structure yields around 1 Å rmsd. Strikingly, some of our predictions are within 1 Å or less of the binding site. However, for 1lza and 2ci2, the rmsd increases by as much as 0.5 Å when restricting consideration to the region within 10 Å of the interface. In these cases, the binding site in the unbound ligand structure is distorted by large misfolded side chains (Arg-45 and Met-40 in 1lza and 2ci2, respectively), thereby increasing the free energy of all near-native conformations. The rmsd for 1mlb/1lza is particularly large (3.51 Å for the restricted set of residues), but the rmsd of 3.03 Å for all C-α atoms indicates that, even for this large local deviation, the relative orientation of the two molecules in the complex is essentially correct. We also note that, even for this most difficult case, the method was able to refine the initial cluster of 12 Å rmsd structures to around 3 Å.
Table 2.
rmsd of predicted complex structures with respect to the unbound ligand aligned as in the complex crystal structure
| Recep./ligand PDB codes | Prediction rmsd, Å | Prediction rmsd*, Å | Opt.Unb.lig. rmsd*, Å |
|---|---|---|---|
| 1ppg/2ovo | 1.85 | 1.36 | 1.19 |
| 5cha/2ovo | 1.58 | 0.86 | 1.13 |
| 2ace/1fsc | 1.59 | 1.78 | 0.97 |
| 1a2p/1a19 | 2.52 | 2.58 | 0.41 |
| 2st1/3ssi | 2.79 | 1.12 | 0.83 |
| 2ptn/6pti | 2.69 | 1.50 | 0.36 |
| 2sbc/2ci2 | 1.28 | 1.92 | 1.13 |
| 1mlb/1lza | 3.03 | 3.51 | 0.88 |
For comparison, we list the rmsd of our predictions, both for all backbone atom and only for the ones in the interface, as well as for the optimal alignment of the unbound ligand to the native structure. rmsd* denotes the rmsd restricted to the C-α atoms within 10 Å of the native interface.
To study the convergence of the method and the robustness of the results, some of the calculations have been repeated by using different clusters of initial structures. We have checked that the predictions of the method are robust in the sense that small changes on the initial set of structures do not change the final rmsd by more than 0.5 Å. More generally, the details of the sampling method are not crucial for our results as long as the local character of the random sampling is preserved. However, the method fails to converge if the initial structures are too far apart from each other (around 20 Å rmsd). Docking tests of structures with cluster centers more than 15 Å away from the native often converge to unique structures, with free energies consistently higher than the docked structures near the binding region.
As already mentioned in Methods, the cluster centers of the systems studied here are typically around 7 Å rmsd away from the native. Reducing 7 Å to about 2 Å as seen in our results is a nontrivial problem, and the modified simplex method clearly performs very well. Assuming that at least some of the cluster points are in the region of attraction of the global minimum, the simplex method will move the entire cluster toward the native state. However, convergence to a false minimum can occur if, along this pathway, the cluster becomes small enough to be fully accommodated by the basin of attraction of a minimum. Three different mechanisms are used to reduce the possibility of such premature convergence. First, restricting consideration to electrostatic and solvation terms in the early stages of the search smoothes the target function, removing most of the local minima that could serve as traps. Second, the randomization in step iii of the algorithm substantially expands the region sampled by the method. Finally, it is important to select initial structures that are on the order of 10 Å from each other. For the small proteins considered in this paper, 10 Å appears to be the length scale of the region of attraction of the native state because of electrostatic and desolvation interactions. As the above discussion implies, despite the various countermeasures, convergence does not necessarily indicate that the global minimum has been found, and thus the reliability of results can be improved by performing repeated minimizations from different initial clusters.
Discussion
We focus on the prediction of bound complexes from independently crystallized (unbound) receptor and ligand structures starting from a set of structures within the basin of attraction of the binding site (10 Å rmsd from the native structure). Despite the structural differences between the cocrystallized complex and unbound structures, our method successfully predicts low rmsd structures, on the order of 2 Å, from the native for eight receptor-ligand systems. We emphasize that exactly the same method was used in all 8 complexes, and that the only presumption is that the initial set of structures are within the basin of attraction of the binding site (i.e., around 10 Å rmsd).
We provide strong evidence suggesting that it is possible to efficiently trail down the free energy binding funnel. Specifically, the method captures the basic mechanism of protein recognition as well as the short range tuning of the surface complementarity in the high affinity complex. The former is governed by the electrostatic and desolvation components of the free energy, which captures the partial affinity of the macromolecules when properly aligned near their binding site (8). As the binding funnel narrows (see standard deviation in Fig. 3), the vdW interactions become the dominant driving force on the biasing field, improving the surface complementarity between molecules. The robustness of our target function can be seen in Figs. 3 and 5, which show how the chemical affinity and vdW interactions complement each other to obtain the best possible fit, and is confirmed by the fact that exactly the same method yields reasonably accurate predictions for all of the eight systems considered here.
We argue that the evolution of the average top 10 ranked structures (+ symbols in the figures) should, to some degree, reflect the typical behavior of productive binding pathways. In accord with Fig. 2, some systems show a characteristic peak on the vdW energy at ≈5 Å rmsd when plotted against “time” (see Fig. 7). This transition peak is also seen sometimes in the full free energy. The peak corresponds to the transition point between the soft binding regime, controlled by the “smooth” chemical affinity, and the tight regime, controlled by the “rugged” vdW interactions. Because these interactions forcefully move the receptor and the ligand to close proximity, rotational and translational entropy plays no role on our docking method.
From a kinetic point of view, Figs. 2 and 7 are consistent with the view that the recognition process is dominated by a weakly bound intermediate whose affinity is mostly determined by the smooth free energy terms. This process can take place in a reasonably fast time scale (≈10−8 s), in accord with recent Brownian dynamic simulations (10) and nonspecific aggregation data (22). On the other hand, the steric barriers arising from the snug fit of the interface should lead to longer time scales for docking to the final complex structure.
Acknowledgments
We are grateful to D. Gatchell for providing the DOT-generated structures. This research has been supported by Grants DBI-9904834 from the National Science Foundation and GM61867-01 from the National Institutes of Health.
Abbreviations
- rmsd
rms deviation
- vdW
van der Waals
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Novotny J, Rashin A A, Bruccoleri R E. Proteins Struct Funct Genet. 1988;4:19–30. doi: 10.1002/prot.340040105. [DOI] [PubMed] [Google Scholar]
- 2.Vajda S, Sippl M, Novotny J. Curr Opin Struct Biol. 1997;7:222–228. doi: 10.1016/s0959-440x(97)80029-2. [DOI] [PubMed] [Google Scholar]
- 3.Lazaridis T, Karplus M. Proteins Struct Funct Genet. 1999;35:132–152. doi: 10.1002/(sici)1097-0134(19990501)35:2<133::aid-prot1>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- 4.Camacho C, Gatchell D, Kimura S, Vajda S. Proteins Struct Funct Genet. 2000;40:525–537. doi: 10.1002/1097-0134(20000815)40:3<525::aid-prot190>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- 5.Jackson R, Gabb H, Sternberg M. J Mol Biol. 1998;276:265–285. doi: 10.1006/jmbi.1997.1519. [DOI] [PubMed] [Google Scholar]
- 6.Vakser I, Aflalo C. Proteins Struct Funct Genet. 1994;20:320–329. doi: 10.1002/prot.340200405. [DOI] [PubMed] [Google Scholar]
- 7.Vakser I, Matar O, Lam C. Proc Natl Acad Sci USA. 1999;96:8477–8482. doi: 10.1073/pnas.96.15.8477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Camacho C, Weng Z, Vajda S, DeLisi C. Biophys J. 1999;76:1166–1178. doi: 10.1016/S0006-3495(99)77281-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chothia C, Janin J. Nature (London) 1975;256:705–708. doi: 10.1038/256705a0. [DOI] [PubMed] [Google Scholar]
- 10.Camacho C, Kimura S, DeLisi C, Vajda S. Biophys J. 2000;78:1094–1105. doi: 10.1016/S0006-3495(00)76668-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Camacho C, DeLisi C, Vajda S. In: Thermodynamics of the Drug-Receptor Interactions. Raffa R, editor. London: Wiley; 2001. [Google Scholar]
- 12.Wawak R J, Pillardy J, Liwo A, Gibson K D, Scheraga H A. J Phys Chem A. 1998;102:2904–2918. [Google Scholar]
- 13.Andricioaei I, Straub J. J Comput Chem. 1998;19:1445–1455. [Google Scholar]
- 14.Dill K, Phillips A, Rosen J. J Comp Biol. 1997;4:227–239. doi: 10.1089/cmb.1997.4.227. [DOI] [PubMed] [Google Scholar]
- 15.Eyck L T, Mandell J, Roberts V, Pique M. In: Proceedings of the 1995 ACM/IEEE Supercomputing Conference. Hayes A, Simmons M, editors. New York: ACM Press; 1995. [Google Scholar]
- 16.McCammon J, Wolynes P, Karplus M. Biochemistry. 1979;18:927–942. doi: 10.1021/bi00573a001. [DOI] [PubMed] [Google Scholar]
- 17.Zhang C, Vasmatzis G, Cornette J, DeLisi C. J Mol Biol. 1997;267:707–726. doi: 10.1006/jmbi.1996.0859. [DOI] [PubMed] [Google Scholar]
- 18.Miyazawa S, Jernigan R. Macromolecules. 1985;18:534–552. [Google Scholar]
- 19.Brooks B, Bruccoleri R, Olafson B, States D, Swaminathan S, Karplus M. J Comp Chem. 1983;4:187–217. [Google Scholar]
- 20.Nelder J A, Mead R. Comput J. 1964;7:308–313. [Google Scholar]
- 21.Kimura S, Brower R, Vajda S, Camacho C. Biophys J. 2001;80:635–642. doi: 10.1016/S0006-3495(01)76044-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Northrup S, Erickson H. Proc Natl Acad Sci USA. 1992;89:3338–3342. doi: 10.1073/pnas.89.8.3338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schreiber G, Fersht A. Nat Struct Biol. 1996;3:427–431. doi: 10.1038/nsb0596-427. [DOI] [PubMed] [Google Scholar]
- 24.Janin J. Proteins Struct Funct Genet. 1997;28:153–161. doi: 10.1002/(sici)1097-0134(199706)28:2<153::aid-prot4>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]