

Abstract
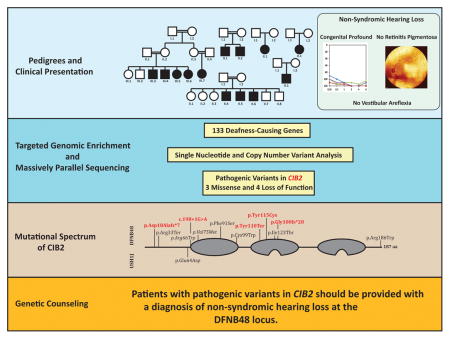
The genetic, mutational and phenotypic spectrum of deafness-causing genes shows great diversity and pleiotropy. The best examples are the group of genes, which when mutated can either cause non-syndromic hearing loss (NSHL) or the most common dual sensory impairment, Usher Syndrome (USH). Variants in the CIB2 gene have been previously reported to cause of hearing loss at the DFNB48 locus and deaf-blindness at the USH1J locus. In this study, we characterize the phenotypic spectrum in a multiethnic cohort with autosomal recessive non-syndromic hearing loss (ARNSHL) due to variants in the CIB2 gene. Of the 6 families we ascertained, 3 segregated novel loss-of-function variants, 2 families segregated missense variants (one novel) and 1 family segregated a previously reported pathogenic variant in trans with a frameshift variant. This report is the first to show that biallelic loss of function variants in CIB2 cause ARNSHL and not USH. In the era of precision medicine, providing the correct diagnosis (NSHL vs USH) is essential for patient care as it impacts potential intervention and prevention options for patients. Here we provide evidence disqualifying CIB2 as an USH-causing gene.
Keywords: Non-syndromic hearing loss, CIB2, Usher syndrome, deafness, DFNB48, USH1J
Graphical Abstract
Introduction
Valid variant identification, classification and phenotypic correlation are at the core of genomic medicine. As next-generation sequencing becomes a routine part of clinical care, the translation of genetic variants into clinically actionable therapies and interventions is tied directly to the accuracy of the interpretation of genomic variants and ultimately the molecular diagnosis. Both missed-diagnosis and misdiagnosis yield non-ventured personalized therapeutic and intervention opportunities for patients. In genes that exhibit pleiotropy, such as the genes associated with Usher syndrome, elucidating the phenotypic outcome based on the genetic data often alters treatment avenues and patient quality of life.
Hereditary hearing loss (HHL) is the most common sensorineural defect, affecting roughly 1 in 500 newborns [1]. It is both genetically and phenotypically heterogeneous with over 150 genes causally associated with a wide range of phenotypic outcomes through a combinatorial effect of type (sensorineural or conductive), severity, frequencies affected, onset, progression and the presence or absence of other system defects (non-syndromic or syndromic). Adding an additional layer of complexity is the pleiotropy exhibited by several deafness-causing genes, best illustrated by Usher syndrome (USH), the most common dual sensory defect affecting hearing and vision. Usher syndrome can be classified into three types depending on the auditory and retinal phenotypes. The most severe, USH type 1 (USH1), is characterized by congenital severe-to-profound hearing loss, vestibular dysfunction and an onset of retinitis pigmentosa (RP) in the first decade of life [2].
Six genes are currently linked to the USH1 phenotype: MYO7A (OMIM# 276903), USH1C (OMIM# 605242), CDH23 (OMIM# 605516), PCDH15 (OMIM# 605514), USH1G (OMIM# 607696) and CIB2 (OMIM# 605564). Variants in each of these genes also give rise to non-syndromic hearing loss (NSHL). This pleiotropy, along with the non-congenital visual phenotype, can make predicting the clinical outcome associated with specific genetic variants challenging. For example, while it is widely recognized that variants predicting null alleles cause USH [3,4], predicting the effect of missense variants is very difficult. It is often unclear which variants or variant combinations lead to a NSHL phenotype versus an USH phenotype.
CIB2 belongs to a highly conserved family of EF-hand domain containing and calcium binding proteins [5,6]. Functionally, their roles are either structural, affecting calcium binding, or regulatory, as part of intercellular calcium-dependent signaling cascades [6–10]. CIB2 comprises three EF-hand domains of which only the two most C-terminal have been shown functionally to bind calcium [10,11]. In humans there are four alternatively spliced transcripts of CIB2, which produce four proteins ranging in size from 138 amino acids (aa) to 192 aa. CIB2 is widely transcribed in many tissues including the inner ear and retina [12]. In the mouse inner ear it is expressed in the hair cells, supporting cells and the vestibular system, where it interacts with two components of the Usher interactome, whirlin (WHRN) and myosin VIIa (MYO7A) [12]. In the mouse retina, CIB2 is broadly expressed in the retina pigment epithelium, inner and outer plexiform layers, the ganglion cell layer and the inner and outer segment of photoreceptors.
The pathogenic mechanism underlying CIB2-related deafness is unclear. Two mechanisms have been proposed: a decrease in CIB2 integrin-binding ability or a diminished capacity to properly buffer calcium [12–14]. Recent studies in several murine mutants suggest both mechanisms could be at play and might not be mutually exclusive [15,16] (Michel et al. 2017 is currently under review). Results from Giese et al suggest that variants in different regions of the CIB2 protein cause distinct effects. Variants in the N-terminal region of CIB2, which interacts with TMC1 and TMC2, alter this interaction but do not affect CIB2 calcium buffering ability; in contrast, variants in the C-terminal region affect only calcium buffering. Variants in the middle region alter both CIB2-TMC1/TMC2 interactions and calcium buffering. Michel and colleagues showed that complete loss of CIB2 results in mislocalization of WHRN and integrin α8, an extracellular matrix and intracellular cytoskeleton linker.
In humans, variants in CIB2 have been linked to hearing loss at the DFNB48 and USH1J loci [12]. Here, we present a multi-ethnic cohort of patients with autosomal recessive NSHL (ARNSHL) due to pathogenic variants in CIB2. We expand the mutational spectrum of CIB2 to include copy number variations (CNVs), splice-site variants and indels, and demonstrate that regardless of variant-type or combination, all pathogenic variants in CIB2 in our cohort give rise to non-syndromic deafness. These data call into question any involvement of CIB2 in the physiopathology of Usher syndrome.
Materials and Methods
Patients and Audiometric evaluations
Two Iranian (L-1644, and L-3156), one Turkish (661) and three European (51550, Trio-B, Trio-C) families segregating ARNSHL were ascertained for this study. Clinical examination of the proband in each family was performed by an otolaryngologist, ophthalmologist and clinical geneticist. Funduscopy examination was also completed on all affected persons in each family. Hearing thresholds were measured by pure tone audiometry at 0.25, 0.5, 1, 2, 3, 4 and 8 kHz. The proband of family Trio-B also underwent physical therapy evaluation for vestibular dysfunction and was evaluated using the Peabody Developmental Motor Scale (PDMS-2). After obtaining written informed consent to participate in this study, blood samples were obtained from all family members and genomic DNA (gDNA) was extracted using routine techniques. All procedures were approved by the human research Institutional Review Boards at the Welfare Science and Rehabilitation University and the Iran University of Medical Sciences, Tehran (Iran), the University of Miami, Miami, Florida (USA), Ankara University Medical School, Ankara (Turkey), and the University of Iowa, Iowa City, Iowa (USA).
Linkage Analysis
Genome-wide SNP genotyping was carried out on genomic DNA of 4 affected and 4 unaffected individuals from family L-3156 using Affymetric GeneChip Mapping 100K arrays (Affymetrix, Santa, CA, USA). Linkage analysis was performed using MERLIN assuming a rare recessive model, and haplotypes were examined with HaploPainter.
Targeted Genomic Enrichment and Massively Parallel Sequencing
Targeted genomic enrichment with massively parallel sequencing (TGE+MPS) using the OtoSCOPE® platform (version 7) was performed in one affected person from families L-1644, 51550, Trio-A and Trio-B to screen all known genes implicated in NSHL and USH for pathogenic and likely pathogenic variants as previously described [17,18]. Whole exome capture was performed with the Agilent SureSelectXT Human All Exon V4 (Agilent Technologies, Santa Clara, CA) on one affected individual from families L-3156 and 661 as previously described [19–22]. Enriched libraries were sequenced on the Illumina HiSeq 2000 (Illumina, Inc., San Diego, CA) using 100bp paired-end reads.
Bioinformatic analysis
Data analysis was performed using either a custom annotation pipeline [17,19,20] or the Illumina CASAVA v1.8 pipeline [21,22]. In each instance, sequencing reads were mapped to the NCBI Build 37 reference genome using BWA [23] and variant calling was performed using Genomic Analysis Tool Kit (GATK) [24]. Variants were annotated and filtered as described [17,19,21]. Briefly, variants were filtered based on depth >10, quality score >30 and minor allele frequency (MAF) <2% in Genome Aggregation Database (gnomAD), 1000 Genomes Project database, the Exome Aggregation Consortium (ExAC) and the National Heart, Lung, and Blood Institute (NHLBI) Exome Sequencing Project Exome Variant Server (EVS). Retained variants were then prioritized based on their conservation (GERP and PhyloP), predicted deleteriousness (SIFT, PolyPhen2, MutationTaster, LRT, and the Combined Annotation Dependent Depletion (CADD)) and variant-type (missense, nonsense, indel or splice-site). Potential effects on splicing were analyzed using Human Splicing Finder 3.0 (HSF) (http://www.umd.be/HSF3/) and the Berkeley NNSPLICE 0.9 (BDGP) (http://www.fruitfly.org/seq_tools/splice.html). In addition, samples captured using the OtoSCOPE® platform were analyzed for CNVs using a sliding window method to asses read-depth ratios [25]. For families 661 and L-3156, visual inspection of the CIB2 gene using Integrated Genomics Viewer (IGV) (Broad Institute) was used to detect CNVs. All reported variants have been submitted to the University of Iowa Deafness Variation Database (http://deafnessvariationdatabase.org/) for curation and integration.
Molecular Modeling
Molecular modeling of the CIB2 is based on the highly homologous CIB1 protein structure PDB: 2L4H. The model was built using Yasara and WHAT IF Twinset [26] via the project HOPE server (http://www.cmbi.ru.nl/hope/).
CNV Breakpoint mapping and Sanger Sequencing
CNV breakpoints were determined using long range PCR, nested PCR and Sanger sequencing. Primers for both PCR and sequencing were designed using Primer3 (http://bioinfo.ut.ee/primer3-0.4.0/) (Table 1S). Breakpoint-spanning PCR was carried out with TaKaRa LA Taq Polymerase (TaKaRa Bio) using the manufacture’s protocols. PCR products were visualized on a 1.5% agarose gel. Validation and segregation of candidate variants were completed by Sanger sequencing on an ABI 3730 Sequencer (Perkin Elmer, Waltham, MA) using gene-specific primers. All sequencing chromatograms were compared to published cDNA sequence (CIB2: NM_006383) and a control DNA. Nucleotide changes were detected using Sequencher v5 (Gene Code Corporation, Ann Arbor, MI).
Results
Phenotypic characterization
The ascertained families originated from 3 ethnic backgrounds - Iranian (L-3156 and L1644), Turkish (661) and European (51550, Trio-B and Trio-C) (Table 1) - with reported consanguinity in the Iranian and Turkish families (Figure 1A–C). Audiometry revealed a bilateral symmetric prelingual severe-to-profound hearing loss across all frequencies in all affected individuals. The proband of family 51550 underwent bilateral cochlear implantation at 13 months and his preoperative temporal bone computed tomography showed normal cochlear anatomy. Although the proband of family Trio-B underwent physical therapy evaluation for vestibular dysfunction at 10 months of age (she scored “average” per the PDMS-2 and had excellent dynamic sitting balance and appropriate head-eye movements), all affected individuals had normal developmental motor milestones with no delays in sitting or walking. Ophthalmological evaluations of all affected individuals revealed absence of RP as demonstrated by funduscopy (Figure 1B). Affected individual II.6 of family L-1644 did have a benign posterior vitreous detachment (Figure 1B). Detailed physical and clinical examination of affected individuals excluded any syndromic features.
Table 1.
Clinical Summary of Patients
| Family ID | L-3156 | L-1644 | 661 | 51550 | Trio-B | Trio-C | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Ethnicity | Iranian | Iranian | Turkish | European | Russian/Danish | European | |||||||
| Patient ID | III.2 | III.3 | III.4 | III.6 | III.7 | II.4 | II.5 | II.6 | II.1 | II.2 | II.1 | II.1 | II.1 |
| Age at Examination | 10 | 17 | 12 | 20 | 18 | 28 | 39 | 49 | 28 | 14 | 10 | 1 | 2 |
| Ophthalmologic examination | NL | NL | NL | NL | NL | NL | |||||||
| Motor milestone | NL | NL | NL | NL | NL | NL | |||||||
| Severity of HL | Profound | Profound | Severe-profound | Severe-profound | Severe-profound | Profound | |||||||
| Onset of HL | Prelingual | Prelingual | Prelingual | Congenital | Congenital | Congenital | |||||||
Italics and underlined indicate individual with funduscopy images. Age is given in years.
Figure 1.

Pedigrees showing haplotypes in the DFNB48 locus, audiometric data, funduscopy images and segregation for variants in CIB2. Filled symbols denote affected individuals and double lines indicate consanguinity. Red and bold represents the CIB2 mutant alleles segregating with the NSHL. Audiograms were obtained using pure tone audiometry with air conduction from frequencies from 250 Hz to 8,000 Hz. A: Pedigree for family L-3156 with six haplotype markers. Solid red markers indicate the morbid haplotype, while other haplotypes are in blue and grey. A “+” indicates the wildtype alleles and the “-“ represents the mutant allele harboring the 3.1kb deletion. Parametric linkage analysis plot of chromosome 15. B: Pedigree, audiograms and funduscopy images for families L-1644. C–F: Pedigrees for families 661, 51550, Trio-B and Trio-C.
Linkage Analysis
Homozygosity mapping and linkage analysis identified a single homozygous region with a LOD score > 3 on chr15q22.2–q25.1. This region, which was shared by all affected siblings and generated a maximum LOD score of 3.6, spans 18.5Mb between rs951265 and rs2902957 and overlaps two known deafness loci: DFNB48 and DFNA30 (Figure 1A). The linked region harbors 246 genes.
Variant Identification and segregation
TGE and MPS using the OtoSCOPEv7® platform on probands from families L-1644, 51550, Trio-B and Trio-C yielded an average of 10 million reads per sample and a coverage of 99.5% at ≥10X and 98.5% at ≥30X. After filtering for quality and MAF, an average of 9 variants per sample was identified. CNVs were not detected in any of the samples. Using a recessive model, we further filtered variants to include only variants that were either homozygous or compound heterozygous.
In family L-1644, only one homozygous variant was identified: c.344A>G in CIB2 (NM_006383). This novel missense variant changes a conserved tyrosine residue to a cysteine, p.Tyr115Cys (NP_006374), and is predicted to be deleterious by PolyPhen-2, SIFT, MutationTaster, LRT and CADD (Table 2). It is located in exon 4, thus affecting all alternatively spliced transcripts (Figure 2A) and the amino acid residue Tyr115, which is positioned in the second EF-hand domain of the protein (Figure 2B). Segregation analysis showed all affected individuals were homozygotes, whereas unaffected individuals were either heterozygous or wild-type for this allele (Figure 1B).
Table 2.
CIB2 Variants and Phenotype Correlation
| Family | Phenotype | Origin | gDNA | cDNA | Protein | Type | Domain | gnomAD (%) |
Conservation | Deleterious | REF | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| GERP | PhyloP | PP2 | SIFT | MT | LRT | CADD | ||||||||||
| L-3156 | NSHL | Iranian | 78413407_78416520del | c.52_86del | p.Asp18Alafs*7 | CNV/frameshift | 0 | - | - | - | - | - | - | - | This study | |
| Trio-B | NSHL | European American | 78403509G>A | c.196C>T | p.Arg66Trp | missense | EF-1 | 0.002 | C | C | D | D | D | D | 34 | This study + [14] |
| 78401614_78401623del | c.300_309del | p.Glu100fs*28 | frameshift | EF-2 | 0.006 | - | - | - | - | - | - | 35 | This study | |||
| Trio-C | NSHL | European | 78401700C>T | c.223G>A | p.Val75Met | missense | EF-1 | 0.003 | C | C | D | D | D | D | 29.7 | This study + [17] |
| 51550 | NSHL | European American | 78403506C>T | c.198+1G>A | splice-site | 0 | C | C | - | - | D | - | 25.3 | This study | ||
| 661 | NSHL | Turkish | 78401593A>T | c.330T>A | p.Tyr110Ter | nonsense | EF-2 | 0 | C | C | - | - | - | - | 36 | |
| L-1644 | NSHL | Iranian | 78401579T>C | c.344A>G | p.Tyr115Cys | missense | EF-2 | 0 | C | C | D | D | D | D | 27 | |
| 07-1069 | NSHL | Dutch | 78403608G>A | c.97C>T | p.Arg33Ter | nonsense | 0.006 | C | C | - | - | - | D | 36 | [14] | |
| PKDF117 | USH1 | Pakistani | 78403513C>G | c.192G>C | p.Glu64Asp | missense | 0.0004 | C | C | D | D | D | D | 24 | [12] | |
| PKDF* | NSHL | Pakistani | 78401651A>G | c.272T>C | p.Phe91Ser | missense | EF-1 | 0.003 | C | C | D | D | D | D | 29.9 | |
| DEM4025 DEM4225 |
NSHL | Pakistani | 78401626G>C | c.297C>G | p.Cys99Trp | missense | EF-1 | 0.001 | C | C | D | D | D | D | 27 | [12,14] |
| 802 | NSHL | Turkish | 78398237A>G | c.386T>C | p.Ile123Thr | missense | EF-2 | 0 | C | C | - | - | - | - | - | [12] |
| JS | NSHL | Hispanic | 78397661G>A | c.556C>T | p.Arg186Trp | missense | 0.005 | C | C | D | D | D | D | 35 | [13] | |
Nucleotide numbering: the A of the ATG translation initiation site is noted as +1 using transcript NM_006383 of CIB2. NSHL, non-syndromic hearing loss; USH1, Usher Syndrome Type 1; C, predicted Conserved; D, predicted Damaging or Deleterious; -, data not available; EF, EF-hand; PP2, PolyPhen-2; MT, MutationTaster. Dashes denote mutations outside functional domains. An “*” indicates a founder mutation identified in a total of 56 Pakistani families. Italics indicate previously described variants also identified in this study.
Figure 2.

A schematic representation of the transcripts, protein and molecular modeling of CIB2. A: Novel and previously described variant alignment to the four coding transcripts of human CIB2. Transcripts 1–4 correspond to RefSeqs NM_006383.3, NM_001271888.1, NM_001271889.1 and NM_001301224.1, respectively. Red and bold variants identified in this study, italics represent the recurrent c.196C>T (p.Arg66Trp) and c.223G>A (p.Val75Met) variants. B: CIB2 has three EF-hand domains (grey), with the first being a non-calcium binding EF-hand and the ladder two having active calcium binding motifs. Previously reported USH1J variant (on bottom) and shown on top are the previously described and the DFNB48 variants identified in this study. Variants are mapped using RefSeq NP_006374.1 numbering. C: Ribbon-presentation of the CIB2 protein. D–E: Locally zoomed wildtype (green) and variants (red) for p.Typ115Cys and p.Val75Met. Yellow and grey balls indicate calcium ions.
An ultra-rare homozygous splice-site variant in CIB2, c.198+1G>A, was identified in the proband of 51550 (Figure 1D). Computation splice-site predictions predicted the loss of the wild-type donor site of exon 3 in transcript 1 (Figure 2A).
Compound heterozygous variants in CIB2: c.196C>T; p.Arg66Trp and c.300_309del; p.Glu100fs*28 were identified in the proband of family Trio-B (Figure 1E). The p.Arg66Trp has been reported pathogenic for ARNSHL and affects the coding sequence of transcripts 1 and 2 (Table 2). It is located in the first EF-hand domain (Figure 2B). The second allele, c.300_309del is a rare frameshift variant that predicts a null allele and affects the coding sequence of all four transcripts (Table 2) (Figure 2A).
A homozygous nonsense variant in CIB2, c.330T>A, p.Tyr110Terwas identified in the proband of family 661 and was found to segregate with the hearing loss in the family (Figure 1C). Variant results for this family have been reported previously without clinical correlation or details [22]. The variant is located in exon 4, produces a protein truncated at codon 110, which is located near the start of the second EF-hand domain (Figure 2B), and affects the coding sequence of all transcripts (Figure 2A).
Ten variants passed filtering criteria for the proband of Trio-C. Of these, a homozygous previously reported pathogenic alteration in CIB2: c.223G>A; p.Val75Met was identified (Figure 1F). This variant is located early in exon 4 and is affects the first EF-hand domain (Figure 2A–B).
Finally family L-3156 underwent whole exome capture. After initial filtering for quality and MAF, 32 homozygous variants were identified. None of the 32 variants occurred in genes associated with non-syndromic hearing loss or common syndromic forms of deafness. Filtering for variants inside the linked region narrowed the list of candidates to 4 missense variants across 4 genes. Variants in the IGDCC3 (OMIM# 604184), IGDCC4 (OMIM# 616810) and ISLR (OMIM# 602059) genes were not predicted to be deleterious by any pathogenicity prediction tool (Supplemental Table 2). A variant in the PEAK1 gene (OMIM# 614248) was identified that is conserved and predicted to be deleterious (Supplemental Table 2). Direct sequencing showed this variant was homozygous in all individuals. Assessing the coverage for each gene in the region using IGV revealed absence of sequence reads mapping to exon 2 of CIB2 (Figure 3A). PCR amplification using primers flanking exon 2 failed to generate a product for all affected individuals. Long-range PCR encompassing ~8.8 kb around exon 2 resulted in 2 PCR products: a ~8.8 kb and ~5.7 kb product. Unaffected individuals have either both products or just the ~8.8 kb product, whereas patients only have the ~5.7 kb product (Figure 3B). The size of the deletion was determined to be 3113 bp with the breakpoints at chr15:78413407 and Chr15:78416520 (Figure 3D). This CNV removes the coding exon 2 (c.52_86delGACTGCACCTTCTTCAATAAGAAGGACATCCTCAA) from transcripts 1 and 4 (Figure 2A) resulting in a frameshift and a truncated protein, p.Asp18Alafs*7 (Figure 2B). Long-range PCR found the deletion to segregate with hearing loss in the extended family (Figure 1A). This CNV was absent in 96 ethnically matched controls and an additional 175 hearing impaired Iranian probands.
Figure 3.

CNV detection and breakpoint mapping analysis. A: Integrated Genome Viewer (IGV) locally zoomed about exon 2 of CIB2 of proband from family L-3156 and a control sample. Reads mapped are represented by gray horizontal bars. No reads were mapped to III.3 of family L-3156 (top panel), whereas reads spanning exon 2 were mapped in the control (bottom panel). B: Detection of the 3.1 kb deletion by long range PCR. Double bands indicate heterozygotes. The upper band corresponds to the wildtype allele (~8.8kb) and the bottom represents the mutant allele (~5.7kb) which is the only band present in affected individuals. C: Breakpoint Sanger Sequence alignments of the deletion.
Discussion
We used targeted genomic enrichment and massively parallel sequencing to implicate CIB2 as the causal gene in 6 families segregating ARNSHL. Of the identified seven variants identified, four are loss-of-function (LOF) variants predicted to produce null alleles and three are missense variants; three of the LOF variants and one missense variant (p.Tyr115Cys) are novel (not reported in the Genome Aggregation Database, gnomAD; Figure 2A, Table 2). All affected individuals from families with reported consanguinity are homozygotes for the variants. To date, of the eight pathogenic variants described in CIB2, only one is a LOF variant, in trans with a missense variant in a Dutch family with ARNSHL [14] (Table 2).
In humans, CIB2 has four alternatively spliced transcripts (Figure 3A). While the expression pattern of each transcript has yet to be elucidated, the variants described here help to define which transcripts are essential for hearing. The p.Tyr110Ter variant affects all four transcripts of CIB2 (Figure 2A) and if it escapes mRNA nonsense mediated decay (NMD), would translate a truncated protein without its calcium binding motifs (Figure 2B), thus producing a peptide lacking calcium binding ability. Previous functional analysis of the p.Arg66Trp variant revealed no effect on CIB2 localization in hair cells or its calcium buffering ability [14]. The c.300_309del frameshift variant affects all four transcripts and results in a null allele or a protein (p.Glu100fs*28) lacking calcium binding ability if translated (Figure 2B).
The c.198+1G>A variant is predicted to affect a canonical splice-site by abolishing the donor site, which would lead to skipping of exon 3 in transcript 1 with a shift in reading frame and a presumed null allele. This variant also affects the first coding exon of transcript 2, but not transcript 3 and 4. A patient carrying this variant underwent successful bilateral cochlear implant implantation, indicating that this type of habilitation is successful for this type of deafness. However, the identification of this variant in CIB2 also opens the possibility for molecular intervention using short antisense oligonucleotides (ASOs), which can be designed to target splicing variants [27,28].
WES for Family L-3156 was done before the causative gene at the DFNB48 locus was discovered. While investigating candidate variants identified in family L-3156, CIB2 was identified as the gene responsible for deafness at the DFNB48 locus. Reexamining the sequencing data uncovered no variants in the coding regions of CIB2 however inspection of the coverage for CIB2 showed no reads mapping to exon 2. The deletion was confirmed using PCR primers spanning exon 2, which failed to generate an amplicon in affected individuals. Thus, this family carries the first deafness-causing CNV associated with CIB2. The CNV is flanked by several repetitive LINE and SINE elements, which favor genomic rearrangement by non-allelic homologous recombination. The large 3.1 kb deletion removes exon 2 from transcripts 1 and 4 and causes a frameshift variant and null alleles, but occurs outside the coding sequence of transcripts 2 and 3. In transcript 2, the deletion occurs in the 5′ UTR intron (Figure 2A), while in transcript 3 it inside of intron 1. While the role of 5′ UTR introns are not well understood, they have been shown to play a role in mRNA nuclear export [29], transcription and translation [30]. It is conceivable that the deletion inside of the 5′ UTR intron of transcript 2 alters transcriptional or translational activity of this isoform. While function studies need to be carried out to evaluate how this CNV affects transcripts 2 and 3, it is clear that transcripts 1 and 4 are most likely not translated into a functional peptide.
Missense variants were identified in L-1644 and Trio-C. The p.Tyr115Cys segregating in family L-1644 occurs in the second EF-hand domain just before the calcium binding site (Figure 2B). The replacement of the large hydrophobic tyrosine with the small sulfide containing cysteine alters the conformation of the calcium binding pocket (Figure 2C–D). Given that there is a native cysteine reside only 9 aa away, the new cysteine may form a new disulfide bond thus changing the conformation of the protein, or alternatively, it may compete with native disulfide bonds to destabilize the protein to alter calcium buffering. The variant is unlikely to affect localization based on data from other missense variants in CIB2, which do not have this type of impact [12–15]. It is unclear whether CIB2 p.Cys115 would affect the interaction with TMC1 or TMC2 [15].
The p.Val75Met variant has been previously described [17] (Figure 1F). The alteration replaces a small valine with the larger and bulkier sulfur-containing methionine (Figure 2E). This variant occurs in the EF-1 hand domain between two reported CIB2 variants, p.Arg66Trp and p.Phe91Ser (Figure 2B). In vitro studies of these two variants revealed they do not affect CIB2’s ability to localize to the tips of the stereocilia or its calcium-buffering property, but most likely perturb the CIB2- αIIβ integrin interaction [14,31,32]. Recent data suggest that the N-terminal region of CIB2 interacts with TMC1 and TMC2, and given the location the p.Val75Met change, it too may share the same deleterious functional characteristics - not affecting localization or calcium buffering, but rather weakening or abolishing CIB2’s ability to interact with TMC1 and TMC2 [15].
Of the 8 variants described in CIB2 to date, only one has been associated with USH1J, a p.Glu64Asp missense variant described in one Pakistani family [12]. The disease locus in the family was mapped to an interval spanning both DFNB48 and USH1H, and CIB2 was designated as the USH1J gene for two reasons. First, no variants were found in the family originally defining USH1H, and second, the critical linkage interval for USH1H did not overlap CIB2 [12,33]. However, we believe the evidence for implicating CIB2 as an USH gene is weak. Of note, the p.Glu64Asp missense variant linked to USH1J is only two amino acids removed from the recurrent p.Arg66Trp missense variant that has been linked exclusively to DFNB48 in three families [14]. In addition, all patients we studied have a normal ocular phenotype irrespective of the variant impact (i.e. null allele or missense variant). If CIB2 played a role in the pathobiology of USH, we would have expected to see a retinal phenotype in those families segregating LOF variants.
We do recognize that patient age precludes definitive exclusion of RP in three families we have studied – families 51550, Trio-B and Trio-C. In the remaining three families, however, the oldest affected persons were 20 years old (family L-3156; pathogenic variant – homozygous for p.Asp18Alafs*7), 49 years old (family L-1644; homozygous for p.Tyr115Cys) and 28 years old (family 661; homozygous for p.Tyr110Ter). These persons all have normal funduscopy, suggesting that pathogenic variants in CIB2, irrespective of variant effect be it missense or truncating, do not cause USH.
That variants in CIB2 are not causally related to USH is further supported by expression studies in the murine inner ear. CIB2 is expressed in the supporting cells, cuticular plate and stereocilia of inner and outer hair cells and co-localizes with and binds both MYO7A and WHRN [12]. However, loss of known USH proteins like MYO7A, PCDH15, harmonin, WHRN, USH2A and PDZD7 do not affect the localization of CIB2 [12,15,16,34], and complete loss of CIB2 does not perturb the localization of the USH2 proteins USH2A, ADGRV1, PDZD7 and WHRN [34]. Missense variants in CIB2 also do not affect protein localization [12–15].
There are also important phenotypic differences. Mouse models of USH caused by MYO7A, USH1C, CDH23, PDCH15 and USH1G (the genes responsible for USH1B, USH1C, USH1D, USH1F and USH1G, respectively, in humans) all share a common phenotype: congenital profound deafness and vestibular dysfunction manifested as a visible head-bobbing and circling phenotype [35–40]. Notably, mice homozygous for the targeted deletion of CIB2 or targeted knock-in animals have profound deafness but no obvious severe vestibular phenotype [15,16]. Ultrastructurally, while all USH1 mouse mutants exhibit hair cell disorganization and stereocilia defects, the CIB2 mutant does not have any gross hair cell abnormalities. Thus, absence of CIB2 in the mouse inner ear does not compare phenotypically to absence of known USH1 proteins [15,16].
In summary, we implicate CIB2 as the cause of congenital profound ARNSHL in 6 families representing 3 ethnicities. In so doing, we expand the genetic spectrum of CIB2 variants to include CNVs, splice-site variants and frameshift variants, and show that the phenotype associated with null alleles of CIB2 is ARNSHL and not USH. Based on this observation and the available murine data, we believe CIB2 is causally related only to NHSL and not Usher Syndrome. This distinction is important as providing the correct molecular interpretation of genetic variants is critical to patient care, and in the case of USH vs NSHL, may define the intervention and prevention avenues available for patients. We acknowledge that more detailed studies are required to understand the role of CIB2 in the inner ear.
Supplementary Material
Acknowledgments
We would like to thank the families reported here for their collaboration in this study. This research was supported in part by Iran National Science foundation. Grant number: 95S47307 to HN, NIDCD RO1s DC009645 to MT, DC003544, DC002842 and DC012049 to RJHS.
Footnotes
Conflict of Interest: The authors declare no competing financial interests.
References
- 1.Morton NE. Genetic Epidemiology of Hearing Impairment. Ann N Y Acad Sci. 1991;630(1):16–31. doi: 10.1111/j.1749-6632.1991.tb19572.x. [DOI] [PubMed] [Google Scholar]
- 2.Lentz J, Keats BJ. Usher Syndrome Type I [Internet] GeneReviews(®) 1993 Available from: http://www.ncbi.nlm.nih.gov/pubmed/20301442.
- 3.Schultz JM, Bhatti R, Madeo aC, Turriff a, Muskett Ja, Zalewski CK, et al. Allelic hierarchy of CDH23 mutations causing non-syndromic deafness DFNB12 or Usher syndrome USH1D in compound heterozygotes. J Med Genet. 2011;48(11):767–75. doi: 10.1136/jmedgenet-2011-100262. [DOI] [PubMed] [Google Scholar]
- 4.Petit C. Usher syndrome: from genetics to pathogenesis. Annu Rev Genomics Hum Genet [Internet] 2001;2(1):271–97. doi: 10.1146/annurev.genom.2.1.271. Available from: http://www.annualreviews.org/doi/abs/10.1146/annurev.genom.2.1.271. [DOI] [PubMed] [Google Scholar]
- 5.Gentry HR, Singer AU, Betts L, Yang C, Ferrara JD, Sondek J, et al. Structural and biochemical characterization of CIB1 delineates a new family of EF-hand-containing proteins. J Biol Chem. 2005;280(9):8407–15. doi: 10.1074/jbc.M411515200. [DOI] [PubMed] [Google Scholar]
- 6.Lewit-Bentley A, Réty S. EF-hand calcium-binding proteins. Curr Opin Struct Biol. 2000;10(6):637–43. doi: 10.1016/s0959-440x(00)00142-1. [DOI] [PubMed] [Google Scholar]
- 7.Bhattacharya S, Bunick CG, Chazin WJ. Target selectivity in EF-hand calcium binding proteins. Biochim Biophys Acta - Mol Cell Res. 2004;1742(1–3):69–79. doi: 10.1016/j.bbamcr.2004.09.002. [DOI] [PubMed] [Google Scholar]
- 8.Grabarek Z. Structural Basis for Diversity of the EF-hand Calcium-binding Proteins. J Mol Biol. 2006;359(3):509–25. doi: 10.1016/j.jmb.2006.03.066. [DOI] [PubMed] [Google Scholar]
- 9.Denessiouk K, Permyakov S, Denesyuk A, Permyakov E, Johnson MS. Two structural motifs within canonical EF-hand calcium-binding domains identify five different classes of calcium buffers and sensors. PLoS One. 2014;9(10) doi: 10.1371/journal.pone.0109287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Blazejczyk M, Sobczak A, Debowska K, Wisniewska MB, Kirilenko A, Pikula S, et al. Biochemical characterization and expression analysis of a novel EF-hand Ca2+ binding protein calmyrin2 (Cib2) in brain indicates its function in NMDA receptor mediated Ca2+ signaling. Arch Biochem Biophys [Internet] 2009;487(1):66–78. doi: 10.1016/j.abb.2009.05.002. Available from: http://dx.doi.org/10.1016/j.abb.2009.05.002. [DOI] [PubMed] [Google Scholar]
- 11.Huang H, Bogstie JN, Vogel HJ. Biophysical and structural studies of the human calcium- and integrin-binding protein family: understanding their functional similarities and differences. Biochem cell Biol. 2012;90(5):646–56. doi: 10.1139/o2012-021. [DOI] [PubMed] [Google Scholar]
- 12.Riazuddin S, Belyantseva IA, Giese APJ, Lee K, Indzhykulian AA, Nandamuri SP, et al. Alterations of the CIB2 calcium- and integrin-binding protein cause Usher syndrome type 1J and nonsyndromic deafness DFNB48. Nat Genet [Internet] 2012;44(11):1265–71. doi: 10.1038/ng.2426. Available from: http://dx.doi.org/10.1038/ng.2426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Patel K, Giese AP, Grossheim JM, Hegde RS, Delio M, Samanich J, et al. A novel C-terminal CIB2 (calcium and integrin binding protein 2) mutation associated with non-syndromic hearing loss in a Hispanic family. PLoS One. 2015;10(10):1–16. doi: 10.1371/journal.pone.0133082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zazo Seco C, Giese AP, Shafique S, Schraders M, Oonk AM, Grossheim M, et al. Novel and recurrent CIB2 variants, associated with nonsyndromic deafness, do not affect calcium buffering and localization in hair cells. Eur J Hum Genet. 2015;24(10):542–9. doi: 10.1038/ejhg.2015.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Giese APJ, Tang Y, Sinha GP, Bowl MR, Goldring AC, Parker A, et al. CIB2 interacts with TMC1 and TMC2 and is essential for mechanotransduction in auditory hair cells. Nat Commun [Internet] 2017 Jun 29;8(1):43. doi: 10.1038/s41467-017-00061-1. Available from: http://dx.doi.org/10.1038/s41467-017-00061-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Michel V, Cortese M, Lelli A, Avan P, Petit C. Profound deafness but no vestibular and retinal defects in mice lacking the CIB2/USH1J protein. EMBO Mol Med. 2017 [Google Scholar]
- 17.Booth KT, Azaiez H, Kahrizi K, Simpson AC, Tollefson WTA, Sloan CM, et al. PDZD7 and hearing loss: More than just a modifier. Am J Med Genet A [Internet] 2015 Dec;167A(12):2957–65. doi: 10.1002/ajmg.a.37274. Available from: http://www.ncbi.nlm.nih.gov/pubmed/26416264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sloan-Heggen CM, Bierer AO, Shearer AE, Kolbe DL, Nishimura CJ, Frees KL, et al. Comprehensive genetic testing in the clinical evaluation of 1119 patients with hearing loss. Hum Genet [Internet] 2016 Apr;135(4):441–50. doi: 10.1007/s00439-016-1648-8. Available from: http://www.ncbi.nlm.nih.gov/pubmed/26969326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Azaiez H, Decker AR, Booth KT, Simpson AC, Shearer AE, Huygen PLM, et al. HOMER2, a stereociliary scaffolding protein, is essential for normal hearing in humans and mice. PLoS Genet [Internet] 2015 Mar;11(3):e1005137. doi: 10.1371/journal.pgen.1005137. Available from: http://www.ncbi.nlm.nih.gov/pubmed/25816005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Azaiez H, Booth KT, Bu F, Huygen P, Shibata SB, Shearer AE, et al. TBC1D24 Mutation Causes Autosomal-Dominant Nonsyndromic Hearing Loss. Hum Mutat. 2014;35(7):819–23. doi: 10.1002/humu.22557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Diaz-Horta O, Duman D, Foster J, Sirmaci A, Gonzalez M, Mahdieh N, et al. Whole-Exome Sequencing Efficiently Detects Rare Mutations in Autosomal Recessive Nonsyndromic Hearing Loss. PLoS One. 2012;7(11):1–5. doi: 10.1371/journal.pone.0050628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bademci G, Foster J, Mahdieh N, Bonyadi M, Duman D, Cengiz FB, et al. Comprehensive analysis via exome sequencing uncovers genetic etiology in autosomal recessive nonsyndromic deafness in a large multiethnic cohort. Genet Med [Internet] 2016;18(4):364–71. doi: 10.1038/gim.2015.89. Available from: http://www.ncbi.nlm.nih.gov/pubmed/26226137%5Cnhttp://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=PMC4733433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li H, Durbin R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics. 2010;26(5):589–95. doi: 10.1093/bioinformatics/btp698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet [Internet] 2011;43(5):491–8. doi: 10.1038/ng.806. Available from: http://dx.doi.org/10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nord AS, Lee M, King M-C, Walsh T. Accurate and exact CNV identification from targeted high-throughput sequence data. BMC Genomics [Internet] 2011;12(1):184. doi: 10.1186/1471-2164-12-184. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3088570&tool=pmcentrez&rendertype=abstract. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Venselaar H, Te Beek TAH, Kuipers RKP, Hekkelman ML, Vriend G. Protein structure analysis of mutations causing inheritable diseases. An e-Science approach with life scientist friendly interfaces. BMC Bioinformatics [Internet] 2010;11(1):548. doi: 10.1186/1471-2105-11-548. Available from: http://bmcbioinformatics.biomedcentral.com/articles/10.1186/1471-2105-11-548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Slijkerman RW, Vaché C, Dona M, García-García G, Claustres M, Hetterschijt L, et al. Antisense Oligonucleotide-based Splice Correction for USH2A-associated Retinal Degeneration Caused by a Frequent Deep-intronic Mutation. Mol Ther Acids [Internet] 2016;5(10):e381. doi: 10.1038/mtna.2016.89. Available from: http://www.nature.com/doifinder/10.1038/mtna.2016.89. [DOI] [PubMed] [Google Scholar]
- 28.Lentz JJ, Gordon WC, Farris HE, MacDonald GH, Cunningham DE, Robbins CA, et al. Deafness and retinal degeneration in a novel USH1C knock-in mouse model. Dev Neurobiol. 2010;70(4):253–67. doi: 10.1002/dneu.20771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cenik C, Chua HN, Zhang H, Tarnawsky SP, Akef A, Derti A, et al. Genome analysis reveals interplay between 5???UTR introns and nuclear mRNA export for secretory and mitochondrial genes. PLoS Genet. 2011;7(4) doi: 10.1371/journal.pgen.1001366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bicknell AA, Cenik C, Chua HN, Roth FP, Moore MJ. Introns in UTRs: Why we should stop ignoring them. BioEssays. 2012;34(12):1025–34. doi: 10.1002/bies.201200073. [DOI] [PubMed] [Google Scholar]
- 31.Yamniuk AP, Vogel HJ. Calcium- and magnesium-dependent interactions between calcium- and integrin-binding protein and the integrin alphaIIb cytoplasmic domain. Protein Sci [Internet] 2005;14(6):1429–37. doi: 10.1110/ps.041312805. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=2253396&tool=pmcentrez&rendertype=abstract. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yamodo IH, Blystone SD. Calcium Integrin Binding Protein Associates with Integrins αVβ3 and αIIbβ3 Independent of β3 Activation Motifs. CellBio [Internet] 2012;1(2):30–7. doi: 10.4236/cellbio.2012.12004. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3807131&tool=pmcentrez&rendertype=abstract. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ahmed ZM, Riazuddin S, Khan SN, Friedman PL, Riazuddin S, Friedman TB. USH1H, a novel locus for type I Usher syndrome, maps to chromosome 15q22–23. Clin Genet. 2009;75(1):86–91. doi: 10.1111/j.1399-0004.2008.01038.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zou J, Chen Q, Almishaal A, Dinesh Mathur P, Zheng T, Tian C, et al. The roles of USH1 proteins and PDZ domain-containing USH proteins in USH2 complex integrity in cochlear hair cells. Hum Mol Genet. 2016;0(0):1–13. doi: 10.1093/hmg/ddw421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Alagramam KN, Murcia CL, Kwon HY, Pawlowski KS, Wright CG, Woychik RP. The mouse Ames waltzer hearing-loss mutant is caused by mutation of Pcdh15, a novel protocadherin gene. Nat Genet. 2001;27(january):99–102. doi: 10.1038/83837. [DOI] [PubMed] [Google Scholar]
- 36.Alagramam KN, Stahl JS, Jones SM, Pawlowski KS, Wright CG. Characterization of vestibular dysfunction in the mouse model for usher syndrome 1F. JARO - J Assoc Res Otolaryngol. 2005;6(2):106–18. doi: 10.1007/s10162-004-5032-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gibson F, Walsh J, Mburu P, Varela A, Brown KA, et al. A type VII myosin encoded by the mouse deafness gene shaker-1 [Internet] Nature. 1995;374:62–4. doi: 10.1038/374062a0. Available from: http://www.nature.com/doifinder/10.1038/374062a0%5Cnhttp://www.ncbi.nlm.nih.gov/pubmed/7870172. [DOI] [PubMed] [Google Scholar]
- 38.Johnson KR, Gagnon LH, Webb LS, Peters LL, Hawes NL, Chang B, et al. Mouse models of USH1C and DFNB18: Phenotypic and molecular analyses of two new spontaneous mutations of the Ush1c gene. Hum Mol Genet. 2003;12(23):3075–86. doi: 10.1093/hmg/ddg332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Di Palma F, Holme RH, Bryda EC, Belyantseva Ia, Pellegrino R, Kachar B, et al. Mutations in Cdh23, encoding a new type of cadherin, cause stereocilia disorganization in waltzer, the mouse model for Usher syndrome type 1D. Nat Genet. 2001;27(january):103–7. doi: 10.1038/83660. [DOI] [PubMed] [Google Scholar]
- 40.Kikkawa Y, Shitara H, Wakana S, Kohara Y, Takada T, Okamoto M, et al. Mutations in a new scaffold protein Sans cause deafness in Jackson shaker mice. Hum Mol Genet. 2003;12(5):453–61. doi: 10.1093/hmg/ddg042. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.