

Abstract
Dominant mutations in MFN2 cause a range of phenotypes, including severe, early-onset axonal neuropathy, “classical CMT2”, and late-onset axonal neuropathies. We report a large family with an axonal polyneuropathy, with clinical onset in the 20s, followed by slow progression.
Keywords: Charcot-Marie-Tooth Disease, CMT, neuropathy
Introduction
Charcot-Marie-Tooth (CMT) disease is the eponym that is usually given to inherited neuropathies that are not an obvious part of more complex syndromes. Mutations in more than 80 different genes cause CMT, which is subdivided according to the pattern of inheritance and nerve conduction values. CMT2A is the most common, dominantly inherited axonal neuropathy and is caused by many different dominant mutations in the dynamin GTPase mitofusin2 (MFN2) gene (Cartoni and Martinou, 2009). More than 100 different, disease-associated mutations have been reported to date, and many variants of unknown significance (http://hihg.med.miami.edu/code/http/cmt/public_html/index.html#/). The clinical manifestations of dominant MFN2 mutations are strikingly varied – ranging from severe, early-onset axonal neuropathy (Feely, et al., 2011), “classical CMT2”, and late-onset axonal neuropathy (Verhoeven, et al., 2006). Motor impairment often predominates the clinical picture, and some patients have additional features, such as optic neuropathy, myelopathy, and white matter changes in imaging. We report a large family with a dominantly inherited axonal CMT with clinical onset around age 20, harboring a previously unreported p.Leu741Trp mutation.
Materials and Methods
Patients
We enrolled 6 family members - 4 affected and 2 unaffected - from a large CMT2 family (Fig. 1 and Fig. S1A). All affected members were seen at the University of Pennsylvania; one unaffected person (II-1) was enrolled remotely. The proband (II-3) was first seen at age 32. She was unaware of any neurological deficits until age 20, when she noticed that she could not stand on her toes. EMG showed an axonal polyneuropathy. Given her family history, the diagnosis of CMT2 was made. Since her diagnosis, her weakness and balance have worsened. When first seen, she had only noticed weakness and numbness in both legs, but not in her hands. At age 32, she had normal strength, bulk and tone in her arms and legs except for 4+ (MRC score) in her first dorsal interosseous (FDI), 4- strength in ankle dorsiflexion bilaterally, 4 strength in ankle plantar flexion. Vibration was reduced to the knees. Pinprick was reduced to the knees. Nerve conduction tests (Table) showed a length-dependent axonal polyneuropathy, and EMG showed severe, chronic denervation in distal muscles, with no spontaneous activity.
Figure 1.
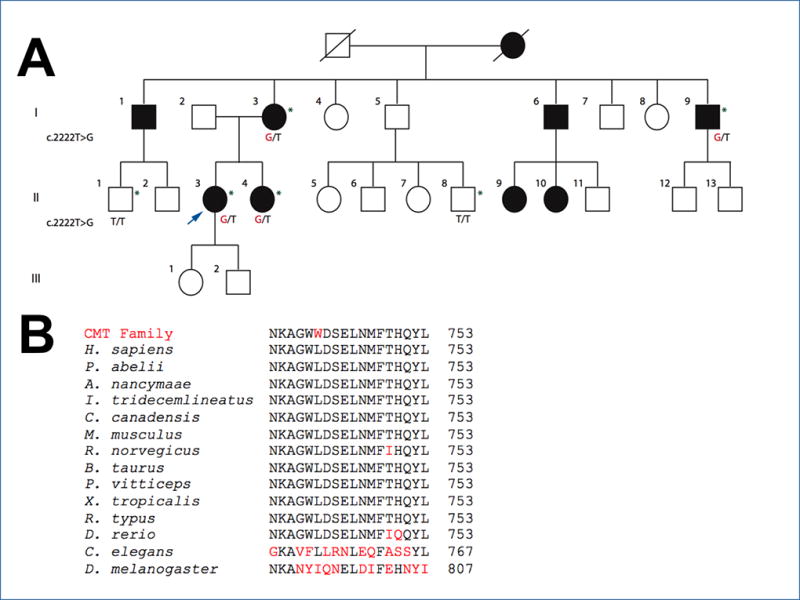
The p.Leu741Trp mutation segregates with CMT, and Leu741 is highly conserved.
(A) All enrolled affected individuals have the c.2222T>G mutation. The proband is indicated by an arrow; genetic results are shown when available; patients who were examined are indicated by an asterisk.
(B) BLAST sequence alignments show conservation of Leu741 in the MFN2/Mfn2 gene across divergent vertebrate species. P. abelii (orangutan); A. nancymaae (monkey); I. tridecemlineatus (squirrel); C. Canadensis (beaver); M. musculus (mouse); R. norvegicus (rat); B. taurus (cow); P. vitticeps (lizard); X. tropicalis (frog); R. typus (shark); D. rerio (zebrafish) conserve these mutations in MFN2-like isoforms. Leu471 is also conserved in the single Mfn gene in divergent invertebrate species, C. elegans (roundworm) but not in D. melanogaster (fruit fly). The letters shown in red are not conserved residues.
Table.
Summary of nerve conductions for affected family members
| Age at EMG |
ulnar MNCV ≥49 m/s |
ulnar CMAP ≥6 mV |
median MNCV ≥49 m/s |
median CMAP ≥4 mV |
peroneal MNCV ≥41 m/s |
peroneal CMAP EDB ≥2 mV |
tibial MNCV ≥41 m/s |
tibial CMAP ≥4 mV |
ulnar SNAP ≥7 μV (O) |
median SNAP ≥10 μV (O) |
radial SNAP ≥15 μV (A) |
sural SNAP ≥6 μV (A) |
CMTNS | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| I-3 | 63 | 53 | 6.8 | 51 | 6.2 | - | 0 | 35 | 0.9 | 0 | 0 | 0 | 0 | 16 |
| I-9 | 54 | 58 | 8 | 51 | 7.8 | - | 0 | 30 | 0.5 | 7.7 | 5.6 | 19.1 | 0 | 13 |
| II-3 | 32 | 62 | 8.6 | 62 | 8.6 | 37 | 1.2 | 37 | 2.9 | 2.1 | 4.7 | 10.3 | 4.2 | 14 |
| II-4 | 32 | 63 | 8.1 | 57 | 7.6 | 41 | 0.2 | 56 | 0.5 | 3.4 | 3.2 | 0 | 0 | 11 |
MNCV = motor nerve conduction velocity; CMAP = compound muscle action potential; SNAP = sensory nerve action potential; O = orthodromic; A = antidromic; CMTNS = CMT Neuropathy Score version 2; (Murphy, et al., 2011)
At age 23, the proband’s sister (II-4) noticed that she had trouble standing on her toes. When evaluated at age 32, she stated that her hands were becoming weak, compromising dexterity, and that her feet felt numb, but still had heat, cold, and pain sensation. Strength, bulk and tone in proximal muscles of arms and legs was normal, with mild weakness in abductor pollicis brevis (APB; 4+), FDI (4+), and extensor hallucis longus (EHL; 4). She was 4+ in ankle dorsiflexion and 4 in ankle plantar flexion and could not stand on toes or heels. Reflexes were absent at the ankles and 1+ at the knees and biceps. Vibration (Rydel-Seiffer tuning fork) was normal at 6 in the toes. Pinprick was reduced to above the ankles. She had a positive Romberg sign. Nerve conductions and needle EMG showed a length-dependent axonal polyneuropathy and chronic denervation that was severe in distal muscles (Table).
The proband’s mother (I-3) was seen at age 63. She has had swallowing difficulties since age 18. At 35, she discovered that she could not stand on her toes. In her 40s, she began to have trouble with fine finger movements. These symptoms have slowly progressed over time, and she developed stress incontinence. She had normal strength, bulk and tone in proximal muscles of arms and legs. She was 4+ in APB, 4 in FDI, and 3 in EHL. Ankle dorsiflexion and plantar flexion were 4−. Reflexes were absent in the legs, and reduced at biceps. Vibration was normal (6 at the toes.) Pinprick was reduced to above the knees. She had a positive Romberg sign. Nerve conductions and needle EMG showed a length-dependent axonal polyneuropathy and chronic denervation that was severe in distal muscles (Table).
An affected uncle (I-9) was seen at age 54. As a child, he frequently dislocated his kneecap. He had balance issues by age 20 and difficulty walking by age 35. There was weakness in distal muscles – 4 in EHL, ankle dorsiflexion, and ankle plantar flexion, 4+ in FDI and APB. Reflexes were present and symmetric at the ankles, knees and biceps. Vibration was reduced in the lower limbs. Pinprick was reduced to above the knees. He had a positive Romberg sign. Nerve conductions and needle EMG showed length-dependent axonal polyneuropathy and chronic denervation that was severe in distal muscles (Table).
Sequencing techniques and analysis
Blood samples were collected from all participants, and DNA was extracted at the University of Miami, where whole exome sequencing (WES) was performed on the proband. GATK software packages created this next generation sequencing data that was analyzed through the program Genomes Management Application (GEM.app), now GENESIS 2.0 (Gonzalez, et al., 2015; Motley, et al., 2016).
Results and Discussion
Analysis of the WES of the index patient identified a missense mutation, c.2222T>G (p.Leu741Trp) in MFN2. Sanger sequencing confirmed this mutation in the proband (Fig. S1B), her mother and sister; it was confirmed in her uncle by next generation sequencing through a commercial panel. The variant segregated in all tested family members (Fig. 1A); it is a novel mutation (https://www.genesis-app.com/#/login) and is not found in GnomAD (http://gnomad.broadinstitute.org) or NHLBI Exome Variant Server (http://evs.gs.washington.edu/EVS/).
Leu741 is located in the heptad repeat 2 (HR2) domain (amino acids 681–757; Fig. S2A), which is a coiled-coil domain and is the chief interaction site for the homo- or heterodimerization of MFN1 or MFN2 monomers (Chandhok, et al., 2017). This domain is essential for the fusion of the outer mitochondrial membranes. The entire HR2 domain is highly conserved (https://www.ebi.ac.uk/interpro/entry/IPR006884), and Leu741 is absolutely conserved across divergent vertebrate and even some invertebrate species (Fig. 1B) with high conservation scores (PhastCons Score > 0.9 and phyloP Score > 4.0). The p.Leu741Trp change is predicted to be highly disruptive and damaging by in silico predictors of mutation impact (SIFT, MutationTaster, PolyPhen-2, FATHMM, LRT, and MutationAssessor).
In the HR2 domain, 34 non-synonymous variants have been described (http://hihg.med.miami.edu/code/http/cmt/public_html/index.html#/). Some of these variants (Ala716Thr, Ala716Pro, Trp740Arg, His750Pro, Asn751Ter, Tyr752Ter) have been reported to cause a severe, early-onset axonal neuropathy, others a more typical later-onset CMT2 phenotype (Arg707_709del, Ala716dup, Trp740Ser), at least two with an older adult-onset, mild neuropathy (Arg707Trp, Trp740Cys, Glu744Asp, Glu744Met, Leu745Pro, and Met747Thr), at least one benign polymorphism (Val705Ile), and many whose clinical characteristics are inadequately described. Thus, the diversity of phenotypes that result from mutations in the HR2 domain mirrors the diversity seen from mutations in the rest of the MFN2 gene. There are not many well described families with a “CMT phenotype” caused by a MFN2 mutation. To the best of our knowledge, ours is the first such family with a mutation at an essential site for interactions with the HR2 domain, which may directly disrupt the tethering process (Fig. S2A).
The HR2 domains of MFN1 and MFN2 are highly homologous (Fig. S2B). The Leu741 in MFN2 corresponds to Leu722 in MFN1, and this residue is one of many repeating residues (20 total, 7 of which are Leu) that form the interface between the heptad repeat 1 (HR1) and HR2 α-helixes (Franco, et al., 2016). The HR2 domain is particularly important in the fusion of mitochondrial outer membranes: tethering is required for the transition from the inactive state (a cis, HR1-HR2 interaction) to the active state (a trans, HR2-HR2 interaction) (Franco, et al., 2016; Koshiba, et al., 2004). The p.Leu691Pro and p.Leu705Pro mutations in Mfn1, which corresponds to Leu710 and Leu724 in Mfn2, reduce the stability of the HR2 coiled-coil and disrupt fusion activity (Franco, et al., 2016; Koshiba, et al., 2004). Based on this model, we predict that the p.Leu741Trp mutation we report here also disrupts the ability of the HR2 domain to bind to HR1 and/or HR2, thereby affecting mitochondrial fusion. If this is the single, common pathogenic mechanism by which all MFN2 mutations cause neuropathy, then we predict that the p.Leu741Trp mutation is less disruptive than those mutations that cause a severe, early-onset axonal neuropathy, and more disruptive than mutations that cause a late-onset axonal neuropathy.
Supplementary Material
Acknowledgments
This work was supported by the Inherited Neuropathy Consortium (INC), Rare Disease Clinical Research Consortium funded by the National Institutes of Health (NINDS/ORD) and the Judy Seltzer Levenson Memorial Fund for CMT Research. The INC (U54 NS065712) is a part of the NCATS Rare Diseases Clinical Research Network (RDCRN). RDCRN is an initiative of the Office of Rare Diseases Research (ORDR), NCATS, funded through a collaboration between NCATS and the NINDS. We thank the family for participating in our work. We thank Tanya Bardakjian and Dr. David Herrmann for their help.
Footnotes
Web Resources
URLs for data presented herein are as follows:
BLAST, https://blast.ncbi.nlm.nih.gov/Blast.cgi
GENESIS, https://genomics.med.miami.edu
GnomAD, http://gnomad.broadinstitute.org
Inherited Neuropathy Variant Browser, http://hihg.med.miami.edu/code/http/cmt/public_html/index.html#/
InterPro, https://www.ebi.ac.uk/interpro/
SIFT GENOME CENTER, http://sift.jcvi.org/www/SIFT_chr_coords_submit.html
GenBank, https://www.ncbi.nlm.nih.gov/genbank/
NHLBI Exome Sequencing Project Variant Server, http://evs.gs.washington.edu/EVS/
References
- Cartoni R, Martinou JC. Role of mitofusin 2 mutations in the physiopathology of Charcot-Marie-Tooth disease type 2A. Exp Neurol. 2009;218:268–273. doi: 10.1016/j.expneurol.2009.05.003. [DOI] [PubMed] [Google Scholar]
- Chandhok G, Lazarou M, Neumann B. Structure, function, and regulation of mitofusin-2 in health and disease. Biol Rev Camb Philos Soc. 2017 doi: 10.1111/brv.12378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feely SME, Laura M, Siskind CE, Sottile S, Davis M, Gibbons VS, Reilly MM, Shy ME. MFN2 mutations cause severe phenotypes in most patients with CMT2A. Neurology. 2011;76:1690–1696. doi: 10.1212/WNL.0b013e31821a441e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franco A, Kitsis RN, Fleischer JA, Gavathiotis E, Kornfeld OS, Gong G, Biris N, Benz A, Qvit N, Donnelly SK, Chen Y, Mennerick S, Hodgson L, Mochly-Rosen D, Dorn GW., II Correcting mitochondrial fusion by manipulating mitofusin conformations. Nature. 2016;540:74–79. doi: 10.1038/nature20156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez M, Falk MJ, Gai X, Postrel R, Schule R, Züchner S. Innovative genomic collaboration using the GENESIS (GEM.app) platform. Hum Mutat. 2015;36:950–956. doi: 10.1002/humu.22836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koshiba T, Detmer SA, Kaiser JT, Chen HDR, McCaffery JM, Chan DC. Structural basis of mitochondrial tethering by mitofusin complexes. Science. 2004;305:858–862. doi: 10.1126/science.1099793. [DOI] [PubMed] [Google Scholar]
- Motley WW, Palaima P, Yum SW, Gonzalez MA, Tao F, Wanschitz JV, Strickland AV, Löscher WN, Vriendt ED, Koppi S, Medne L, Janecke A, Jordanova A, Züchner S, Scherer SS. De novo PMP2 mutations in families with type 1 Charcot-Marie-Tooth disease. Brain. 2016;139:1649–1656. doi: 10.1093/brain/aww055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy SM, Herrmann DN, McDermott MP, Scherer SS, Shy ME, Reilly MM, Pareyson D. Reliability of the CMT neuropathy score (second version) in Charcot-Marie-Tooth disease. J Periph Nerv Syst. 2011;16:191–198. doi: 10.1111/j.1529-8027.2011.00350.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verhoeven K, Claeys KG, Züchner S, Schroder JM, Weis J, Ceuterick C, Jordanova A, Nelis E, De Vriendt E, Van Hul M, Seeman P, Mazanec R, Saifi GM, Szigeti K, Mancias P, Butler IJ, Kochanski A, Ryniewicz B, De Bleecker J, Van den Bergh P, Verellen C, Van Coster R, Goemans N, Auer-Grumbach M, Robberecht W, Rasic VM, Nevo Y, Tournev I, Guergueltcheva V, Roelens F, Vieregge P, Vinci P, Moreno MT, Christen HJ, Shy ME, Lupski JR, Vance JM, De Jonghe P, Timmerman V. MFN2 mutation distribution and genotype/phenotype correlation in Charcot-Marie-Tooth type 2. Brain. 2006;129:2093–2102. doi: 10.1093/brain/awl126. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.