

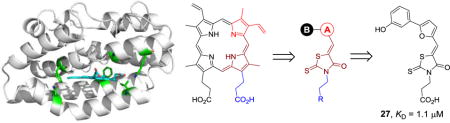
Graphical abstract
Pseudomonas aeruginosa is an opportunistic Gram-negative bacterial and one of the most common causes of hospital-acquired infections worldwide.1,2 It is the major human pathogen that leads to severe lung deterioration in cystic fibrosis (CF) patients.3 The treatment of Pseudomonas is further complicated by its resistance to most available antibiotics.4 For P. aeruginosa infections polymyxins B and E (colistin) are the only choices of antibiotics despite their high toxicity.5 Therefore, it is urgent to develop new therapeutic agents for MDR P. aeruginosa.
Iron is an essential nutrient for survival and virulence of P. aeruginosa.6 It regulates a number of virulence factors in P. aeruginosa including exotoxins, proteases, and biofilm formation.7–10 Within the host iron is not readily available due to sequestration by iron binding proteins such as transferrin and ferritin or in the form of heme.11 However Pseudomonas overcomes this situation through a variety of mechanisms including the secretion of siderophores (pyoverdine and pyochelin),12,13 ferrous iron uptake (Feo),14 and its unique heme acquisition systems.15–19
As part of the heme acquisition system, heme degradation by the enzyme heme oxygenase (HemO) is essential for P. aeruginosa to acquire iron from the host.20 P. aeruginosa HemO (pa-HemO) catalyzes the reaction to breakdown heme and release iron, along with CO and β- and δ-biliverdin.20 It has also been shown by the Wilks Lab that the catalytic activity of pa-HemO was pivotal to drive the metabolic flux of heme into the cell.21–23 Furthermore, the P. aeruginosa ΔhemO isogenic mutant, or a strain complemented with a non-functional pa-HemO, showed significant attenuation of infection in a mouse lung infection model, when compared to the wild type strain (unpublished results). This data suggests pa-HemO inhibitors, by blocking a key mechanism of the iron acquisition system, represents a promising therapeutic target for P. aeruginosa infections.
Small molecule inhibitors of pa-HemO have been reported.24–26 These inhibitors achieved activities by binding either to the heme-binding active site25 or an allosteric site26 depending on their structural features. Most of these known inhibitors are close derivatives of high throughput screen (HTS) hits, which commonly suffer from major drawbacks such as modest potency (KD 20–50 μM) and poor pharmacological properties. For instance, we recently reported pa-HemO inhibitor 1 (Fig. 1), with the KD value of 18.4 μM and limited chemical stability.25
Fig. 1.
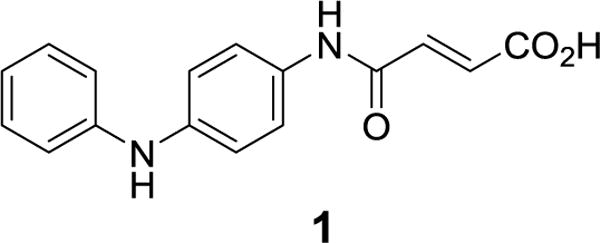
Chemical structure of the pa-HemO inhibitor 1.
The pa-HemO has a unique heme-binding active site. The solvent accessible surface of pa-HemO (~7.5 Å3) is much smaller than the mammalian enzyme HO1 (43.6–59.7 Å3).27–30 More interestingly, different to the binding mode in human HO1 (Fig. 2A) and other known isozymes, heme binds in the pa-HemO active site with a dramatically rotated (~100 degrees) orientation28 as a result of the unique amino acid network present in the active site (Fig. 2B). These structural differences provide opportunities for selective inhibition of the pa-HemO over other HemOs such as HO1.31
Fig. 2.
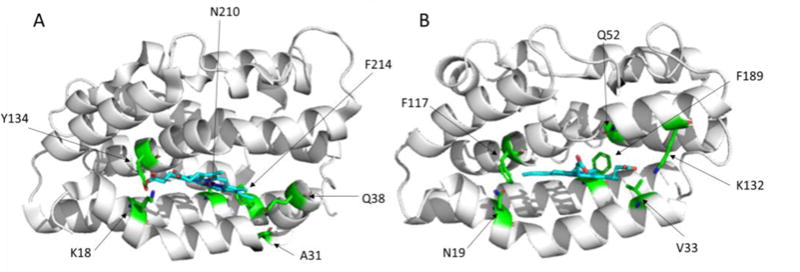
Heme-binding active sites of A) HO1 (PDB 1N45) and B) pa-HemO (PDB 1SK7).
To take advantage of the unique active site of pa-HemO, we designed a series of inhibitors (2–31) based on a 3-(4-oxo-2-thioxothiazolidin-3-yl)propanoic acid scaffold (Fig. 3). New compound design was directed by the computer-aided drug design (CADD) Site Identification by Ligand Competitive Saturation (SILCS) method as reported previously.26 The carboxylic acid or tetrazole groups of compounds 2–31 mimic one of the two acid groups of heme, to form an electrostatic interaction with the basic residue Lys132 of pa-HemO. The 4-oxo-2-thioxothiazolidin-3-yl fragment (shown in brown) is chosen to mimic one of the pyrrole rings of the porphyrin, to fit into the hydrophobic binding pocket around Val33 (Fig. 2B). The same moiety could also form potential interaction with the iron-chelating residue His26 of pa-HemO. We note the controversial reports on the application of rhodanine in medicinal chemistry;32,33 studies in our lab to modify the rhodanine are on-going. The substituted A-ring is selected to interact with the hydrophobic pocket next to residues Phe189 and Q52, while the extended tail B-ring is employed to extend the compound to interact with residues Phe117 and Asn19 of pa-HemO.
Fig. 3.
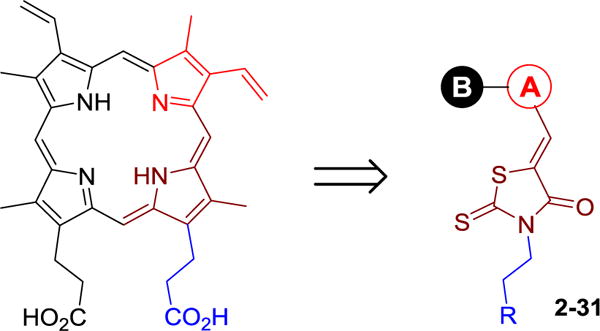
Design and general structure of new inhibitors 2–31.
To test our hypothesis, we first synthesized two pyridinyl compounds 2 and 3 (Fig. 4A). Compounds 2 and 3 indicated modest affinities to pa-HemO with the KD values of 51 μM and 27 μM, respectively, using the previously established fluorescence quenching assay.26 The potency of the tetrazole analog 3 was almost 2-fold higher than that of the carboxylic inhibitor 2. We reasoned that the enhancement in potency might due to the presence of residue Phe128 next to Lys132 (Fig. 2B), which can form additional π–π stacking interaction with the tetrazole fragment of inhibitor 3. We then performed saturation transfer difference-nuclear magnetic resonance (STD-NMR) experiments to confirm the binding of compound 3 to pa-HemO, and found that both the pyridine moiety and aliphatic tail of the inhibitor show clear binding to the enzyme (Fig. 4B). To further characterize the interaction between inhibitor 3 and pa-HemO, we carried out 1H,15N-HSQC NMR experiments (Fig. 4C). The presence of compound 3 caused multiple chemical shift perturbations (CSPs) in the heme binding site of pa-HemO. Interestingly residues with CSP over 3 fold of standard deviation (σ), including Leu32, Ser35, Lys36, Phe39, Arg85, Glu104, Ser119, Glu137, Asn141, Phe189, Gly190 and Arg195, are mostly located in the vicinity of the heme-binding active site (Fig. 4C). Of them only Arg85 and Glu104 locate on the back surface of the enzyme. The strong CSPs of these two residues can be results of the configuration change of the enzyme upon inhibitor binding. Overall, the results show that compound 3 binds to the active site with modest affinity, suggesting that the scaffold can be suitable developing inhibitors for pa-HemO.
Fig. 4.
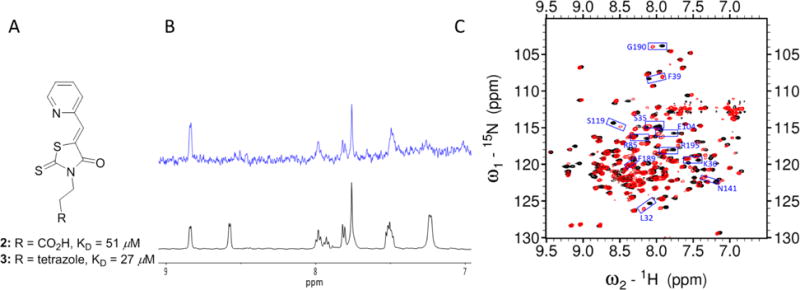
Characterization of initial compounds 2–3. A) Chemical structures and KD values of compounds 2–3 in the fluorescence quenching assays. B) STD-NMR experiment results: 1H spectra (black) and the STD spectrum (blue) of compound 3. Note that the 1H NMR of compound 3 indicated a mixture of two rotamers with estimation of 1:1 ratio. One of the two rotamers binds favorably to apo pa-HemO. C) 1H,15N-HSQC NMR results of compound 3. Black: apo pa-HemO; Red: compound 3 mixed with apo pa-HemO.
As shown in Scheme 1, Knoevenagel condensation of selected aromatic aldehydes (32) with 3-(4-oxo-2-thioxothiazolidin-3-yl)ethylene derivatives (33a-d) in the presence of 10 equivalents of sodium acetate (NaOAc) in acetic acid (AcOH) gave compounds 2–23 and 28–31 in good to excellent yields upon heating. The synthesis of compounds 24–27 has been achieved using a two-step procedure. Suzuki coupling of boronic acid 34 with substituted iodobenzenes in the presence of Pd(0) catalyst generated compound 35 in modest to good yields. Aldehyde 35 was then submitted to Knoevenagel condensation to provide inhibitors 24–27 in good yields.
Scheme 1.
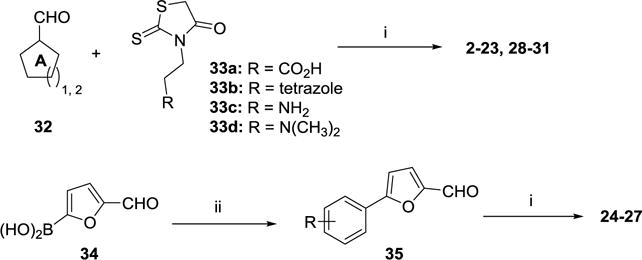
Synthesis of compounds 2–31. Reagents and conditions: (i) 3-(4-oxo-2-thioxothiazolidin-3-yl)propanoic acid, NaOAc:AcOH (1:10),105 °C, 6–14 h, 47–87%; (ii) iodobenzene, Pd(PPh3)2Cl2, K2CO3, dioxane:H2O (3:1), 100 °C, overnight, 71–87%.
Compared to inhibitor 2, the 2-furan analog 4 was slightly more potent, while the catechol analog 5 indicated similar activity (Table 1). Similar to the results of the tetrazole inhibitor 3, slightly enhanced affinities were observed when the carboxylic acid groups of compounds 4 and 5 were replaced by the aromatic acid bioisostere tetrazole. Introduction of an electrodonating methoxy substitution at either the m- (8) or p-position (9) generated new inhibitors with ~2-fold improved inhibitory potency. The p-F substituted analog 10 indicated similar affinity to that of inhibitor 2. Larger halogen Cl-substitution at the same position yielded compound 11 with > 3-fold increased affinity, although the diCl-substituted analog 12 didn’t further improve the potency. The aliphatic p-iPr analog 13 showed a 5-fold improvement in potency compared to compound 2. Despite of the decreased potency of the 1-naphthyl analog 14, the 2-naphthyl analog 15 showed a KD value of 3.9 μM. Similarly good potency was observed with the indole analog 16.
Table 1.
Structure features, calculated properties, KD values of compounds 4–16
| |||||
|---|---|---|---|---|---|
| cmpd | A | R | LGFEa (kcal/mol) | LEa | KDb(μM) |
| 4 |
|
−CO2H | −22.7 | −1.26 | 38 ± 10 |
| 5 | tetrazole | −20.8 | −1.04 | 26 ± 6 | |
| 6 |
|
−CO2H | −23.9 | −1.14 | 57 ± 8 |
| 7 | tetrazole | −23.7 | −1.03 | 45 ± 6 | |
| 8 |
|
−CO2H | −26.1 | −1.24 | 25 ± 8 |
| 9 |
|
−CO2H | −26.7 | −1.27 | 23 ± 4 |
| 10 |
|
−CO2H | −23.9 | −1.19 | 44 ± 9 |
| 11 |
|
−CO2H | −26.6 | −1.33 | 15 ± 2 |
| 12 |
|
−CO2H | −27.7 | −1.32 | 14 ± 4 |
| 13 |
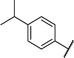
|
−CO2H | −26.9 | −1.22 | 10 ± 4 |
| 14 |
|
−CO2H | −29.6 | −1.28 | 13 ± 2 |
| 15 |
|
−CO2H | −30.0 | −1.30 | 3.9 ± 1 |
| 16 |
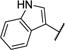
|
−CO2H | −26.6 | −1.21 | 6.4 ± 1.4 |
Ligand Grid Free Energy (LGFE) and Ligand Efficiency (LE) calculated with SILCS.26
The listed result was the average of three independent experiments.
To explore inhibitors with potential interactions to the hydrophobic pocket formed by Phe117 and Asn19, a collection of 11 new inhibitors (17–27) were synthesized with a B-ring included in their chemical structures (Table 2). We first tested structurally rigid bi-Ph compounds 17 and 18. The m-Ph analog 17 indicated a significant decrease of the inhibitory potency, however, the p-Ph compound 18 indicated a promising KD value of 3.3 μM. With the introduction of a flexible linker between the A- and B-rings, compounds 19–22 were synthesized and tested. The results indicated that the m-PhNH analog 21 gave the best inhibitory potency with a KD value of 1.2 μM. We have also studied inhibitors with extended chemical structures based upon the furan analog 4 (23–27). While the p-methylester-Ph substitution (23) significantly diminished the inhibitory potency, the same substituent at the m-position led to compound 25 with a KD value of 7.6 μM. The CN group (26) at the same position was detrimental to the binding affinity. The m-OH substituted analog gave the most potent compound 27 with a KD value of 1.1 μM, which is similar to the value of the substrate heme.24
Table 2.
Structure features, calculated properties, KD values of compounds 17–27
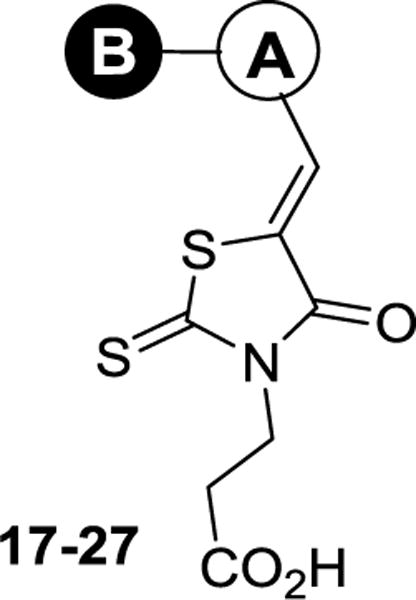
| ||||
|---|---|---|---|---|
| cmpd | A–B | LGFEa(kcal/mol) | LEa | KDb(μM) |
| 17 |
|
−31.1 | −1.24 | 20±2 |
| 18 |
|
−29.8 | −1.19 | 3.3 ± 0.9 |
| 19 |
|
−31.8 | −1.22 | 7.0 ± 2.7 |
| 20 |
|
−30.5 | −1.17 | 6.5 ± 1.3 |
| 21 |
|
−31.4 | −1.21 | 1.2 ± 0.2 |
| 22 |
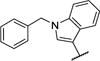
|
−32.6 | −1.13 | 7.4 ± 0.6 |
| 23 |
|
−30.9 | −1.10 | 28 ± 6 |
| 24 |
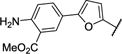
|
−28.9 | −1.00 | 6.5 ± 1.0 |
| 25 |
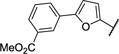
|
−30.7 | −1.10 | 7.6 ± 1.1 |
| 26 |
|
−30.1 | −1.16 | 13 ± 1.4 |
| 27 |
|
−28.7 | −1.15 | 1.1 ± 0.2 |
Ligand Grid Free Energy (LGFE) and Ligand Efficiency (LE) calculated with SILCS.26
The listed result was the average of three independent experiments.
Given the availability of the assay results, further analysis of the utility of the SILCS LGFE scores was undertaken (Fig. 5). Correlation analysis and calculation of the predictive index (PI)34 with respect to LGFE scores were performed. Based on all compounds in Tables 1–2, this analysis results in a high PI ~ 0.70 and R2 ~ 0.53 for the correlation between LGFE and KD as shown in Fig. 5. This indicates FragMaps are of utility with respect to both qualitatively directing ligand design and yield a reasonable level of quantitative predictability, indicating the utility of the approach in the further refinement of the presented compounds.
Fig. 5.
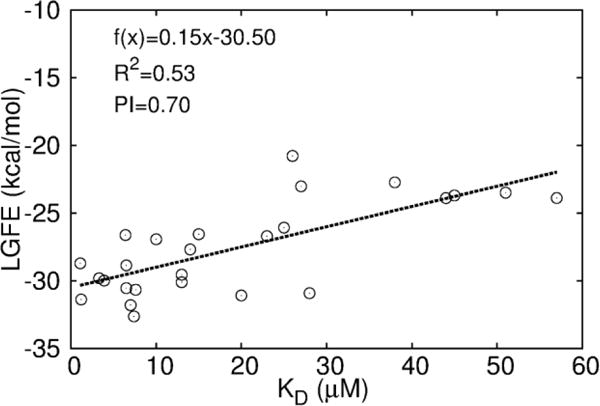
Correlation plots between LGFE and KD. Coefficient of determination (R2) and predictive index (PI) are shown for the quality of correlation.
The binding sites of inhibitors 18, 21, and 27 were studied using both SILCS calculation (Fig. S1) and 1N,15N-HSQC NMR experiments (Fig. 6 and Tables S2–S5). With the presence of inhibitor 18, significant CSPs (> 3σ) were observed for residues N19, L32, S35, K36, Q52, E104, V118, S119, S183, A185, and R195 (Fig. 6A and S2). All these residues are located in the vicinity to the heme binding active site of pa-HemO. Perturbation of six residues, L32, S35, K36, E104, S119 and R195 were also detected for inhibitor 3, suggesting that these inhibitors may bind to the same binding site of the enzyme. Compared to inhibitor 18, the addition of inhibitor 21 led to significant CSPs for less residues including L32, Q52, E104, L116, S119, L177, S183, L193 and R195 (Fig. 6B and S3). Most of these residues overlap with those for compounds 3 and 18, indicating that compound 21 likely adapts similar binding mode in the heme binding site of the pa-HemO. The presence of inhibitor 27 caused significant CSP for pa-HemO residues L18, E37, A40, Q56, L68, S77, L113, G190, and F197 (Fig. 6C and S4). In addition, 1N,15N-HSQC peaks of heme binding site residues L32, Q52, S119 and R195 disappeared due to their fast exchange rate. These results indicated that upon the binding of inhibitor 27, the heme binding site of pa-HemO becomes more flexible.
Fig. 6.
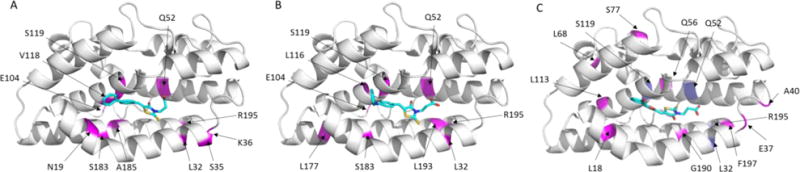
A representation of the pa-HemO based on Protein Data Bank structure (PDB: 1SK7) is shown, indicating residues perturbed upon binding of compounds 18 (A), 21 (B), and 27 (C). (A) In magenta, residues with >3σ chemical shift perturbations listed as the following in increasing order: A84, Q52, L116, L193, L177, E104, S183, R195, L32, and S119. (B) In magenta, residues with >3σ chemical shift perturbations listed as the following in increasing order: K36, Q52, A185, V118, R195, L32, S35, S119, N19, S119 and E104. (C) In magenta, residues with >3σ chemical shift perturbations listed as the following in increasing order: A185, E37, S77, G190, Q56, L113, L68, A40, L18 and F197. In blue, residues L32, Q52, S119 and R195 (shown in purple) disappeared due to the fast exchange rate.
The anti-pseudomonas activity of the three most potent compounds, 21, 27, and 18 were evaluated using both the MIC50 by growing P. aeruginosa in minimal media supplemented with heme or free iron, and biofilm formation assays (Table 3). Our results indicated all three compounds showed poor anti-bacterial effects on pseudomonas in both assays. We reasoned that the presence of the carboxylic acid group in these compounds might prohibit the in vivo effects of inhibitors, as evidenced in a number of previous studies.34,35 It has been well documented that compounds, with in vivo activities toward pseudomonas, usually employs basic amino groups.35 Therefore, we have synthesized and tested compounds 30 and 31, which showed KD values of 43 and 18 μM, respectively. However, these compounds also did not have significant antibacterial activity.
Table 3.
Structure features and properties of compounds 28–31
| cmpd | structure |
KDa (μM) |
MIC50 (μg/mL) |
Biofilm assay (μg/mL) |
|---|---|---|---|---|
| 21 |
|
1.2 ± 0.2 | > 300 | > 300 |
| 27 |
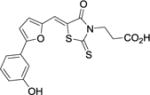
|
1.1 ± 0.2 | > 300 | > 300 |
| 18 |
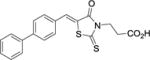
|
3.3 ± 0.9 | > 300 | > 300 |
| 28 |
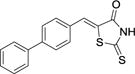
|
2.3 ± 0.5 | NAb | > 300 |
| 29 |
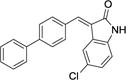
|
5.2 ± 0.7 | NAb | > 300 |
| 30 |
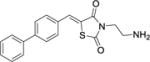
|
43 ± 11 | > 300 | > 300 |
| 31 |
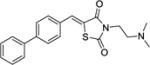
|
18 ± 3 | NAb | > 300 |
The listed result was the average of three independent experiments.
NA refers to no microbial inhibition activity occurred.
In summary, 4-Oxo-2-thioxothiazolidin-3-yl-propanoic acid based inhibitors of pa-HemO are described to interact with the unique network of residues in the heme-binding active site of the enzyme. SAR efforts of the series resulted in analogs 21 and 27, with around 1 μM affinities. NMR studies confirmed the binding site of selected inhibitors of the family, which is consistent with the results obtained from computational analyses. Further structure optimization is undergoing for anti-microbial activities of inhibitors.
Supplementary Material
Acknowledgments
We thank the National Health Institute grant #AI102883 to A.W., and Dr. Amanda Oglesby-Sherrouse for advice on the biofilm formation assays.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and Notes
- 1.Kerr KG, Snelling AM. J Hosp Infect. 2009;73:338–344. doi: 10.1016/j.jhin.2009.04.020. [DOI] [PubMed] [Google Scholar]
- 2.Salter SJ. Nat Rev Microbiol. 2015;13:69. doi: 10.1038/nrmicro3422. [DOI] [PubMed] [Google Scholar]
- 3.Costerton JW. Trends Microbiol. 2001;9:50–52. doi: 10.1016/s0966-842x(00)01918-1. [DOI] [PubMed] [Google Scholar]
- 4.Obritsch MD, Fish DN, MacLaren R, Jung R. Antimicrob Agents Chemother. 2004;48:4606–4610. doi: 10.1128/AAC.48.12.4606-4610.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Falagas ME, Kasiakou SK. Clin Infect Dis. 2005;40:1333–1341. doi: 10.1086/429323. [DOI] [PubMed] [Google Scholar]
- 6.Minandri F, Imperi F, Frangipani E, Bonchi C, Visaggio D, Facchini M, Pasquali P, Bragonzi A, Visca P. Infect Immun. 2016;84:2324–2335. doi: 10.1128/IAI.00098-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cox CD. Infect Immun. 1982;36:17–23. doi: 10.1128/iai.36.1.17-23.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Meyer JM, Neely A, Stintzi A, Georges C, Holder IA. Infect Immun. 1996;64:518–523. doi: 10.1128/iai.64.2.518-523.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Takase H, Nitanai H, Hoshino K, Otani T. Infect Immun. 2000;68:4498–4504. doi: 10.1128/iai.68.8.4498-4504.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Takase H, Nitanai H, Hoshino K, Otani T. Infect Immun. 2000;68:1834–1839. doi: 10.1128/iai.68.4.1834-1839.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Otto BR, Verweij-van Vught AM, MacLaren DM. Crit Rev Microbiol. 1992;18:217–233. doi: 10.3109/10408419209114559. [DOI] [PubMed] [Google Scholar]
- 12.Heinrichs DE, Young L, Poole K. Infect Immun. 1991;59:3680–3684. doi: 10.1128/iai.59.10.3680-3684.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Royt PW. Biol Met. 1990;3:28–33. doi: 10.1007/BF01141174. [DOI] [PubMed] [Google Scholar]
- 14.Marlovits TC, Haase W, Herrmann C, Aller SG, Unger VM. Proc Natl Acad Sci U S A. 2002;99:16243–16248. doi: 10.1073/pnas.242338299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ochsner UA, Johnson Z, Vasil ML. Microbiology. 2000;146:185–198. doi: 10.1099/00221287-146-1-185. [DOI] [PubMed] [Google Scholar]
- 16.Bhakta MN, Wilks A. Biochemistry-Us. 2006;45:11642–11649. doi: 10.1021/bi060980l. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Block DR, Lukat-Rodgers GS, Rodgers KR, Wilks A, Bhakta MN, Lansky IB. Biochemistry-Us. 2007;46:14391–14402. doi: 10.1021/bi701509n. [DOI] [PubMed] [Google Scholar]
- 18.Lansky IB, Lukat-Rodgers GS, Block D, Rodgers KR, Ratliff M, Wilks A. J Biol Chem. 2006;281:13652–13662. doi: 10.1074/jbc.M600824200. [DOI] [PubMed] [Google Scholar]
- 19.O’Neill MJ, Bhakta MN, Fleming KG, Wilks A. Proc Natl Acad Sci U S A. 2012;109:5639–5644. doi: 10.1073/pnas.1121549109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Choby JE, Skaar EP. J mol biol. 2016;428:3408–3428. doi: 10.1016/j.jmb.2016.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Barker KD, Barkovits K, Wilks A. J Biol Chem. 2012;287:18342–18350. doi: 10.1074/jbc.M112.359265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kaur AP, Lansky IB, Wilks A. J Biol Chem. 2009;284:56–66. doi: 10.1074/jbc.M806068200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.O’Neill MJ, Wilks A. ACS Chem Biol. 2013;8:1794–1802. doi: 10.1021/cb400165b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Furci LM, Lopes P, Eakanunkul S, Zhong SJ, MacKerell AD, Wilks A. J Med Chem. 2007;50:3804–3813. doi: 10.1021/jm0700969. [DOI] [PubMed] [Google Scholar]
- 25.Hom K, Heinzl GA, Eakanunkul S, Lopes PEM, Xue F, MacKerell AD, Wilks A. J Med Chem. 2013;56:2097–2109. doi: 10.1021/jm301819k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Heinzl GA, Huang W, Yu W, Giardina BJ, Zhou Y, MacKerell AD, Wilks A, Xue F. J Med Chem. 2016;59:6929–6942. doi: 10.1021/acs.jmedchem.6b00757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Friedman J, Lad L, Deshmukh R, Li H, Wilks A, Poulos TL. J Biol Chem. 2003;278:34654–34659. doi: 10.1074/jbc.M302985200. [DOI] [PubMed] [Google Scholar]
- 28.Friedman J, Lad L, Li H, Wilks A, Poulos TL. Biochemistry-Us. 2004;43:5239–5245. doi: 10.1021/bi049687g. [DOI] [PubMed] [Google Scholar]
- 29.Schuller DJ, Wilks A, de Montellano PRO, Poulos TL. Nat Struct Biol. 1999;6:860–867. doi: 10.1038/12319. [DOI] [PubMed] [Google Scholar]
- 30.Schuller DJ, Zhu W, Stojiljkovic I, Wilks A, Poulos TL. Biochemistry-Us. 2001;40:11552–11558. doi: 10.1021/bi0110239. [DOI] [PubMed] [Google Scholar]
- 31.Ratliff M, Zhu W, Deshmukh R, Wilks A, Stojiljkovic I. J Bacteriol. 2001;183:6394–6403. doi: 10.1128/JB.183.21.6394-6403.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Baell JB, Holloway GA. J Med Chem. 2010;53:2719–2740. doi: 10.1021/jm901137j. [DOI] [PubMed] [Google Scholar]
- 33.Mendgen T, Steuer C, Klein CD. J Med Chem. 2012;55:743–753. doi: 10.1021/jm201243p. [DOI] [PubMed] [Google Scholar]
- 34.Wagner S, Sommer R, Hinsberger S, Lu C, Hartmann RW, Empting M, Titz A. J Med Chem. 2016;59:5929–5969. doi: 10.1021/acs.jmedchem.5b01698. [DOI] [PubMed] [Google Scholar]
- 35.Richter MF, Drown BS, Riley AP, Garcia A, Shirai T, Svec RL, Hergenrother PJ. Nature. 2017;545:299–304. doi: 10.1038/nature22308. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.