

Abstract
Despite the development of promising direct oral anticoagulants, which are all orthosteric inhibitors, a sizable number of patients suffer from bleeding complications. We have hypothesized that allosterism based on the heparin-binding exosites presents a major opportunity to induce sub-maximal inhibition of coagulation proteases, thereby avoiding/reducing bleeding risk. We present the design of a group of sulfated benzofuran dimers that display heparin-binding site-dependent partial allosteric inhibition of thrombin against fibrinogen (ΔY = 55–75%), the first time that a small molecule (MW <800) has been found to thwart macromolecular cleavage by a monomeric protease in a controlled manner. The work leads to the promising concept that it should be possible to develop allosteric inhibitors that reduce clotting, but do not completely eliminate it, thereby avoiding major bleeding complications that beset anticoagulants today.
Keywords: Allosteric inhibition, Anticoagulants, Thrombin, Heparin-binding site, Enzyme regulation
Graphical abstract
It is estimated that nearly 10% of the adult population will be treated with anticoagulants at least once in their lifetime. Orally bioavailable agents constitute frequently used anticoagulants these days but their risk of bleeding remains substantial. A number of patients on oral anticoagulants suffer from bleeding consequences,1 which raises considerable harm.
All current direct oral anticoagulants (DOACs) target the active site of a coagulation enzyme, e.g., thrombin or factor Xa.2 In this orthosteric inhibition mechanism, the only parameter available for regulation of clotting activity is the inhibitory potency (either IC50 or KI). In contrast, allosteric inhibition mechanism offers two parameters for regulating clotting activity, i.e., potency and efficacy (ΔY). Such dual parameter regulation is not possible for orthosteric inhibitors because of their all (ΔY = 100%) or none (ΔY = 0%) property.
Allosteric inhibition also promises to offer enhanced level of selectivity because allosteric sites typically tend to be less homologous.3,4 This has certainly been found to be true for a group of allosteric inhibitors of thrombin that we have been studying for the past few years. We had reasoned that appropriate sulfated non-saccharide glycosaminoglycan mimetics (NSGMs) could be designed to target exosite 2 of thrombin and induce allosteric inhibition (Fig. 1). To realize this, we first developed sulfated low molecular weight lignins5 as polymeric mimetics of the highly sulfated polysaccharide, heparin, which binds to an exosite of certain serine proteases with 10–20 μM affinity.6 However, the sulfated lignins inhibited several coagulation proteases.7 To enhance selectivity, we designed sulfated benzofuran monomers (SBMs), which preferentially inhibited thrombin and factor Xa,8 albeit displaying a high IC50. Further improvement in design led to sulfated benzofuran dimers (SBDs, Fig. 1) that selectively inhibited thrombin by interacting with Arg173 on the periphery of exosite 2.9,10 This was a major advance. Yet, the allosteric SBDs displayed efficacies ≥90%, which does not truly afford an opportunity for efficacy-based regulation (i.e., based on ΔY).
Figure 1.
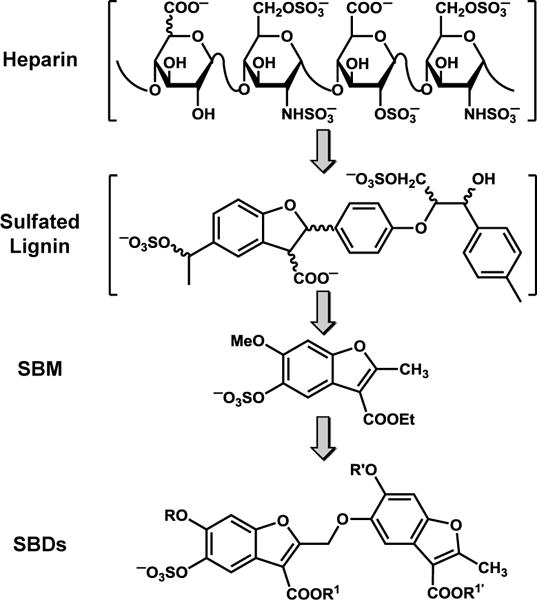
The development of allosteric partial inhibitors of thrombin that target exosite 2 of thrombin. SBMs = sulfated benzofuran monomers; SBDs = sulfated benzofuran dimers. R, R′, R1 and R1′ groups represent alkyl or aryl substituents (see Table 1).
Allosteric agents that display efficacies, or alternatively responses, in the intermediate range (e.g., 30–70%) have been well-known for receptors and oligomeric proteins. Such agents are referred to as partial antagonists. However, partial allosterism has been extremely challenging to discover for monomeric proteins or proteases. We recently discussed the first example of an allosteric partial inhibitor of a monomeric protease, a sulfated coumarin analog, which displayed sub-maximal efficacy.11 The analog reduced thrombin’s hydrolysis of a small chromogenic substrate with a maximal efficacy of only about 50%. However, this partial allosterism was lost for fibrinogen, thrombin’s natural macromolecular substrate. We reasoned that alternative small, allosteric partial inhibitors of thrombin that function even against the macromolecular substrate should be possible to design/discover considering that exosite 2 displays multiple hydrophobic sub-sites that are coupled with its catalytic triad.12
To realize such agents, we focused on a prototypic SBD, 1 (Fig. 1), which had been designed earlier but displayed ΔY of 75%.9 We posited that modifications in aromatic and ester substituents of 1 may lead to a more favorable partial inhibition characteristics (ΔY ~30 – 70%). We synthesized a library of 16 SBDs with variations in R, R’, R1 and R1′ substituents (Fig. 1). We decided that whereas predecessor 1 carries methyls or ethyls at these positions, the analogs would carry larger lipophilic substituents, which could possibly engage the hydrophobic sub-domains present in exosite 2 better.13 The synthesis of these SBDs was achieved in 7–14 steps involving protection–deprotection, nucleophilic substitution, free radical bromination and sulfation reactions (see Supplementary Material and Schemes S1–S7). We also synthesized 12 new SBMs (see Supplementary Table S1), which were also studied.
Thrombin inhibition was studied using chromogenic substrate hydrolysis assay at pH 7.4 and 25 °C in the presence of 2.5 mM CaCl2, as described elsewhere.9-12 Of the 13 SBDs studied, 11 were found to be 2–3-fold more potent than 1 identified earlier (Table 1).10 Of these, 2c carrying a PhCH2CH2 group at the R′ position, instead of a CH3 present in prototype 1, displayed the best potency (IC50 = 1.8 μM, Fig. 2). In contrast, 2o carrying sulfate groups and 2p at the R′ position failed to inhibit thrombin at concentrations lower than 300 μM suggesting a sensitive structure–activity relationship (SAR).
Table 1.
Inhibition of human thrombin by sulfated benzofuran dimers (SBDs).
| R | R′ | R1 | R1′ | IC50 (μM)a |
ΔY (%)a |
|
|---|---|---|---|---|---|---|
| 1b | Me | Me | Et | Et | 6.2±2.7c | 75±1c |
| 2a | Me | C6H11CH2 | Et | Et | 2.6±0.1 | 63±1 |
| 2b | Me | Bnd | Et | Et | 2.0±0.1 | 57±1 |
| 2c | Me | BnCH2 | Et | Et | 1.8±0.1 | 58±1 |
| 2d | Me | pOMOM-Bn | Et | Et | 3.3±0.2 | 61±2 |
| 2e | Me | mOMOM-Bn | Et | Et | 4.0±0.5 | 59±5 |
| 2f | Me | pO-iPr-Bn | Et | Et | 2.3±0.1 | 74±3 |
| 2g | Me | mO-iPr-Bn | Et | Et | 2.6±0.1 | 57±1 |
| 2h | C6H11CH2 | C6H11CH2 | Et | Et | 8.7±0.8 | 93±4 |
| 2i | C6H11CH2 | Bn | Et | Et | 3.6±0.1 | 90±2 |
| 2j | C6H11CH2 | Me | Et | Et | 3.8±0.3 | 77±3 |
| 2k | Me | Me | Et | Bn | 4.9±0.2 | 68±2 |
| 2l | Me | Me | Et | BnCH2 | 3.2±0.2 | 58±3 |
| 2m | Me | Me | Et | pOMOM-Bn | 2.6±0.1 | 66±2 |
| 2n | Me | Bn | Xe | Et | 14±0.5 | 74±2 |
| 2o | Me | Yf | Et | Et | >300g | –h |
| 2p | Me | SO3Na | Et | Et | >300g | – |
| Hirudin peptide (54 – 75) | 1.3i | |||||
| Fibrinogen peptide (410 – 427) | 130i | |||||
IC50 and ΔY were measured spectrophotometrically using a chromogenic substrate hydrolysis assay in 20 mM Tris-HCl buffer, pH 7.4, containing 25 mM CaCl2, 100 mM NaCl and 0.1% PEG 8000 at 25 °C.
IC50 and ΔY taken from ref.10.
Errors represent ± 1 S.E.
Bn=benzyl.
X= —CH2CH2OSO3Na.
Y=Bn-mOSO3Na.
No inhibition was observed at 300 μM.
Not applicable.
Values taken from ref.18.
Figure 2.
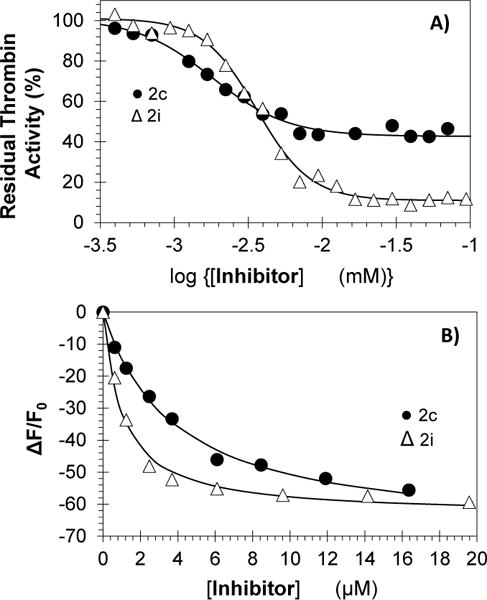
(A) Direct thrombin inhibition by sulfated benzofuran dimers (SBDs) 2c (●) and 2i (Δ) at pH 7.4 and 25 °C. Solid lines represent sigmoidal fits to calculate IC50 and ΔY. (B) Affinity titrations of 2c (●) and 2i (Δ) at pH 7.4 and 25 °C binding to fluorescein-tagged thrombin (λEM = 490 nm, λEX = 520 nm). Solid lines represent the nonlinear fits using quadratic equation 3. See details in Supporting Information.
Although the discovery of higher potency was interesting, the most exciting finding was the property of partial inhibition for this series of agents. Of the 11 active inhibitors, nine displayed ΔY in the range of 55 to 75% (Table 1, Fig. 2a). Inhibitor 2c inhibited thrombin with an efficacy of 58% at saturation, a characteristic of possibly major consequences with regard to regulation of enzyme function. Such partial inhibition profile is not possible for orthosteric agents. Interestingly, two agents, i.e., 2h and 2i, inhibited thrombin with ~90% efficacy suggesting intricate SAR involving inhibition efficacy too.
To confirm that this phenomenon arises from direct binding, we measured thrombin affinity of SBDs using spectrofluorimetry. The fluorescence of FPRCK-thrombin, monitored as a function of two representative SBDs 2c and 2i at pH 7.4 and 25 °C decreased in a classic hyperbolic manner (Fig. 2b) to yield KDs of 3.7±0.3 and 1.0±0.1 μM, respectively. In a similar manner, the affinity of wild-type plasma thrombin for 2c, determined using intrinsic fluorescence, was found to be 2.8±0.3 μM. The similarity of the KDs and IC50s is expected on the basis of allosteric inhibition mechanism.
To ascertain the mechanism of thrombin inhibition, we resorted to Michaelis-Menten kinetic studies. For both 2c and 2i, the VMAX of Spectrozyme TH hydrolysis decreased nearly 2-fold in a dose-dependent manner, as expected (see Supplementary Fig. S1 and Table S2). However, their effect on KM was distinctly different. For 2c, KM remained essentially constant at 8.2±1.1 μM, whereas for 2i, it increased from 8.6 μM to 61 μM as the inhibitor concentration increased to 40 μM (Table S2). Thus, although 2c and 2i are structural analogs, they exhibit slightly different mechanism of inhibition. SBD 2c functions as a noncompetitive inhibitor, while 2i displays mixed inhibition. Yet, both 2c and 2i do not compete with the substrate alluding to an allosteric inhibition process.
To identify the allosteric site engaged by 2c and 2i, we utilized prototypic ligands hirudin peptide (HirP) and unfractionated heparin (UFH) that bind to exosites 1 and 2, respectively. Figure 3 shows the thrombin inhibition profiles at varying levels of HirP and UFH. The profiles further clarify the difference between the two related allosteric inhibitors. The potency of thrombin inhibition by both 2c and 2i remains essentially unaffected by the presence of HirP (IC50 = 1.8±0.6 μM (2c) or 2.8±0.6 μM (2i), see Table S3) suggesting that these molecules do not engage exosite 1. For competition by UFH, the potency of 2c decreases by a factor of 11-fold, whereas that of 2i remains constant at 2.5±0.4 μM (Table S3). This implies that 2c competes with UFH, while 2i does not.
Figure 3.
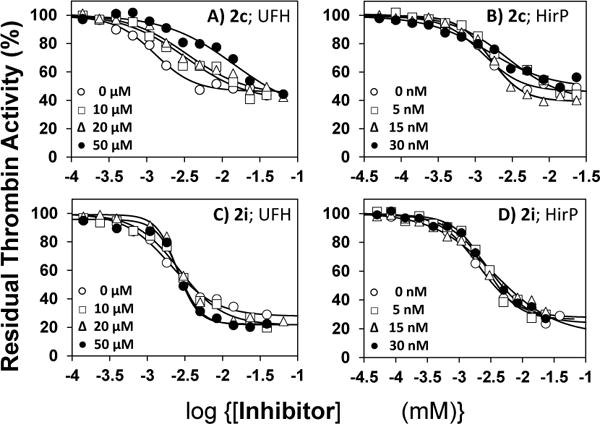
Thrombin inhibition by 2c in the presence of UFH (A) and HirP (B), and by 2i in the presence of UFH (C) and HirP (D). Residual thrombin activity was measured by determining Spectrozyme TH hydrolysis in 20 mM Tris-HCl buffer, pH 7.4, containing 100 mM NaCl, 2.5 mM CaCl2, and 0.1%PEG8000 at 25 °C in the presence of 0 to 50 nM HirP and 0–50 μM of UFH or 0–30 nM of HirP. Solid lines represent fits using a logistic equation (see Supporting Information) to obtain the apparent IC50, as described in experimental procedures.
To further pinpoint the site of binding, we utilized a group of 11 recombinant thrombin mutants containing replacement of a single electropositive residue of exosite 2 by alanine. These mutants have been studied earlier in the Rezaie laboratory14,15 and cover nearly all lysines and arginines known to interact with UFH.6 For 2c, there was virtually no difference in IC50 between wild-type and mutant thrombin except for Arg233Ala and Lys235Ala proteins (Fig. 4a). This implies that whereas the prototypic SBD 1 was found to engage Arg173, 2c behaved in a variant fashion. Considering that 1 and 2c differ only in the type of substituent at the R′ position, these differences bring forth additional subtle facets of thrombin allostery.16,17
Figure 4.
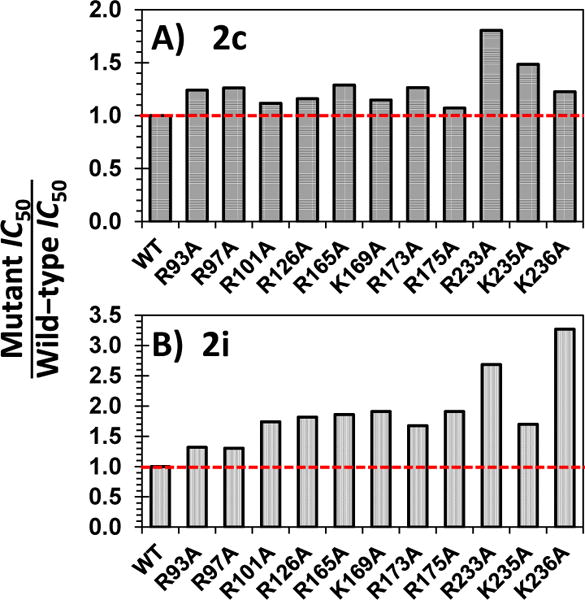
Comparison of inhibition potencies (IC50) of mutant versus recombinant wild-type(WT) thrombins by 2c (A) and 2i (B) at pH 7.4 and 25 °C. The IC50s were measured using Spectrozyme TH substrate hydrolysis assay, as described earlier.
For 2i, the potency changed significantly for several mutants including Arg165Ala, Lys169Ala, Arg175Ala, Arg233Ala and Lys236Ala (Fig. 4b). This is an unusual result if one takes into account the absence of competition with UFH (Fig. 3c, Table S3). Yet, the result can be explained by noting that 2i cannot possibly engage each of these residues simultaneously if it binds in a highly selective manner. Most probably it samples many different binding modes in which each of these residues are engaged part of the time. Such a non-selective interaction process not only explains non-competition with UFH but also helps understand the mixed inhibition mechanism identified above (Fig. S1). These differences also help rationalize the considerable difference in efficacy of inhibition observed between 2c and 2i (see Fig. 2).
In terms of drug discovery, the above results present 2c and 2i, two small molecules with significant difference in efficacy of inhibition, as tools to evaluate the concept of allosteric partial inhibition. As stated in the introductory section, partial allosterism against small chromogenic substrate may not necessarily hold against a macromolecular substrate.11 In fact, it is to be expected that the binding energy gained upon complexation with a macromolecule (e.g., fibrinogen), which is typically high, may help disengage the small molecule from its binding site, thereby releasing the partial allosteric conformation. This is the key reason behind the difficulty of discovering such small inhibitors of monomeric proteases.
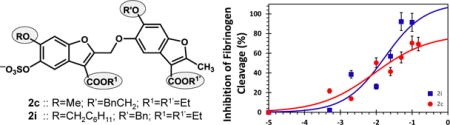
So to assess whether the submaximal inhibition, observed against the chromogenic substrate, would be transferred to the primary thrombin substrate in vivo, we utilized polyacrylamide gel electrophoresis (PAGE) for monitoring fibrinogen cleavage. Thrombin cleavage of fibrinogen results in cleaved fibrinogen of ~320 kDa and two fibrinopeptides of much lower molecular weights (which cannot be followed easily using PAGE). We performed densitometry analysis of bands at 320 kDa to measure levels of uncleaved fibrinogen in the presence and absence of 2c and 2i (Fig. 5). The results show that cleavage of fibrinogen by thrombin reduces as the concentration of inhibitors increase, as expected. However, at saturating levels of 2c, inhibition of fibrinogen cleavage reached a maximum of about 70±8% (Fig. 5B), whereas in the presence of inhibitor 2i, inhibition was found to be greater than 90%.
Figure 5.
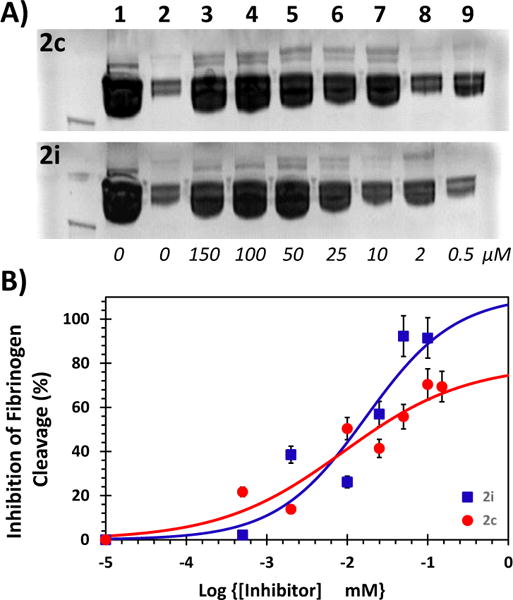
(A) A representative PAGE analysis of inhibition of thrombin cleavage of human fibrinogen in the presence of varying concentrations (0 to 150 μM) of 2c (upper image) and 2i (lower image) in 20 mM Tris-HCl buffer, pH 7.4, containing 100 mM NaCl, 2.5 mM CaCl2, and 0.1%PEG 8000 at 25 °C. Experiments were performed 4.7 μM fibrinogen and 6 nM thrombin. Lanes 1 and 2 are from otherwise identical experiments, except for the absence of thrombin or inhibitor, respectively. (B) Uncleaved fibrinogen, as shown by bands at 320 KDa in PAGE, were quantified using densitometric imaging. Multiple PAGE images were averaged to deduce standard errors (~15%) and plotted on a semi-log plot to estimate inhibition potency and efficacy of fibrinogen cleavage.
These results with small molecules can be compared with allosteric peptide-based inhibitors of proteins. For example, thrombin’s activity is known to be modulated by allosteric peptides and substrates18-20 as well as by nucleic acids.21 However generally, partial allosteric inhibition has not been documented to date. Thus, the sulfated benzofurans being presented in this work as partial inhibitors represent a novel class of allosteric agents.
Another point worth mentioning is that the results imply that a molecule as small as 2c, which is less than 800 Da in size, induces a conformational change in thrombin that cannot be reversed by the binding of a large macromolecule (320 kDa) such as fibrinogen. To the best of our knowledge, this is the first observation of induction of conformational rigidity in a monomeric protease by a small molecule that even a 400-fold larger molecule fails to undo.
Most probably, partial allosterism arises from as yet-identified specific coupling between the allosteric site and the protease active site. This is supported by the observation that 2c and 2i appear to bind to different sites on thrombin, although being structurally very similar. The results also suggest that SBD inhibition of thrombin is likely to be a thermodynamic property, and not a kinetic property. This is important because if allosteric inhibition is a kinetic property, it is unlikely to benefit in terms of drug development. Finally, whereas this concept of partial allosterism has been known and easily applied for multimeric proteins, e.g., receptors, realizing it for monomeric, soluble proteins has been extremely difficult.
A key outcome of the concept being demonstrated here is that it may help solve the problem of bleeding risk associated with DOACs. Current anticoagulants completely shut down clotting at saturation and inadvertent use of higher doses leads to bleeding. In contrast, partial allosteric inhibition of coagulation proteases would only reduce clotting signal even at saturation, or in the event of an inadvertent excessive dose. For example, in the case of 2c, approximately 30% fibrinogen cleavage continues to proceed even in the presence of saturating levels of inhibitor.
In summary, we present the first SBDs that display partial allosterism against fibrinogen (ΔY = 55–70%), the first time a small molecule (MW <800) has been found to thwart macromolecular cleavage of a monomeric protein in a regulated manner. We believe that partial allosterism of thrombin is a valuable concept because it affords the possibility of maintaining basal level of a pro-clotting signal. Such basal level of fibrinogen cleavage can be expected to retard bleeding and thereby reduce risk of major bleeding, which beset all current DOACs. This work also indicates that this promising concept should be possible to explore for other monomeric coagulation proteases, e.g., factor Xa.
Supplementary Material
Acknowledgments
This work was supported by NIH grants HL090586, HL107152 and HL128639 to URD. We thank Ms. Ashley Boice of VCU for recording HRMS profiles of molecules studied in this work; Dr. Mohini Ghatge of VCU for help with gel electrophoresis experiments; Dr. Akul Mehta of Harvard Medical School for technical support in characterization of SBMs and SBDs, and Dr. Aurijit Sarkar of High Point University for helpful discussions.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Supplementary Material
The Supporting Information is available free of charge on the Bioorg. Med. Chem. Lett. web-site. It includes experimental procedures used in the study.
References and notes
- 1.Ruff CT, Giugliano RP, Braunwald E, Hoffman EB, Deenadayalu N, Ezekowitz MD, Camm AJ, Weitz JI, Lewis BS, Parkhomenko A, Yamashita T, Antman EM. Lancet. 2014;383:955. doi: 10.1016/S0140-6736(13)62343-0. [DOI] [PubMed] [Google Scholar]
- 2.Henry BL, Desai UR. In: Burger’s Medicinal Chemistry, Drug Discovery and Development. 7th. Abraham DJ, Rotella DP, editors. John Wiley; Hoboken, NJ: 2010. pp. 365–408. [Google Scholar]
- 3.Merdanovic M, Mönig T, Ehrmann M, Kaiser M. ACS Chem Biol. 2013;8:19. doi: 10.1021/cb3005935. [DOI] [PubMed] [Google Scholar]
- 4.Hauske P, Ottmann C, Meltzer M, Ehrmann M, Kaiser M. Chembiochem. 2008;9:2920. doi: 10.1002/cbic.200800528. [DOI] [PubMed] [Google Scholar]
- 5.Henry B. J Biol Chem. 2007;282:31891. doi: 10.1074/jbc.M704257200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Carter W. J Biol Chem. 2005;280:2745. doi: 10.1074/jbc.M411606200. [DOI] [PubMed] [Google Scholar]
- 7.Henry B. Biochem Biophys Res Commun. 2012;417:382. doi: 10.1016/j.bbrc.2011.11.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Verghese J, Liang A, Sidhu P. Bioorg Med Chem Lett. 2009;19:4126. doi: 10.1016/j.bmcl.2009.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Abdel Aziz M. J Med Chem. 2012;55:6888. doi: 10.1021/jm300670q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sidhu P. J Med Chem. 2011;54:5522. doi: 10.1021/jm2005767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Verespy S, Mehta A. Sci Rep. 2016;6:24043. doi: 10.1038/srep24043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sidhu P. J Med Chem. 2013;56:5059. doi: 10.1021/jm400369q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Abdel Aziz M. Biochem Biophys Res Commun. 2011;413:348. doi: 10.1016/j.bbrc.2011.08.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.He X, Ye J, Esmon CT, Rezaie AR. Biochemistry. 1997;36:8969. doi: 10.1021/bi9704717. [DOI] [PubMed] [Google Scholar]
- 15.Yang L, Rezaie AR. Thromb Haemostasis. 2007;97:899. doi: 10.1160/th06-12-0697. [DOI] [PubMed] [Google Scholar]
- 16.Lechtenberg B. Biol Chem. 2012;393:889. doi: 10.1515/hsz-2012-0178. [DOI] [PubMed] [Google Scholar]
- 17.Di Cera E. Mol Aspects Med. 2008;29:203. doi: 10.1016/j.mam.2008.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hortin G. Biochem Biophys Res Commun. 1990;169:437. doi: 10.1016/0006-291x(90)90350-v. [DOI] [PubMed] [Google Scholar]
- 19.Hortin G. J Biol Chem. 1991;266:6866. [PubMed] [Google Scholar]
- 20.Mengwasser K. J Biol Chem. 2005;280:26997. doi: 10.1074/jbc.M502678200. [DOI] [PubMed] [Google Scholar]
- 21.Nimjee SM, Oney S, Volovyk Z, Bompiani KM, Long SB, Hoffman M, Sullenger BA. RNA. 2009;15:2105. doi: 10.1261/rna.1240109. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.