

Abstract
Connexins play vital roles in hearing, including promoting cochlear development and sustaining auditory function in the mature cochlea. Mutations in connexins expressed in the cochlear epithelium, Cx26 and Cx30, cause sensorineural deafness and in the case of Cx26, is one of the most common causes of non-syndromic, hereditary deafness. Connexins function as gap junction channels and as hemichannels, which mediate intercellular and transmembrane signaling, respectively. Both channel configurations can play important, but very different roles in the cochlea. The potential roles connexin hemichannels can play are discussed both in normal cochlear function and in promoting pathogenesis that can lead to hearing loss.
Keywords: hemichannels, connexins, deafness, pannexins, ATP
Introduction
Genetic studies have linked mutations in two connexin (Cx) genes, GJB2 and GJB6, which encode Cx26 and Cx30, respectively, to inherited forms of autosomal, recessive, nonsyndromic sensorineural deafness [1, 2]. Uniquely among ion channels, Cxs can adopt two channel configurations that impact cellular functions in very different ways. One configuration is that of a gap junction (GJ) channel, formed by the docking of two hexameric hemichannels, which functions in direct intercellular signaling. The other configuration is that of an undocked hemichannel, which functions in transmembrane and paracrine signaling.
Mutations in GJB2 are common and account for as much as 50% of severe-to-profound autosomal, recessive deafness across diverse ethnic populations [3]. These mutations are generally deletions, truncations and frameshifts that result in loss of GJ- and hemichannel-mediated functions. However, there are dominant mutations in GJB2 linked to syndromes in which deafness is accompanied by infectious and neoplastic skin disorders [4, 5]. Syndromic deafness mutations are generally missense mutations and aberrant hemichannel function is a common feature. Since individuals with Cx26 loss-of-function mutations lack skin pathology, Cx26-linked deafness syndromes appear to be gain-of-function disorders. Moreover, the lack GJ channel function in some syndromic mutants suggests that gain of function pertains to hemichannels.
The cochlear epithelia and expression Cx26 and Cx30
The cochlea has three fluid-filled compartments (Fig. 1), the scala tympani and the scala vestibuli, which contain perilymph similar in composition to plasma and the scala media, which contains endolymph that is high in K+ and low in Na+ and Ca2+. The hair cells and the surrounding supporting cells of the sensory organ of Corti are largely bathed in perilymph except at their apical surfaces where they face endolymph. The endolymph-filled scala media also rests at a high positive potential, the endolymphatic potential (EP), which is generated by the specialized epithelia, the stria vascularis, of the lateral wall. The EP is critical for proper signal transduction in hairs cells by creating a sufficient driving force for K+ entry, depolarization and transmitter release onto the central processes of the spiral ganglion neurons, the primary afferents in the auditory pathway.
Figure 1.
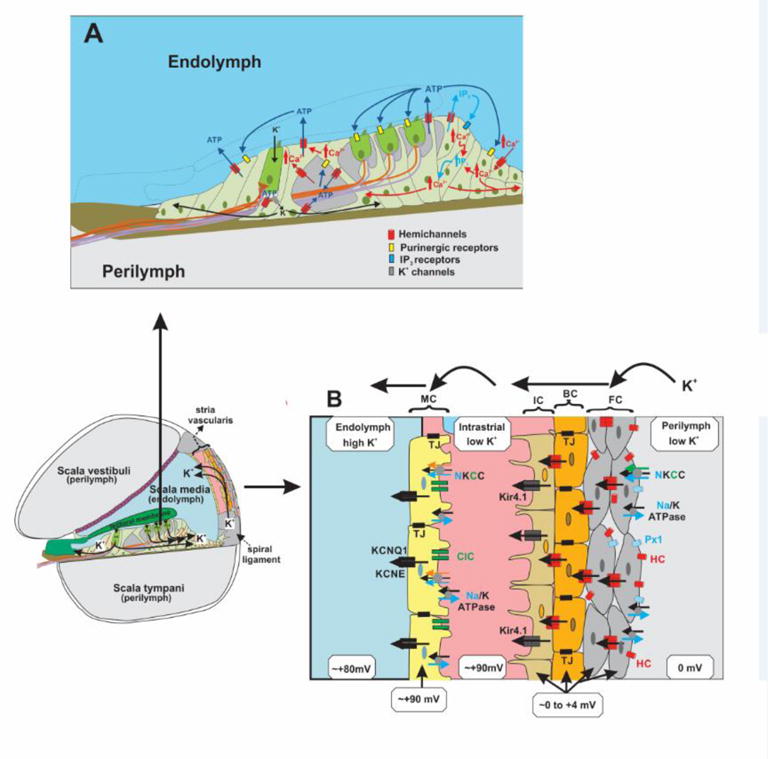
Illustrations of the cochlear ducts and epithelial cell networks depicting proposed functions of hemichannels. A) Expanded view of the sensory epithelium. Hemichannels in supporting cells can release ATP to modulate hair cell electromotility, activate K+ channels, reduce K+ accumulation and initiate Ca2+ waves. Ca2+ signaling could also involve hemichannel-mediated IP3 release and direct Ca2+ entry. B) Expanded view of the stria vascularis in the lateral wall. Hemichannels can depolarize the multi-cell layer consisting of fibrocytes (FC), basal cells (BC) and intermediate cells (IC). These cells are separated from the marginal cells (MC) by an electrically-insulated intrastrial space. Shown are the key pumps, transporters, ion channels, the potentials and K+ concentrations of the cellular and extracellular compartments that move K+ from perilymph to endolymph; Na/K ATPases, N/K/Cl (NKCC) co-transporters, ClC chloride channels, Kir4.1 and KCNQ1/KCNE K+ channels, Cx hemichannels (HC) and gap junctions (GJs), Panx1 channels (Px1) and tight junctions (TJs).
Immunostaining has shown widespread and overlapping expression of both Cx26 and Cx30 in supporting cells of the sensory epithelium and in the lateral wall; expression is absent in hairs cells [6–8]. Although coupling is extensive, there are two independent coupled cellular networks, one comprised of the supporting cells of the organ of Corti and the cells that extend into the spiral ligament and the other comprised of fibrocytes, basal cells and intermediate cells in the lateral wall [9].
Cx26 and Cx30 have been shown to co-localize to the same GJ plaques suggesting that heteromeric channels form. Functional studies that have examined properties of GJ channels in cell lines expressing Cx26 and/or Cx30 as well as in cochlear supporting cells in rodents indicate that Cx26 and Cx30 can and do form heteromeric channels [10–12], although homomeric channels in the cochlea also likely occur. A recent study in human cochlea using super-resolution fluorescence microscopy showed that Cx26 and Cx30 reside in separate GJ plaques in the lateral wall of [13]. Thus, the degree of heteromerization and its function in human cochlea remains unknown.
Cx hemichannels and the cochlear sensory epithelium
Immunofluorescence staining using antibodies directed against the extracellular loops of Cx26 was reported in supporting cells in mouse organotypic cultures, notably in the apical membranes exposed to endolymph [14]. These hemichannels would be exposed to a low “extracellular” Ca2+ environment (~20uM), a condition that favors hemichannel opening. Functional studies have shown increased uptake of fluorescent dyes in several different supporting cell types isolated from guinea pig cochleae upon exposure to low extracellular Ca2+ [15]. Dye uptake was blocked by 18-AGA, a non-specific inhibitor of GJ channels, but not by inhibitors of P2x receptors.
Various roles for hemichannels in the cochlea have been proposed, mainly linked to release of signaling molecules such as ATP and IP3 (summarized in Fig. 1A). Mechanical stimulation was suggested to induce hemichannel opening linking ATP release to acoustic stimulation-induced basilar membrane vibrations. ATP can dampen the electromotility of outer hair cells through activation of P2 receptors providing a negative feedback mechanism that can regulate hearing sensitivity and protect against cell damage at high sound intensities [16]. Propagation of intercellular Ca2+ waves through the cochlear epithelium also was attributed to ATP release through hemichannels [17]. Focally applied ATP in mouse cochlear organotypic cultures was shown to activate purinergic receptors in neighboring cells, initiating Ca2+ release and propagated Ca2+ waves. Ca2+ responses failed to propagate in cultures from mice lacking Cx26, but continued to propagate in cultures from mice lacking P2x7 receptors or pannexin-1 (Panx1) channels. Although the role Ca2+ waves play is unclear, Ca2+ signaling may affect connexin expression thereby affecting cochlear homeostasis [18]. In addition to ATP, hemichannels were reported to release IP3 [19]. Immunofluorescent labeling showed staining of IP3 receptors in the sensory epithelium both intracellularly and at the cell surface. Release of IP3 increased upon exposure to Ca2+-free solutions and was blocked by several Cx channel inhibitors, but not with the P2x receptor antagonist, PPADS. Because extracellular IP3 can be stable due to a lack of degradative enzymes, it could provide an additional mechanism by which Ca2+ signals can be initiated and spread over distances through regenerative release of IP3. Finally, ATP release through hemichannels was suggested to activate inward K+ currents that promote K+ entry into supporting cells, thereby potentially functioning to reduce extracellular accumulation of K+ near hair cells [20].
Cx hemichannels and the lateral wall
The stria vascularis with the lateral wall is responsible for generating the EP and is comprised of a multi-cell layer consisting of fibrocytes, basal cells and intermediate cells and a monolayer of marginal cells whose apical ends face the scala media; an intrastrial space separates the two cell layers (Fig. 1B). Cxs are expressed in fibrocytes, basal cells and intermediate cells and the abundant GJs between these cells establishes an electrical syncytium. Cx expression is lacking in marginal cells. Na+/K+ ATPases and Na+/K+/Cl− (NCCK) cotransporters in fibrocytes mediate uptake of K+ from the perilymph, which diffuses to the basal and intermediate cells via GJs and exits into the intrastrial space via Kir4.1 channels [21]. Tight junctions electrically isolate the intrastrial space from the perilymph, endolymph and capillary blood. The intrastrial space has been shown to have low K+ concentration and to reside at a high positive potential [22]. K+ in the intrastrial space is taken up by Na+/K+-ATPases and NCCKs into the marginal cells, which moves into the endolymph via KCNQ1/KCNE1 channels.
An important factor in the net movement of K+ from perilymph to endolymph is the unusually depolarized membrane potentials of the fibrocytes, basal cells and intermediate cells, which are in the range of 0 mV to +4 mV. This potential contributes to a favorable electrochemical gradient that moves K+ out of the intermediate cells into the intrastrial space. The origin of this depolarized membrane potential remains unknown, but it is feasible that Cx hemichannels serve this role, although this possibility has not been directly tested. Conditional deletion of Cx26 using different Cre lines showed either little or modest reductions in EP that did not correlate with the degree of hearing loss [23]. Cx30 null mice showed a substantial reduction in EP [24], but reduced Cx26 expression was also evident in these mice and deafness could be prevented when Cx26 expression was maintained [25, 26]. Thus, maintenance of either Cx26 or Cx30 expression appears to largely preserve the EP. A reduced EP with reductions in expression of both Cxs could result in insufficient numbers of hemichannels to maintain depolarization of the syncytial cells and reduced coupling that could produce deficient metabolic communication and/or signaling. Cx30 null mice also showed possible damage to endothelial cells of blood vessels in the stria vascularis, which would disrupt the electrical isolation of intrastrial space and lead to a collapse of the EP separate from GJ or hemichannel function [27].
Cx hemichannels and the developing cochlea
Prior to the onset of hearing, inner hair cells in rodent cochleae release glutamate onto the processes of spiral ganglion neurons suggesting the presence of spontaneous activity, which can support neuronal survival and guide wiring of the auditory pathways [28]. Bursts of spontaneous spiking in spiral ganglion neurons were attributed to ATP released from supporting cells that, in turn, depolarized inner hair cells to cause glutamate release. Moreover, spontaneous ATP release could be enhanced by low extracellular Ca2+ and blocked by Cx channel inhibitors octanol and carbenoxolone. ATP release elicited synchronized activities of neighboring inner hair cells suggesting those encoding similar frequencies could be synchronized, helping refine the tonotopic segregation of neuronal projections in auditory pathways.
An alternative view as to the origin of spontaneous activity suggests that currents through the transduction channels in the apical membranes of hair cells, enhanced by the low-Ca2+ environment of the endolymph, can provide a sufficient depolarizing influence [29]. It has also been suggested that ATP concentrations produced by release from supporting cells may be insufficient to generate a depolarizing response [18]. In fact, endogenous levels of ATP around inner hair cells were shown to have a hyperpolarizing effect through the activation of SK type K+ channels that may be linked to P2x3 receptors, which respond to nanomolar concentrations of ATP [30]. P2x3 receptors show an apical-to-basal gradient of expression in the cochlea [31], which could explain tonotopic differences in inner hair cell activity observed along the cochlea [30]. Thus, ATP release from hemichannels in supporting cells may not be necessary for spontaneous activity in hair cells, but could still play an important role in modulating electrical activity, thereby contributing to the refining of the auditory neural circuits in the brainstem.
Conditional deletion of Cx26 was also shown to have effects on cochlear development and to impair postnatal development of the organ of Corti (reviewed in [32]). Whether hemichannels specifically contribute to early cochlear development is unknown.
Enter Pannexins
Pannexins function as membrane channels with a similar capacity as Cx hemichannels to mediate the transmembrane flux of larger molecules such as ATP [33]. Although they bear no sequence similarity to Cxs, pannexins and Cxs adopt similar membrane topologies and are inhibited by many of the same pharmacological agents suggesting they share common structural motifs. Thus, attributing dye uptake and release of ATP to Cx hemichannels based pharmacological inhibition can be problematic [34]. This issue now affects interpretation of previous data in the cochlea with pannexins, particularly Panx1, which is expressed in some supporting cells in the sensory epithelium and in the lateral wall [35].
Using a Panx1 cKO mouse and a foxg1-Cre mouse line to delete Panx1 [36], ATP release was shown to be substantially reduced in tissue extracted from the lateral wall. Cx26 cKO mice generated with pax2-Cre as well as Cx30 KO mice showed no changes in ATP release. However, previous examination of Cx26 cKO mice generated using pax-2 Cre showed no statistical difference in Cx26 protein expression in the stria vascularis [37]. The lack of an effect on ATP release in mice null for Cx30, which also show reduced levels of Cx26 [25], argues in favor of Panx1 as the principle source of ATP release in the lateral wall.
Additional considerations for assessing contributions of Panx1 channels and Cx hemichannels in cochlea include the reported increases in ATP release and dye uptake in cochlear supporting cells with reduced extracellular Ca2+ [16]. Open probability of Cx hemichannels, but not Panx1 channels, increases in low extracellular Ca2+ [38]. However, Panx1 can exhibit Ca2+ sensitivity indirectly through its activation by P2x7 receptors, but is inconsistent with the lack of an effect of P2x channel blockers in ATP-mediated Ca2+ signaling. Also, Ca2+ responses in supporting cells failed to propagate in cochlear organotypic cultures from mice lacking Cx26 expression, but continued to propagate in cultures from mice globally null for Panx1or P2x7 receptors [17]. Of note, Cx hemichannels can be activated at physiological Ca2+ levels in the 1–2 mM range [39]. Thus, various conditions may activate Cx hemichannels, separate from reductions in extracellular Ca2+.
A final point on pannexins in the cochlea, conditional deletion of Panx1 using mice from KOMP (UC Davis, CA) and foxg1-Cre or pax2-Cre mouse line to drive deletion was reported to cause moderate to severe hearing loss [40]. However, another study found no hearing deficits in Panx1 null mice, generated by Genentech (San Francisco, CA) and a ubiquitously active Cre deleter mouse to achieve widespread deletion of Panx1[41]. Global deletion of Panx1 did not alter expression of Cx26 or Cx30. Thus, unlike Cx26, in which all mouse models agree that Cx26 is essential for hearing, the importance of Panx1 in hearing requires further examination.
Syndromic deafness mutations and hemichannels
There is a growing list of missense mutations in Cx26 associated with syndromic deafness in which aberrant hemichannel function is a common feature [5, 42]. The most studied Cx-linked deafness syndrome is keratitis-ichthyosis-deafness (KID). Mutations at 8 positions in Cx26 have been identified [42] and most of them cluster in the N-terminal domain (NT) and at the border of the first transmembrane (TM1) and first extracellular loop (E1) domains (Fig. 2A), which contribute to the aqueous pore and to channel gating and regulation by voltage, Ca2+ and pH [43]. When superimposed on the crystal structure of Cx26 [44], KID mutations localize to the pore and to interfaces between subunits (Figs. 2B).
Figure 2.
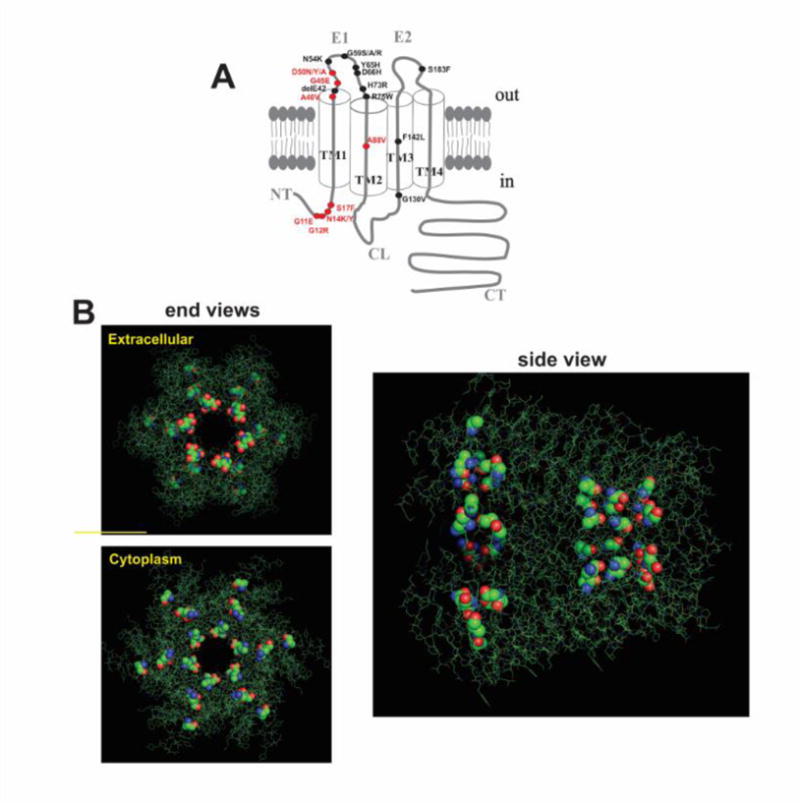
Cx26 syndromic deafness mutants. A) Topology of a Cx subunit consisting of four transmembrane domains (TM1-TM4), two extracellular loops (E1, E2), a cytoplasmic loop (CL) and amino (NT) and carboxy (CT) termini. Denoted are the 18 residues with mutations causing syndromic deafness; those associated with KID syndrome are indicated in red. B) Representation of six subunits (in green) around a central aqueous pore from the atomic structure of Cx26 (Protein Data Bank accession no. 2ZW3) published by [44]. The structure is displayed using the PyMOL Molecular Graphics System, Version 1.8 Schrödinger, LLC. KID mutant residues are shown as spherical renditions. Shown are end views from the extracellular and cytoplasmic sides a side view. The KID mutations often occur at residues facing the pore and at subunit interfaces.
Several KID mutations have been shown to weaken Cx26 hemichannel inhibition by extracellular Ca2+, suggesting a mechanism for cellular dysfunction caused by excessive hemichannel opening. However, the G45E mutation, which is associated with a fatal form of KID syndrome [45], maintains robust Ca2+ inhibition [46]. Biophysical studies demonstrated that G45 is a pore-lining residue and that the G45E mutation increases hemichannel permeability to Ca2+ [46]. This gain-of-function provides a compelling explanation for the serious phenotype G45E patients’ experience, namely excessive Ca2+ entry leading to cell dysfunction and/or death triggered by Ca2+-dependent signaling cascades.
Using substituted cysteine accessibility, several KID mutations, like G45E, have been shown to occur at pore-lining residues [46–48], suggesting that altered hemichannel permeability may be a common mechanism of disease pathogenesis. Effects on permeability would depend on the nature of the mutation, which together with other aberrant hemichannel properties could result in heterogeneous phenotypes among patients carrying different KID mutations.
Aberrant Cx26 hemichannel function has not been explicitly tested in the cochlea. Although the lack of skin disorders with loss of Cx26 implicates a gain of function in syndromic deafness, both loss and gain of Cx26 function could be detrimental to hearing. Loss of Cx26 function is a leading cause of nonsyndromic deafness, but it is unknown whether loss of hemichannel function is a contributing factor. In syndromic cases, gain of hemichannel function is a possible contributor to hearing loss, but some mutants function as GJ channels, which also may behave aberrantly and contribute to disease. Mutants that do not function as homomeric GJ channels may function when co-expressed with WT Cx26 and/or with Cx30. Aberrant heteromization has been described in the S17F KID syndrome mutant, which leads to gain of function via heteromeric hemichannels that normally do not form [49].
Conclusions
Several roles for Cx hemichannels in the cochlea have been proposed, mainly linked to ATP release from supporting cells. The more recent reports of Panx1 expression in supporting cells requires re-analyses of the origins of ATP release. Reports of the effects of deleting Panx1 in mice, both with respect to ATP release and hearing loss, are not in agreement and may be due to differences in the mouse lines and the mechanisms used to generate Panx1 and Cx26 KO animals. In disease, a convincing argument can be made for aberrantly-functioning Cx26 hemichannels as contributors to hearing loss, whether or not hemichannels have a significant role to play physiologically. Unequivocal experimental evidence for aberrant hemichannels in hearing loss, however, is lacking and awaits the development of specific tools to distinguish contributions of hemichannels, GJs and pannexin channels in cochlear physiology and pathology.
Highlights.
Connexin hemichannels composed of Cx26 and/or Cx30 have been proposed to play multiple important roles in developing and mature cochlea
ATP release by hemichannels may play a prominent role in cochlear function, but controversy has emerged between hemichannels and Panx1 channels as principal contributors to ATP release
Aberrant Cx26 hemichannel function likely contributes to cochlear pathogenesis in syndromic sensorineural deafness
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Kelsell DP, Dunlop J, Stevens HP, Lench NJ, Liang JN, Parry G, Mueller RF, Leigh IM. Connexin 26 mutations in hereditary non-syndromic sensorineural deafness. Nature. 1997;387:80–83. doi: 10.1038/387080a0. [DOI] [PubMed] [Google Scholar]
- 2.Grifa A, Wagner CA, L DA, Melchionda S, Bernardi F, Lopez-Bigas N, Rabionet R, Arbones M, Monica MD, Estivill X, Zelante L, Lang F, Gasparini P. Mutations in GJB6 cause nonsyndromic autosomal dominant deafness at DFNA3 locus [letter] Nat Genet. 1999;23:16–18. doi: 10.1038/12612. [DOI] [PubMed] [Google Scholar]
- 3.Angeli S, Lin X, Liu XZ. Genetics of hearing and deafness. Anat Rec (Hoboken) 2012;295:1812–1829. doi: 10.1002/ar.22579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Richard G. Connexins: a connection with the skin. Exp Dermatol. 2000;9:77–96. doi: 10.1034/j.1600-0625.2000.009002077.x. [DOI] [PubMed] [Google Scholar]
- 5.Lee JR, White TW. Connexin-26 mutations in deafness and skin disease. Expert Rev Mol Med. 2009;11:e35. doi: 10.1017/S1462399409001276. [DOI] [PubMed] [Google Scholar]
- 6.Lautermann J, ten Cate WJ, Altenhoff P, Grummer R, Traub O, Frank H, Jahnke K, Winterhager E. Expression of the gap-junction connexins 26 and 30 in the rat cochlea. Cell Tissue Res. 1998;294:415–420. doi: 10.1007/s004410051192. [DOI] [PubMed] [Google Scholar]
- 7.Forge A, Becker D, Casalotti S, Edwards J, Marziano N, Nevill G. Gap junctions in the inner ear: comparison of distribution patterns in different vertebrates and assessement of connexin composition in mammals. J Comp Neurol. 2003;467:207–231. doi: 10.1002/cne.10916. [DOI] [PubMed] [Google Scholar]
- 8.Zhao HB, Yu N. Distinct and gradient distributions of connexin26 and connexin30 in the cochlear sensory epithelium of guinea pigs. J Comp Neurol. 2006;499:506–518. doi: 10.1002/cne.21113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kikuchi T, Kimura RS, Paul DL, Takasaka T, Adams JC. Gap junction systems in the mammalian cochlea. Brain Res Brain Res Rev. 2000;32:163–166. doi: 10.1016/s0165-0173(99)00076-4. [DOI] [PubMed] [Google Scholar]
- 10.Ahmad S, Chen S, Sun J, Lin X. Connexins 26 and 30 are co-assembled to form gap junctions in the cochlea of mice. Biochem Biophys Res Commun. 2003;307:362–368. doi: 10.1016/s0006-291x(03)01166-5. [DOI] [PubMed] [Google Scholar]
- 11.Sun J, Ahmad S, Chen S, Tang W, Zhang Y, Chen P, Lin X. Cochlear gap junctions coassembled from Cx26 and 30 show faster intercellular Ca2+ signaling than homomeric counterparts. Am J Physiol Cell Physiol. 2005;288:C613–623. doi: 10.1152/ajpcell.00341.2004. [DOI] [PubMed] [Google Scholar]
- 12.Yum SW, Zhang J, Valiunas V, Kanaporis G, Brink PR, White TW, Scherer SS. Human connexin26 and connexin30 form functional heteromeric and heterotypic channels. Am J Physiol Cell Physiol. 2007;293:C1032–1048. doi: 10.1152/ajpcell.00011.2007. [DOI] [PubMed] [Google Scholar]
- 13.Liu W, Edin F, Blom H, Magnusson P, Schrott-Fischer A, Glueckert R, Santi PA, Li H, Laurell G, Rask-Andersen H. Super-resolution structured illumination fluorescence microscopy of the lateral wall of the cochlea: the Connexin26/30 proteins are separately expressed in man. Cell Tissue Res. 2016;365:13–27. doi: 10.1007/s00441-016-2359-0. [DOI] [PubMed] [Google Scholar]
- 14.Majumder P, Crispino G, Rodriguez L, Ciubotaru CD, Anselmi F, Piazza V, Bortolozzi M, Mammano F. ATP-mediated cell-cell signaling in the organ of Corti: the role of connexin channels. Purinergic Signal. 2010;6:167–187. doi: 10.1007/s11302-010-9192-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhao HB. Connexin26 is responsible for anionic molecule permeability in the cochlea for intercellular signalling and metabolic communications. Eur J Neurosci. 2005;21:1859–1868. doi: 10.1111/j.1460-9568.2005.04031.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhao HB, Yu N, Fleming CR. Gap junctional hemichannel-mediated ATP release and hearing controls in the inner ear. Proc Natl Acad Sci U S A. 2005;102:18724–18729. doi: 10.1073/pnas.0506481102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Anselmi F, Hernandez VH, Crispino G, Seydel A, Ortolano S, Roper SD, Kessaris N, Richardson W, Rickheit G, Filippov MA, Monyer H, Mammano F. ATP release through connexin hemichannels and gap junction transfer of second messengers propagate Ca2+ signals across the inner ear. Proc Natl Acad Sci U S A. 2008;105:18770–18775. doi: 10.1073/pnas.0800793105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mammano F. ATP-dependent intercellular Ca2+ signaling in the developing cochlea: facts, fantasies and perspectives. Semin Cell Dev Biol. 2013;24:31–39. doi: 10.1016/j.semcdb.2012.09.004. [DOI] [PubMed] [Google Scholar]
- 19.Gossman DG, Zhao HB. Hemichannel-mediated inositol 1,4,5-trisphosphate (IP3) release in the cochlea: a novel mechanism of IP3 intercellular signaling. Cell Commun Adhes. 2008;15:305–315. doi: 10.1080/15419060802357217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhu Y, Zhao HB. ATP-mediated potassium recycling in the cochlear supporting cells. Purinergic Signal. 2010;6:221–229. doi: 10.1007/s11302-010-9184-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Takeuchi S, Ando M, Kakigi A. Mechanism generating endocochlear potential: role played by intermediate cells in stria vascularis. Biophys J. 2000;79:2572–2582. doi: 10.1016/S0006-3495(00)76497-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hibino H, Nin F, Tsuzuki C, Kurachi Y. How is the highly positive endocochlear potential formed? The specific architecture of the stria vascularis and the roles of the ion-transport apparatus. Pflugers Arch. 2010;459:521–533. doi: 10.1007/s00424-009-0754-z. [DOI] [PubMed] [Google Scholar]
- 23.Chen J, Chen J, Zhu Y, Liang C, Zhao HB. Deafness induced by Connexin 26 (GJB2) deficiency is not determined by endocochlear potential (EP) reduction but is associated with cochlear developmental disorders. Biochem Biophys Res Commun. 2014;448:28–32. doi: 10.1016/j.bbrc.2014.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Teubner B, Michel V, Pesch J, Lautermann J, Cohen-Salmon M, Sohl G, Jahnke K, Winterhager E, Herberhold C, Hardelin JP, Petit C, Willecke K. Connexin30 (Gjb6)-deficiency causes severe hearing impairment and lack of endocochlear potential. Hum Mol Genet. 2003;12:13–21. doi: 10.1093/hmg/ddg001. [DOI] [PubMed] [Google Scholar]
- 25.Ahmad S, Tang W, Chang Q, Qu Y, Hibshman J, Li Y, Sohl G, Willecke K, Chen P, Lin X. Restoration of connexin26 protein level in the cochlea completely rescues hearing in a mouse model of human connexin30-linked deafness. Proc Natl Acad Sci U S A. 2007;104:1337–1341. doi: 10.1073/pnas.0606855104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Boulay AC, del Castillo FJ, Giraudet F, Hamard G, Giaume C, Petit C, Avan P, Cohen-Salmon M. Hearing is normal without connexin30. J Neurosci. 2013;33:430–434. doi: 10.1523/JNEUROSCI.4240-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cohen-Salmon M, Regnault B, Cayet N, Caille D, Demuth K, Hardelin JP, Janel N, Meda P, Petit C. Connexin30 deficiency causes instrastrial fluid-blood barrier disruption within the cochlear stria vascularis. Proc Natl Acad Sci U S A. 2007;104:6229–6234. doi: 10.1073/pnas.0605108104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tritsch NX, Yi E, Gale JE, Glowatzki E, Bergles DE. The origin of spontaneous activity in the developing auditory system. Nature. 2007;450:50–55. doi: 10.1038/nature06233. [DOI] [PubMed] [Google Scholar]
- 29.Johnson SL, Kennedy HJ, Holley MC, Fettiplace R, Marcotti W. The resting transducer current drives spontaneous activity in prehearing mammalian cochlear inner hair cells. J Neurosci. 2012;32:10479–10483. doi: 10.1523/JNEUROSCI.0803-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Johnson SL, Eckrich T, Kuhn S, Zampini V, Franz C, Ranatunga KM, Roberts TP, Masetto S, Knipper M, Kros CJ, Marcotti W. Position-dependent patterning of spontaneous action potentials in immature cochlear inner hair cells. Nat Neurosci. 2011;14:711–717. doi: 10.1038/nn.2803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Huang LC, Ryan AF, Cockayne DA, Housley GD. Developmentally regulated expression of the P2X3 receptor in the mouse cochlea. Histochem Cell Biol. 2006;125:681–692. doi: 10.1007/s00418-005-0119-4. [DOI] [PubMed] [Google Scholar]
- 32.Wingard JC, Zhao HB. Cellular and Deafness Mechanisms Underlying Connexin Mutation-Induced Hearing Loss - A Common Hereditary Deafness. Front Cell Neurosci. 2015;9:202. doi: 10.3389/fncel.2015.00202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Esseltine JL, Laird DW. Next-Generation Connexin and Pannexin Cell Biology. Trends Cell Biol. 2016;26:944–955. doi: 10.1016/j.tcb.2016.06.003. [DOI] [PubMed] [Google Scholar]
- 34.Lohman AW, Isakson BE. Differentiating connexin hemichannels and pannexin channels in cellular ATP release. FEBS Lett. 2014;588:1379–1388. doi: 10.1016/j.febslet.2014.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang XH, Streeter M, Liu YP, Zhao HB. Identification and characterization of pannexin expression in the mammalian cochlea. J Comp Neurol. 2009;512:336–346. doi: 10.1002/cne.21898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chen J, Zhu Y, Liang C, Chen J, Zhao HB. Pannexin1 channels dominate ATP release in the cochlea ensuring endocochlear potential and auditory receptor potential generation and hearing. Scientific reports. 2015;5:10762. doi: 10.1038/srep10762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang Y, Chang Q, Tang W, Sun Y, Zhou B, Li H, Lin X. Targeted connexin26 ablation arrests postnatal development of the organ of Corti. Biochem Biophys Res Commun. 2009;385:33–37. doi: 10.1016/j.bbrc.2009.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Johnson RG, Le HC, Evenson K, Loberg SW, Myslajek TM, Prabhu A, Manley AM, O’Shea C, Grunenwald H, Haddican M, Fitzgerald PM, Robinson T, Cisterna BA, Saez JC, Liu TF, Laird DW, Sheridan JD. Connexin Hemichannels: Methods for Dye Uptake and Leakage. J Membr Biol. 2016;249:713–741. doi: 10.1007/s00232-016-9925-y. [DOI] [PubMed] [Google Scholar]
- 39.Saez JC, Schalper KA, Retamal MA, Orellana JA, Shoji KF, Bennett MV. Cell membrane permeabilization via connexin hemichannels in living and dying cells. Exp Cell Res. 2010;316:2377–2389. doi: 10.1016/j.yexcr.2010.05.026. [DOI] [PubMed] [Google Scholar]
- 40.Zhao HB, Zhu Y, Liang C, Chen J. Pannexin 1 deficiency can induce hearing loss. Biochem Biophys Res Commun. 2015;463:143–147. doi: 10.1016/j.bbrc.2015.05.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Abitbol JM, Kelly JJ, Barr K, Schormans AL, Laird DW, Allman BL. Differential effects of pannexins on noise-induced hearing loss. Biochem J. 2016;473:4665–4680. doi: 10.1042/BCJ20160668. [DOI] [PubMed] [Google Scholar]
- 42.Sanchez HA, Verselis VK. Aberrant Cx26 Hemichannels and Keratitis-Ichthyosis-Deafness Syndrome: Insights into Syndromic Hearing Loss. Frontiers in Cellular Neuroscience. 2014;8 doi: 10.3389/fncel.2014.00354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kronengold J, Srinivas M, Verselis VK. The N-terminal half of the connexin protein contains the core elements of the pore and voltage gates. J Membr Biol. 2012;245:453–463. doi: 10.1007/s00232-012-9457-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Maeda S, Nakagawa S, Suga M, Yamashita E, Oshima A, Fujiyoshi Y, Tsukihara T. Structure of the connexin 26 gap junction channel at 3.5 A resolution. Nature. 2009;458:597–602. doi: 10.1038/nature07869. [DOI] [PubMed] [Google Scholar]
- 45.Janecke AR, Hennies HC, Gunther B, Gansl G, Smolle J, Messmer EM, Utermann G, Rittinger O. GJB2 mutations in keratitis-ichthyosis-deafness syndrome including its fatal form. Am J Med Genet A. 2005;133A:128–131. doi: 10.1002/ajmg.a.30515. [DOI] [PubMed] [Google Scholar]
- 46.Sanchez HA, Mese G, Srinivas M, White TW, Verselis VK. Differentially altered Ca2+ regulation and Ca2+ permeability in Cx26 hemichannels formed by the A40V and G45E mutations that cause keratitis ichthyosis deafness syndrome. J Gen Physiol. 2010;136:47–62. doi: 10.1085/jgp.201010433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sanchez HA, Villone K, Srinivas M, Verselis VK. The D50N mutation and syndromic deafness: altered Cx26 hemichannel properties caused by effects on the pore and intersubunit interactions. J Gen Physiol. 2013;142:3–22. doi: 10.1085/jgp.201310962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sanchez HA, Slavi N, Srinivas M, Verselis VK. Syndromic deafness mutations at Asn 14 differentially alter the open stability of Cx26 hemichannels. J Gen Physiol. 2016;148:25–42. doi: 10.1085/jgp.201611585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Garcia IE, Maripillan J, Jara O, Ceriani R, Palacios-Munoz A, Ramachandran J, Olivero P, Perez-Acle T, Gonzalez C, Saez JC, Contreras JE, Martinez AD. Keratitis-ichthyosis-deafness syndrome-associated Cx26 mutants produce nonfunctional gap junctions but hyperactive hemichannels when co-expressed with wild type Cx43. J Invest Dermatol. 2015;135:1338–1347. doi: 10.1038/jid.2015.20. [DOI] [PMC free article] [PubMed] [Google Scholar]