

Abstract
Background
The purpose of this study was to evaluate the participation of satellite glial cells (SGC), microglia and astrocytes in a model of streptozotocin-induced diabetes initiated in neonatal rats (nSTZ) and to determine the pharmacological profile for pain relief.
Methods
nSTZ was used to induce experimental diabetes. Von Frey filaments were used to assess tactile allodynia. Drugs were given by systemic administration. Western blotting and immunohistochemistry were used to determine protein expression and cellular localization.
Results
nSTZ produced mild hyperglycemia, weight loss, glucose intolerance, and reduction of nerve conduction velocity of C fibers. Moreover, nSTZ enhanced activating transcription factor 3 (ATF3) immunoreactivity in dorsal root ganglia (DRG) and sciatic nerve of adult rats. ATF3 was found in SGC (GFAP+ cells) surrounding DRG at week 16. Late changes in ATF3 immunoreactivity in DRG correlated with up-regulation of ATF3 and GFAP protein expression. nSTZ increased GFAP and OX-42 immunoreactivity and percentage of hypertrophied and ameboid microglia in the spinal dorsal horn. These changes correlated with the presence of mechanical hypersensitivity (tactile allodynia). Administration of gabapentin (30–100 mg/kg, po) and metformin (200 mg/kg/day, po for 2 weeks) alleviated tactile allodynia, whereas morphine (1-3 mg/kg, ip) had a modest effect.
Conclusions
Results suggest that nSTZ leads to activation of SGC, microglia and astrocytes in DRG and spinal cord. Pharmacological profile in the nSTZ model resembles diabetic neuropathic pain in humans. Our findings support the conclusion that the nSTZ rat model has utility for the study of a long-lasting diabetic neuropathic pain.
Keywords: Astrocytes, Diabetes, Microglia, nSTZ model, Satellite glial cells
Introduction
Type 2 diabetes is a chronic metabolic disorder that results from early defects in insulin receptor function and progresses from hyperinsulinemia to catastrophic beta cell failure. Diabetic patients develop several complications including nephropathy, retinopathy, vascular complications and neuropathy. Diabetic neuropathy affects nerve fibers of the peripheral nervous system in at least 50% of diabetic patients [1]. Extensive clinical studies have implicated hyperglycemia as a leading causative factor of diabetic neuropathy [2]. It has been proposed that hyperglycemia leads to nerve injury by activating several mechanisms as increased aldose reductase activity, non-enzymatic glycation and oxidative–nitrosative stress [3]. Moreover, it has been suggested that an impairment of neurotrophic support [4] and mitochondrial dysfunction may also participate [5].
The majority of studies of diabetic neuropathy and neuropathic pain have been performed using animals treated with the beta cell toxin streptozotocin (STZ) [6] which, when given to adult rodents, produces type 1 diabetes with severe insulinopenia and hyperglycemia that requires insulin administration to ensure survival for longer periods [7]. This model produces mechanical and cold allodynia at about 4–8 weeks [8, 9]. However, the pertinence of models of type 1 diabetes to the more common type 2 form of the disease is undergoing increased scrutiny and it has been suggested that the pathogenesis of neuropathy differs in the two forms of diabetes [10]. There is therefore a need to establish the neuropathy phenotype in models of type 2 diabetes.
It has been reported that the neonatal administration of STZ (nSTZ) may better mimic the long-lasting complications and slower evolution of type 2 diabetes in humans [11, 12]. Administration of STZ 4–5 days after birth leads to mild hyperglycemia, abnormal glucose tolerance and mild hyperinsulinemia [13]. Despite the fact that this model has been used to study several complications of the disease, investigations concerning pain mechanisms have been scarcely reported [14]. The purpose of this study was three-fold. First aim was to develop a model of long-lasting diabetic neuropathic pain. Our second aim was to study whether activation of satellite glial cells in the DRG or activation of microglia and astrocytes in the spinal dorsal horn coincides with the development of nSTZ-induced tactile allodynia. Third aim was to determine the antiallodynic effects of several analgesic drugs that are currently in use for the treatment of painful diabetic neuropathy.
Materials and Methods
Drugs and reagents
Streptozotocin (Cat. # 572201), metformin hydrochloride (Cat. # 1396309) and di-n-butyl phthalate-polystyrene-xylene (DPX, Cat. # 06522), gabapentin (Cat. # G154) and diclofenac (Cat. # D6899) were purchased from Sigma-Aldrich (St. Louis, MO). Morphine was a gift from Health Ministery. Morphine, diclofenac, gabapentin and metformin were dissolved in saline solution (0.9%). Streptozotocin was dissolved in 0.1 M citrate buffer, pH 4.5.
Rabbit anti-ATF3 antibody (Cat. # sc-188) and rabbit anti-OX-42 (Cat. # MCA275G) were obtained from Santa Cruz Biotechnology (Dallas, TX) and Bio-Rad (Hercules, CA), respectively. Rabbit anti-GFAP antibody (Cat. # AB5804) and mouse anti-β-actin antibody (Cat. # MAB1501R) were purchased from Millipore (Bellerica, MA). Horseradish peroxidase-conjugated secondary antibody donkey anti-rabbit Cy3 (Cat. # 711-165-152) and Cy2 (Cat. # 711-225-152) were obtained from Jackson ImmunoResearch (West Grove, PA). Amersham ECL western blotting detection reagent (Cat. # RPN2232) was obtained from GE Healthcare Life Sciences (Chicago, IL). Rat insulin ELISA kit (Cat. # 80-INSRT-E01) was purchased from ALPCO (Salem, NH).
Neonatal STZ-induced diabetic rat model
The nSTZ model was performed in accordance with previous reports [12, 15]. Briefly, neonate male Wistar rats (3-4 days old) were divided in two groups. Control rats received an intraperitoneal (ip) injection of vehicle (0.1 M citrate buffer, pH 4.5), whereas that diabetic rats received a single STZ dose (70 mg/kg, ip). Following vehicle or STZ injection, neonatal rats were kept with their mothers under a 12-hour light/dark cycle at 23°C. Three weeks after STZ injection, rats were weaned and separated in individual cages to perform further behavioral and molecular experiments. All experiments were conducted according to the EC Directive 86/609/EEC for animal experiments, and Guidelines on Ethical Standards for Investigation of Experimental Pain in Animals [16]. In addition, the Institutional Animal Care and Use Committee approved the study.
Body weight and capillary glucose measurement
Body weight and capillary glucose were measured weekly in all groups. Capillary glucose was measured using a glucometer (Accu-Chek Active, Glucotrend, Roche®) under both fasted (14 h) and non-fasted conditions in the same animals. Data are expressed as mean (n=10) ± SEM of the capillary glucose (mg/dl) and weight (g).
Oral glucose tolerance test and insulin secretion
Oral glucose tolerance test and insulin secretion was performed in conscious control and diabetic rats at 4, 8, 12, and 16 weeks after vehicle or nSTZ administration. Animals were fasted during 14 hours. Then, some of them received a 50% D-glucose solution (2 g/kg) by oral gavage. Blood samples were collected by puncturing tail vein sequentially before and 30, 60, 90 and 120 min after glucose administration. Capillary glucose was measured using a glucometer and insulin secretion was measured with a rat insulin ELISA kit following the manufacturer´s instructions (ALPCO, Salem, NH). Glucose tolerance data are expressed as mean (n=5) ± SEM of the capillary glucose (mg/dl) and plasma insulin (ng/ml).
Recording of compound action potential and conduction velocity
Compound action potential (cAP) of the putative C fibers was determined as previously described [17]. Control and diabetic rats were anesthetized with a mixture of ketamine (50 mg/kg, ip) and xylazine (10 mg/kg, ip). A laminectomy from L1 to S1 vertebra was performed to expose the dorsal roots. The L5 ganglia attached to the dorsal root and spinal nerve were removed. Tissue was transferred to a recording chamber and bathed with oxygenated (5% CO2 and 95% O2) artificial cerebrospinal fluid at pH 7.6 containing (in mM): 117 NaCl, 3.6 KCl, 1.2 MgCl2, 2.3 CaCl2, 1.2 NaH2PO4, 25 NaHCO3 and 11 glucose at room temperature. To evoke action potentials, electrical stimulation (0.3 ms of duration) was applied to the peripheral cut end of the spinal nerve with a suction electrode. The cAP was recorded from the dorsal root with a suction electrode. The recording electrode was connected to a DC amplifier (World Precision Instruments, Sarasota, FL) with a bandwidth of DC to 10 kHz. Unless otherwise noted, recordings shown represent the average of 10 stimuli applied every 10 s. To record synchronized C component the length of the spinal nerve in continuity with the dorsal root ganglia was about 12 mm.
The average conduction velocity of the cAP was calculated as the conduction distance divided by the conduction time. Conduction distance was measured from the stimulation electrode to the recording electrode. Conduction time was measured as the time elapsed from the electrical stimulus artifact to the maximal amplitude of the cAP. Fibers with conduction velocity lower than 1.4 m/s were considered C fibers. Data are reported as the mean (n=6) ± SEM of the normalized area under the curve of the C component of the cAP and its respective conduction velocity.
Immunofluorescence staining and analysis
Control and diabetic rats were sacrificed with CO2 delivered via compressed gas cylinder and perfused intracardially with 200 ml of 0.1 M phosphate buffered saline (pH=7.4 at 4°C, 137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4 and 2 mM KH2PO4) followed by 250 ml of 4% p-formaldehyde/12% picric acid in 0.1 M phosphate buffered saline. The sciatic nerve, lumbar DRG (L4–L6) and lumbar spinal cord were removed following perfusion. Tissue was post-fixed for 2 h in the perfusion fixative and cryoprotected for 72 h in 30% sucrose buffered with 0.1 M phosphate, at 4°C, and then processed for immunohistochemistry. Frozen DRG (16 μm thick) and lumbar spinal cord (30 μm thick) were cut on a cryostat (Leyca Biosystems, Wetzlar) and thaw-mounted on gelatin-coated slides for processing. Preparations were allowed to dry at room temperature for 30 min, washed in 0.1 M phosphate buffered saline three times for 10 min each (3×10), blocked with 10% normal donkey serum (Jackson ImmunoResearch, West Grove, PA) in phosphate buffered saline with 0.3% Triton-X 100 for 2 h and then incubated overnight with rabbit-anti ATF3 (1:500, Santa Cruz Biotechnology, Dallas, TX), OX-42 (1:500, Bio-Rad, Hercules, CA) or rabbit anti-GFAP (1:300, Millipore, Bellerica, MA) in 1% normal donkey serum and 0.1% Triton-X 100 in 0.1 M phosphate buffered saline at 4°C. The following day, preparations were washed 3×10 min each in phosphate buffered saline and incubated for 2 h at room temperature with secondary antibody conjugated to fluorescent marker (Cy3, 1:600/Cy2, 1:200; Jackson ImmunoResearch, West Grove, PA). Finally, sections were washed 3×10 min each in phosphate buffered saline and dehydrated through a graded alcohol series (70, 80, 90, and 100%), cleared in xylene, and coverslipped with DPX (Sigma-Aldrich, St. Louis, MO). Preparations were allowed to dry at room temperature for 12 h before imaging. Images from L4-L6 DRG were taken on an inverted microscope with epifluorescence (Nikon TE2000-U, Chiyoda, Japan) using a 20× objective at a digital size of 1920×1536 pixel. Images from L4-L6 spinal dorsal horn were taken on an Olympus Fluoview FV 1200 laser scanning confocal microscope (Olympus, Allentown, PA) and analyzed using the co-localization tool within Olympus FV software. In case of double-labeled immunohistochemistry, images of each channel were acquired by sequential scanning. The percentage of ATF3-labeled neurons in the DRG was calculated by dividing the number of ATF3+ neurons by the total number of neurons counted X 100. For microglial quantification in dorsal horn, at least five slices from each rat were used. For this purpose, microglia morphology was classified as previously described [18]. The percentage of a specific morphological type in the dorsal horn was obtained by calculating the ratio of the total cell type morphology to the total number of microglial cells observed from lamina I to lamina VI. Finally, ImageJ software was used to calculate the GFAP-mean fluorescence intensity in L4-L6 spinal dorsal horn from at least five slices from a group of 3 rats.
Western blot
Western blot analysis was used to detect changes in ATF3 and GFAP in the lumbar DRG of control and diabetic rats. Rats were anaesthetized under isoflurane anesthesia and sacrificed by decapitation to get the lumbar DRG (L4-L5). Tissues from individual animals were homogenized in ice-cold lysis buffer (in mM: 150 NaCl, 50 Tris-HCl, 5 EDTA), pH 7.4 for 3 min at 4°C containing PMSF (1 mM), aprotinin (10 μg/ml), leupeptin (10 μg/ml), pepstatin A (10 μg/ml) and 0.1% Triton X-100 (Sigma-Aldrich, St. Louis, MO). The homogenate was then centrifuged (Eppendorf, Hamburg) at 14,000 rpm for 10 min to remove cellular debris. The resultant supernatant was used to measure protein concentration by the Bradford’s method (Bio-Rad, Hercules, CA).
Fifty micrograms of total protein were resolved by denaturing on a 12% SDS-polyacrylamide gel electrophoresis and transferred to PVDF membranes. Membranes were incubated in 5% non-fat milk in phosphate-buffered saline at pH 7.4 (in mM: 137 NaCl, 2.7 KCl, 10 Na2HPO4 and 2 KH2PO4) with 0.1% Tween 20 for 1 h to block nonspecific proteins. After that, they were incubated overnight at 4 °C in 5% non-fat dry milk/PBS containing rabbit anti-ATF3 (1:500, Santa Cruz Biotechnology, Dallas, TX) and rabbit anti-GFAP (1:1000, Millipore, Bellerica, MA) antibodies. The following day, membranes were incubated for 1 h at room temperature in 1% non-fat milk/PBS containing the horseradish peroxidase-conjugated secondary antibody (Donkey anti-rabbit, 1:10000, Jackson ImmunoResearch, West Grove, PA). Protein signal detection was achieved with the ECL chemiluminescence system (GE Healthcare Life Sciences, Chicago, IL). After that, blots were stripped and incubated with a monoclonal antibody directed against β-actin (1:10000, Millipore, Billerica, MA), which was used as an internal control to normalize protein expression levels. Scanning of the immunoblots was performed and the bands were quantified by densitometry using an image analysis program (Image Lab Software Version 5.2.1, Bio-Rad, Hercules, CA). Protein expression data are expressed as mean (n=4) ± SEM.
Assessment of tactile allodynia
Tactile allodynia in control and diabetic rats was determined as previously described [19]. Briefly, rats were placed in cages with a mesh grid floor and allowed to acclimate for a minimum of 30 min before performing the experiment. Von Frey filaments (Stoelting, Wood Dale, IL) were used to determine the 50% paw withdrawal threshold.
Pharmacological experiments
We used four major categories of agents to evaluate the pharmacological profile of the mechanical hypersensitivity in the nSTZ model: gabapentin (30–100 mg/kg, po), morphine (1–3 mg/kg, ip), diclofenac (1–10 mg/kg, po) and the adenosine monophosphate activated protein kinase (AMPK) activator metformin (200 mg/kg, po). Doses were selected from former reports [20–23]. Greater doses of gabapentin, morphine, and diclofenac were not assessed to avoid side effects [24–26]. The first 3 drugs were administered 16 weeks after nSTZ injection. Metformin was given once daily for 2 weeks starting at week 14. Time course of the antiallodynic effects was followed for 6-8 h after drug administration. Data are expressed as the mean ± SEM of 6 independent animals.
Statistical analyses
Statistical differences between two groups were determined by unpaired t-test. Differences between more than two groups were determined by one- or two-way analysis of variance (ANOVA), followed by the Bonferroni’s multiple comparisons test. A value of p less than 0.05 was considered significant.
Results
nSTZ produces hyperglycemia, glucose intolerance and weight loss
nSTZ, but not vehicle, enhanced capillary glucose from 13 to 16 weeks after administration in fasted rats (n=10, F = 127.9, p < 0.001, Figure 1A). In contrast, nSTZ produced hyperglycemia from 4 to 16 weeks in non-fasted animals (n=10, F = 470.2, p < 0.001, Figure 1A). Regarding body weight, nSTZ reduced body weight gain from 12 to 16 week after injection (n=6, F = 78.45, p < 0.001, Figure 1B). In addition, nSTZ reduced glucose clearance after a glucose load (glucose intolerance) (n=5, F = 33.26, p < 0.001, Figure 1C; n=5, F = 129, p < 0.001, 1E; n=5, F = 1181, p < 0.001, 1G; n=5, F = 12, p < 0.001, 1I). Plasma insulin levels were not different between groups prior to glucose challenge at any time point but in the nSTZ group plasma insulin did not increase during glucose challenge (n=5, F = 17.12, p < 0.001 Figure 1D; n=5, F = 9.379, p = 0.035, 1F; n=5, F = 40.69, p < 0.001, 1H; n=5, F = 61.39, p = 0.0001, 1J). We observed mild hyperinsulinemia in basal conditions at 8 weeks (Fig. 1F).
Figure 1.
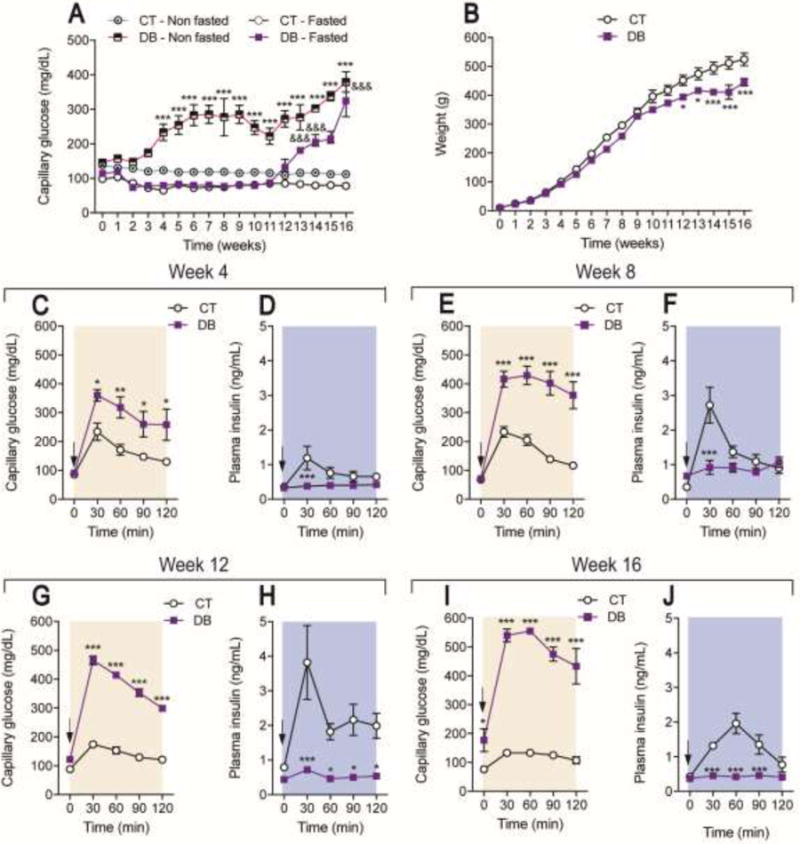
Characteristics of the neonatal streptozotocin (nSTZ) induced diabetic rat model. (A) Time course of capillary glucose concentrations and (B) body weight from control (CT) and diabetic (DB) rats. Data in panels A and B are the mean of 10 animals ± SEM. Capillary glucose levels (C, E, G, I) and (D, F, H, J) serum insulin concentrations before and during an oral glucose tolerance test in control and diabetic rats. Black arrows indicate the time of glucose (2 g/kg) administration. Data in panels C-J are the mean of 5 animals ± SEM. (A) ***p < 0.001, significantly different from the control-non fasted group and &&&p < 0.001, significantly different from the CT-fasted group, as determined by two-way ANOVA followed by the Bonferroni test. (B-J) *p < 0.05, **p < 0.01 and ***p < 0.001 significantly different from the CT-group, as determined by two-way ANOVA followed by the Bonferroni test.
nSTZ produces changes on nerve conduction velocity and compound action potential of C fibers
In this study, we recorded the velocity of conduction of C fibers as well as the C component of the cAP in the dorsal root attached to its ganglion after peripheral stimulation of the spinal nerves (Figure 2A). nSTZ reduced, in a time-dependent manner, the conduction velocity of C fibers (p = 0.0175, Figure 2B). Moreover, nSTZ decreased the area under the curve of the C component of the cAP at 12 (n=6, p = 0.0465, Figure 2C) and 16 weeks (n=6, p = 0.0175, Figure 2C).
Figure 2.
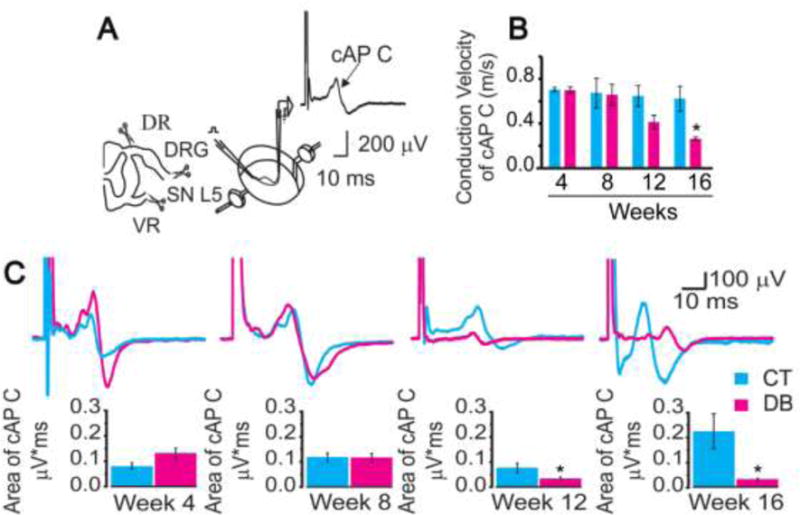
Effects of nSTZ on nerve conduction velocity and compound action potential of C fibers (cAP). (A) Illustration of the preparation and recording of the cAP of C fibers. (B) Effect of nSTZ in nerve conduction velocity of C fibers in control (CT) and diabetic (DB) rats. (C) Effect of nSTZ in the C fibers cAP from control (CT) and diabetic (DB) rats. Upper panel: Representative traces of C fibers cAP recorded from control and diabetic rats. Lower panel: Quantification of normalized C fibers cAP area from control and diabetic rats. Data are the mean of 6 animals ± SEM. *p < 0.05, significantly different from the control group, as determined by Student´s t-test.
ATF3 expression in the DRG and sciatic nerve
We examined the expression of ATF3 in the DRG and sciatic nerve from control and nSTZ-treated rats by immunohistochemistry. As expected, we did not observe ATF3 immunoreactivity in DRG at 4, 8, 12 or 16 weeks after vehicle administration (Figure 3A, C, E, G). Likewise, ATF3 was not expressed in sciatic nerve of control rats (Figure 3I). In contrast, nSTZ enhanced, in a time-dependent fashion, ATF3 immunoreactivity in DRG (Figure 3B, D, F, H). Of note, late changes in ATF3 immunoreactivity were also present in the sciatic nerve (Figure 3J). Interestingly, ATF3 was mostly observed in neuronal cells at earlier stages following nSTZ administration (from week 8 to week 12). In marked contrast, ATF3 was mainly found in satellite glial cells (GFAP+ cells) surrounding DRG at late stages (week 16) (Figure 3K–M). Late changes in ATF3 immunoreactivity in DRG correlated with an up-regulation of ATF3 (n=4) and GFAP (n=4) protein expression quantified by Western blot (Figure 3N). The percentage of neuronal cells that expressed ATF3 in STZ-treated rats was 0.50 ± 0.2% (week 4), 1.6 ± 0.4% (week 8), 2.1 ± 0.6% (week 12), and 8.0 ± 1.9% (week 16). In contrast, the percentage of GFAP+ cells that expressed ATF3 was 36.6 ± 2.8% (week 16) (see Figure 3O for details). These % were determined in five slices (from a group of 3 rats).
Figure 3.
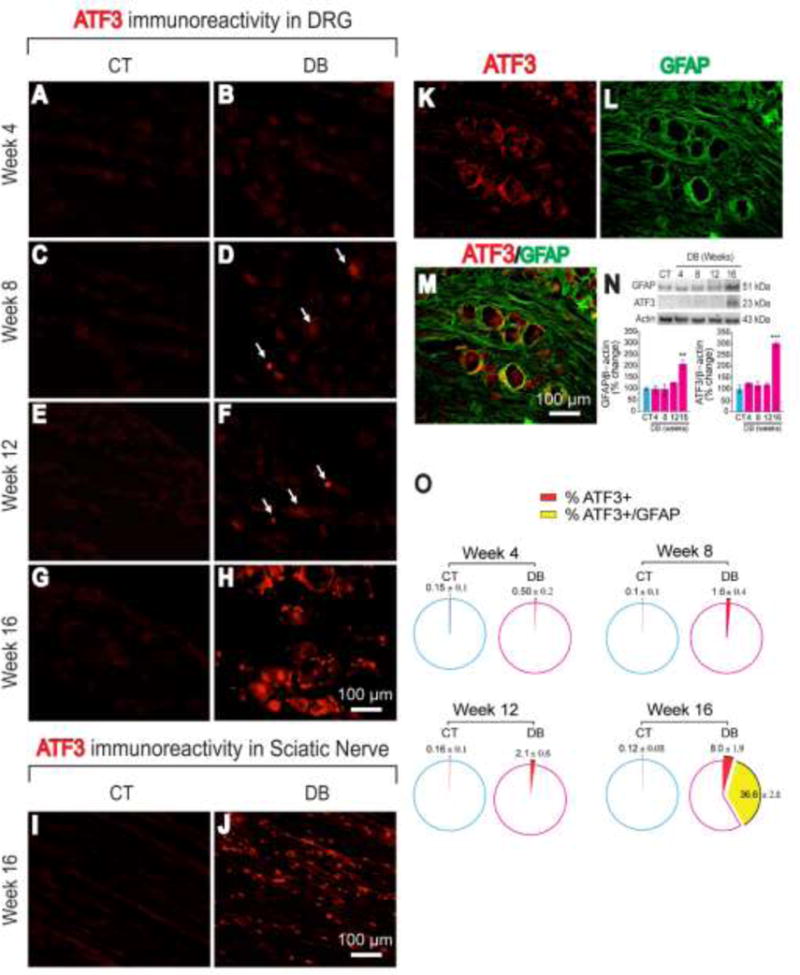
Effects of the nSTZ on immunoreactivity and expression of ATF3 in lumbar DRG and sciatic nerve. (A-H) Representative images of ATF3 immunoreactivity in L4-L6 DRG from control (CT) and diabetic (DB) rats. (I-J) Representative images of ATF3 immunoreactivity in sciatic nerve in CT and DB rats at week 16. (K-M) representative images of ATF3 immunoreactivity co-localization with satellite glial cells (GFAP+) in L4-L6 DRG at week 16. (N) Time-course of ATF3 and GFAP expression in L4-L6 DRGs by western blot (n=4). (O) Quantification of ATF3-Immunoreactivity in L4-L6 DRG (5 slices from a group of 3 rats). **p < 0.01, ***p < 0.001 Significantly different from the control group (CT), as determined by one-way ANOVA followed by the Bonferroni test.
Glial cell activation in spinal dorsal horn and mechanical hypersensitivity
Microglial immunoreactivity in the spinal dorsal horn was evaluated using the specific marker CD11b/c (OX-42). Administration of nSTZ, but not vehicle, decreased, in a time-dependent fashion the percentage of ramified microglia from 4 to 16 weeks (Figure 4A-I). These changes correlated with an increase in the percentage of microglia with a hypertrophied and ameboid morphology (five slices from a group of 3 rats, Figure 4A-I). Moreover, nSTZ, but not vehicle, increased expression of GFAP-immunoreactivity in the dorsal horn of the spinal cord (five slices from a group of 3 rats, Figure 4J-O). These pathological changes in microglia and astrocytes in the spinal dorsal horn correlated with the presence of mechanical hypersensitivity measured by von Frey testing (n=12, F = 196.3, p < 0.001, Figure 4P).
Figure 4.
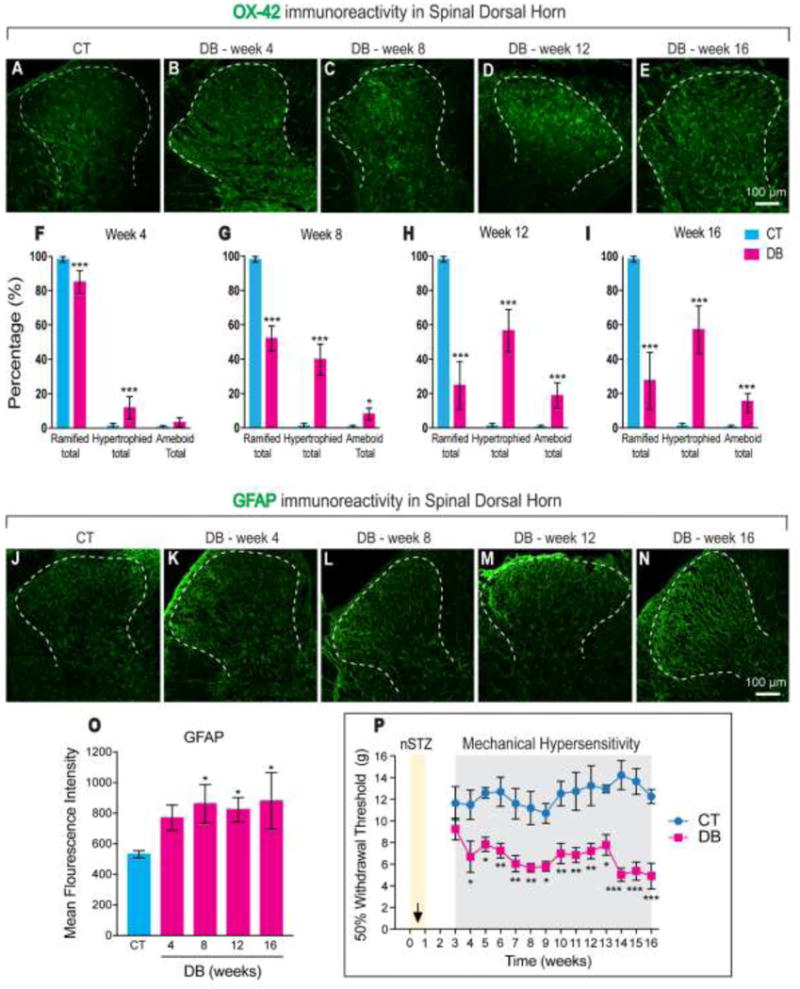
Effects of nSTZ on glial cells activation and mechanical pain hypersensitivity. (A-E) Representative images of OX-42 staining in the spinal dorsal horn from control (CT) and diabetic (DB) rats. (F-I) Quantification of microglial morphology in the spinal dorsal horn from CT and DB rats (5 slices from a group of 3 rats). (J-N) Representative images of GFAP staining in the spinal dorsal horn from CT and DB rats. (O) Quantification of GFAP fluorescence intensity in CT and DB conditions (5 slices from a group of 3 rats). (P) Time course of mechanical hypersensitivity produced by vehicle or nSTZ administration. Data are the mean of 12 rats ± SEM. *p < 0.05, **p < 0.01 and ***p < 0.001 significantly different from the CT group, as determined by one- or two-way ANOVA followed by the Bonferroni test.
Pharmacological profile of the hypersensitivity in the nSTZ model
Oral administration of gabapentin (30-100 mg/kg) 16 weeks after nSTZ treatment dose-dependently reversed nSTZ-induced tactile allodynia (n=6, F = 157, p < 0.001, Figure 5A). Morphine (1-3 mg/kg) (n=6, F = 17.23, p < 0.001, Figure 5B) and, to a lesser extent, diclofenac (n=6, F = 5.801, p < 0.0041, Figure 5C) produced a mild attenuation of tactile allodynia in the nSTZ-treated rats when given at this same point. These antiallodynic effects were independent of sedation, as at these doses, drugs did not affect motor coordination assessed in the rota-rod apparatus at 2 h. Greater doses of gabapentin (300 mg/kg) or morphine (10 mg/kg) attenuated tactile allodynia but also altered motor coordination (data not shown). Greater doses of diclofenac (30 mg/kg) were not assessed as this drug produces ulcers and bleeding [24]. On the other hand, once a day administration of metformin, starting 14 weeks after nSTZ treatment, significantly attenuated mechanical hypersensitivity 6 days after beginning treatment (n=6, F = 82.48, p < 0.001, Figure 5D). This antiallodynic effect continued through 15 days of metformin treatment without affecting glucose blood levels (220.8 ± 24.4 versus 194.5 ± 37.6 mg/dl, week 16).
Figure 5.
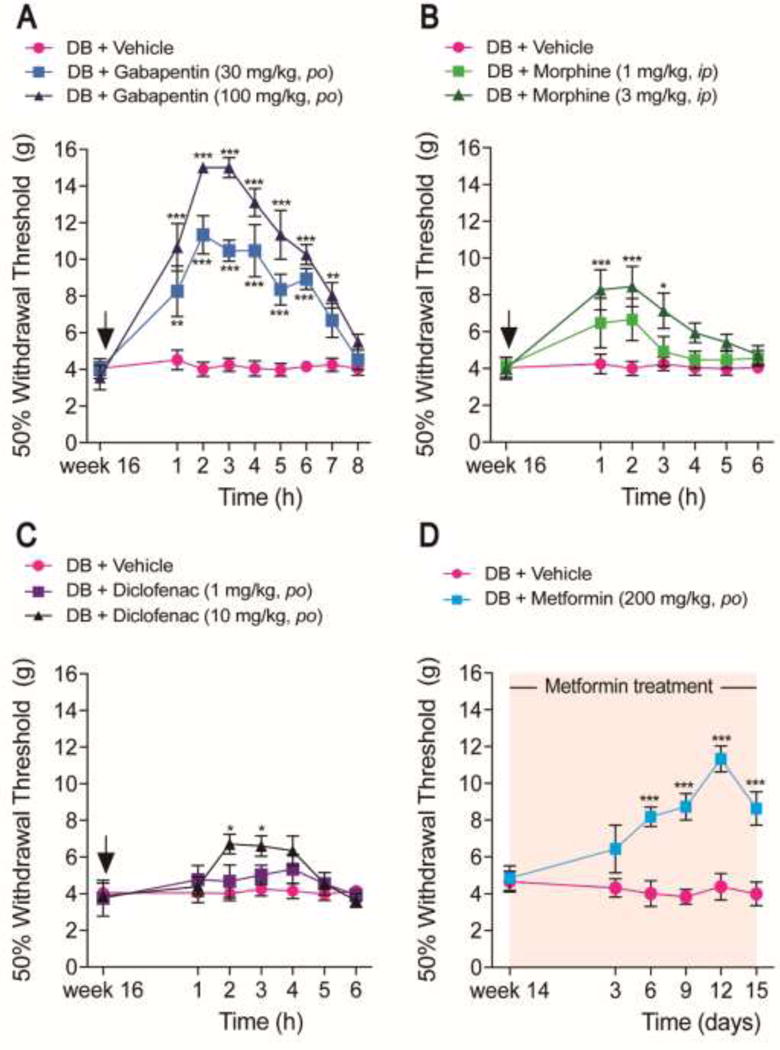
Pharmacological effects of drugs. (A) Antinociceptive effects of gabapentin (30–100 mg/kg, po), (B) morphine (1–3 mg/kg, ip), (C) diclofenac (1–10 mg/kg, po), and (D) metformin (200 mg/kg, po, daily from week 14 to week 16) in the mechanical hypersensitivity produced by nSTZ. Data are the mean of 6 rats ± SEM. *p < 0.05, **p < 0.01 and ***p < 0.001 significantly different from the CT group, as determined by two-way ANOVA followed by the Bonferroni test.
Discussion
nSTZ-induced diabetes model
n-STZ produced hyperglycemia, polyphagia, polydipsia, polyuria, weight loss and glucose intolerance. Interestingly, nSTZ produced hyperglycemia from 4 to 16 weeks in non-fasting conditions. In marked contrast, nSTZ only produced hyperglycemia from 12 to 16 weeks in fasting conditions. In any case, glucose levels were below 300 mg/dl during 15 weeks. These data indicate that nSTZ induces alterations in glucose levels lower than those produced in adult rats treated with STZ (about 500 mg/dl). Although nSTZ-induced diabetes model has been widely described as a model that mimics human type 2 diabetes [13, 15], our data do not support such a conclusion. However, nSTZ certainly produces many of the alterations observed in type 2 diabetes. There is evidence that STZ injection in adult rats reduces motor nerve conduction velocity at 4-8 weeks post-administration [27, 28]. We found that nSTZ reduced nerve conduction velocity in C fibers at 12–16 weeks. Thus, our data agree with these studies by showing that nSTZ reduces nerve conduction velocity of C fibers. Taken together, our findings suggest that nSTZ leads to an insidious process that is almost asymptomatic at its start but manifests at the adult age (about 12–16 weeks) as tactile allodynia.
nSTZ enhances expression of ATF3 and GFAP in satellite glial cells
We found that nSTZ injection enhances ATF3 immunoreactivity in neurons at 8 and 12 weeks, whereas ATF3 immunoreactivity was found mainly in the DRG and sciatic nerve at 16 weeks. The fact that induction of ATF3 expression at later time points is found in neuronal nuclei but also co-localizes with GFAP suggests that ATF3 is additionally expressed in satellite glial cells as neuropathy progresses in this model. Expression in the sciatic nerve is certainly coming from Schwann cells as these are the major nucleated cell type in the nerve. Since ATF3 is considered a marker of nerve injury in the DRG [29], our data support the idea that nSTZ leads to nerve injury. Reinforcing this, nerve injury in rats increases ATF3 expression in DRG [29, 30]. To our knowledge, this is the first report about the effect of nSTZ on ATF3 expression in DRG and satellite glial cells in rats. We also found that nSTZ enhances the expression of GFAP in satellite glial cells at 16 weeks. Our study contrasts with a previous report indicating that STZ administration in adult rats increases GFAP immunoreactivity in satellite glial cells at 2 weeks [31]. Differences could be due to the different way to induce hyperglycemia. The enhanced expression of ATF3 and GFAP in satellite glial cells has also been reported in paclitaxel- or chronic constriction-induced neuropathic pain [32]. Taking together, our findings indicate that nSTZ increases ATF3 and GFAP immunoreactivity and protein expression leading to activation of satellite glial cells and nerve injury.
nSTZ leads to activation of microglia and astrocytes
We found that nSTZ enhances OX-42 immunoreactivity in the spinal dorsal horn from 4 to 16 weeks, suggesting that nSTZ leads to microglia activation. To our knowledge, this is the first report about the microglial activation by nSTZ. Our data agree with other studies demonstrating that STZ injection in adult rats produces activation of microglia in the dorsal horn [33, 34]. Moreover, several studies have found that STZ in adult rats leads to spinal microglia activation at 4 weeks [35, 36]. Accordingly, peripheral nerve injury promotes microglia activation in the spinal dorsal horn of rodents (from 1 day to 3 weeks) [37]. Of note, nSTZ-induced microglia activation paralleled the development of tactile allodynia.
nSTZ also enhanced expression of GFAP immunoreactivity in the spinal dorsal horn from 8 to 16 weeks suggesting that activation of astrocytes may also be linked to neuropathic pain in this paradigm. Our data are in accordance with a previous report showing that db/db diabetic mice (type 2 diabetes model) develop tactile allodynia and activation of astrocytes in the spinal dorsal horn from 8 to 16 weeks [38]. However, it has also been reported that STZ administration in adult rats increases GFAP immunoreactivity in the spinal cord during the first 3–4 weeks [34]. The fact that astrocytic activation has been observed in the spinal dorsal horn in models of neuropathic pain [37, 39], supports the conclusion that nSTZ produces nerve injury which could be linked to activation of glial cells in the spinal cord. Microglia and astrocytes could contribute to the development and maintenance of tactile allodynia as previously reported [38, 40].
Effects of gabapentin and metformin on nSTZ-induced tactile allodynia
We observed that gabapentin and morphine reverted nSTZ-induced tactile allodynia in rats. In contrast, diclofenac was practically inactive in the model. This pharmacological profile suggests a neuropathic-like pain [25, 41]. Moreover, metformin was also effective in nSTZ rats suggesting a diabetic neuropathic-like pain [42]. Our data agree with other studies showing that metformin reduces STZ-induced increase in advanced glycation end-products [43] as well as tactile allodynia [44]. Furthermore, metformin diminishes tactile allodynia in other neuropathic pain models [23, 45]. It is interesting that despite metformin was not able to modify glucose blood levels in nSTZ-treated rats, it showed antiallodynic effect in those rats indicating that its anti-neuropathic effect is independent of a glucose lowering effect. These data are similar to those previously reported for metformin [44].
In conclusion, nSTZ produces mild hyperglycemia, glucose intolerance, weight loss as well as activation of satellite glial cells, microglia and astrocytes. All these features are accompanied by tactile allodynia. Administration of gabapentin, metformin and, to a lesser extent, morphine relieves tactile allodynia suggesting that nSTZ produces nerve injury and neuropathic pain. Unlike the type 1 diabetes model (STZ in adult rats), rats in the nSTZ model have relatively intact overall health indicating that this model could be a more appropriate option for the study of pain mechanisms in diabetic neuropathic pain.
HIGHLIGHTS.
nSTZ produced hyperglycemia, weight loss and glucose intolerance in neonatal rats.
nSTZ enhanced ATF3 expression in DRG, satellite glial cells and sciatic nerve.
nSTZ increased GFAP and OX-42 immunoreactivity in the spinal dorsal horn.
nSTZ produced mechanical hypersensitivity (tactile allodynia).
Gabapentin and metformin alleviated tactile allodynia.
Acknowledgments
We greatly appreciate the technical assistance of Guadalupe C. Vidal-Cantú, José Rodolfo Fernández-Calderón and Guadalupe Raya-Tafoya. Paulino Barragán-Iglesias and Emmanuel Loaeza-Alcocer received a postdoctoral fellowship from Conacyt (Grant CB-2012/179294). Victor Hugo Oidor-Chan, Jorge Baruch Pineda-Farias, Isabel Velazquez-Lagunas and Ana Belen Salinas-Abarca are Conacyt fellows. This work was supported by Conacyt (CB-2012/179294 to VG-S and RD-L) and NIH (NS065926 to TJP).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflicts of interest
None.
References
- 1.Tesfaye S, Boulton AJ, Dickenson AH. Mechanisms and management of diabetic painful distal symmetrical polyneuropathy. Diabetes Care. 2013;36:2456–65. doi: 10.2337/dc12-1964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pop-Busui R, Martin C. Neuropathy in the DCCT/EDIC-What was done then and what we would do better now. Int Rev Neurobiol. 2016;127:9–25. doi: 10.1016/bs.irn.2016.02.020. [DOI] [PubMed] [Google Scholar]
- 3.Yagihashi S. Glucotoxic mechanisms and related therapeutic approaches. Int Rev Neurobiol. 2016;127:121–49. doi: 10.1016/bs.irn.2016.03.006. [DOI] [PubMed] [Google Scholar]
- 4.Tomlinson DR, Gardiner NJ. Glucose neurotoxicity. Nat Rev Neurosci. 2008;9:36–45. doi: 10.1038/nrn2294. [DOI] [PubMed] [Google Scholar]
- 5.Fernyhough P. Mitochondrial dysfunction in diabetic neuropathy: a series of unfortunate metabolic events. Curr Diab Rep. 2015;15:89. doi: 10.1007/s11892-015-0671-9. [DOI] [PubMed] [Google Scholar]
- 6.Biessels GJ, Bril V, Calcutt NA, Cameron NE, Cotter MA, Dobrowsky R, et al. Phenotyping animal models of diabetic neuropathy: A consensus statement of the diabetic neuropathy study group of the EASD (Neurodiab) J Peripher Nerv Syst. 2014;19:77–87. doi: 10.1111/jns5.12072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wei M, Ong L, Smith MT, Ross FB, Schmid K, Hoey AJ, et al. The streptozotocin-diabetic rat as a model of the chronic complications of human diabetes. Heart Lung Circ. 2003;12:44–50. doi: 10.1046/j.1444-2892.2003.00160.x. [DOI] [PubMed] [Google Scholar]
- 8.Courteix C, Eschalier A, Lavarenne J. Streptozocin-induced diabetic rats: Behavioural evidence for a model of chronic pain. Pain. 1993;53:81–8. doi: 10.1016/0304-3959(93)90059-X. [DOI] [PubMed] [Google Scholar]
- 9.Calcutt NA, Jorge MC, Yaksh TL, Chaplan SR. Tactile allodynia and formalin hyperalgesia in streptozotocin-diabetic rats: effects of insulin, aldose reductase inhibition and lidocaine. Pain. 1996;68:293–9. doi: 10.1016/s0304-3959(96)03201-0. [DOI] [PubMed] [Google Scholar]
- 10.Callaghan BC, Hur J, Feldman EL. Diabetic neuropathy: One disease or two? Curr Opin Neurol. 2012;25:536–41. doi: 10.1097/WCO.0b013e328357a797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Portha B, Blondel O, Serradas P, McEvoy R, Giroix MH, Kergoat M, et al. The rat models of non-insulin dependent diabetes induced by neonatal streptozotocin. Diabete Metab. 1989;15:61–75. [PubMed] [Google Scholar]
- 12.Blondel O, Bailbe D, Portha B. Insulin resistance in rats with non-insulin-dependent diabetes induced by neonatal (5 days) streptozotocin: evidence for reversal following phlorizin treatment. Metabolism. 1990;39:787–93. doi: 10.1016/0026-0495(90)90120-2. [DOI] [PubMed] [Google Scholar]
- 13.Takada J, Machado MA, Peres SB, Brito LC, Borges-Silva CN, Costa CE, et al. Neonatal streptozotocin-induced diabetes mellitus: a model of insulin resistance associated with loss of adipose mass. Metabolism. 2007;56:977–84. doi: 10.1016/j.metabol.2006.05.021. [DOI] [PubMed] [Google Scholar]
- 14.Ariza L, Pages G, Garcia-Lareu B, Cobianchi S, Otaegui PJ, Ruberte J, et al. Experimental diabetes in neonatal mice induces early peripheral sensorimotor neuropathy. Neuroscience. 2014;274:250–9. doi: 10.1016/j.neuroscience.2014.05.015. [DOI] [PubMed] [Google Scholar]
- 15.Portha B, Levacher C, Picon L, Rosselin G. Diabetogenic effect of streptozotocin in the rat during the perinatal period. Diabetes. 1974;23:889–95. doi: 10.2337/diab.23.11.889. [DOI] [PubMed] [Google Scholar]
- 16.Zimmermann M. Ethical guidelines for investigations of experimental pain in conscious animals. Pain. 1983;16:109–10. doi: 10.1016/0304-3959(83)90201-4. [DOI] [PubMed] [Google Scholar]
- 17.Pineda-Farias JB, Barragán-Iglesias P, Loeza-Alcocer E, Torres-López JE, Rocha-González HI, Pérez-Severiano F, et al. Role of anoctamin-1 and bestrophin-1 in spinal nerve ligation-induced neuropathic pain in rats. Mol Pain. 2015;11:41. doi: 10.1186/s12990-015-0042-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ayoub AE, Salm AK. Increased morphological diversity of microglia in the activated hypothalamic supraoptic nucleus. J Neurosci. 2003;23:7759–66. doi: 10.1523/JNEUROSCI.23-21-07759.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chaplan SR, Bach FW, Pogrel JW, Chung JM, Yaksh TL. Quantitative assessment of tactile allodynia in the rat paw. J Neurosci Methods. 1994;53:55–63. doi: 10.1016/0165-0270(94)90144-9. [DOI] [PubMed] [Google Scholar]
- 20.Field MJ, McCleary S, Hughes J, Singh L. Gabapentin and pregabalin, but not morphine and amitriptyline, block both static and dynamic components of mechanical allodynia induced by streptozocin in the rat. Pain. 1999;80:391–8. doi: 10.1016/s0304-3959(98)00239-5. [DOI] [PubMed] [Google Scholar]
- 21.Ortega-Varela LF, Herrera JE, Caram-Salas NL, Rocha-González HI, Granados-Soto V. Isobolographic analyses of the gabapentin-metamizol combination after local peripheral, intrathecal and oral administration in the rat. Pharmacology. 2007;79:214–22. doi: 10.1159/000101390. [DOI] [PubMed] [Google Scholar]
- 22.Ortiz MI, Lozano-Cuenca J, Granados-Soto V, Castañeda-Hernández G. Additive interaction between peripheral and central mechanisms involved in the antinociceptive effect of diclofenac in the formalin test in rats. Pharmacol Biochem Behav. 2008;91:32–7. doi: 10.1016/j.pbb.2008.06.008. [DOI] [PubMed] [Google Scholar]
- 23.Melemedjian OK, Asiedu MN, Tillu DV, Sanoja R, Yan J, Lark A, et al. Targeting adenosine monophosphate-activated protein kinase (AMPK) in preclinical models reveals a potential mechanism for the treatment of neuropathic pain. Mol Pain. 2011;7:70. doi: 10.1186/1744-8069-7-70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Reyes-García G, Déciga-Campos M, Médina-Santillán R, Granados-Soto V. Comparison of antinociceptive efficacy and gastroprotection between celecoxib and diclofenac plus misoprostol in rats. Proc West Pharmacol Soc. 2007;50:69–71. [PubMed] [Google Scholar]
- 25.Kiso T, Watabiki T, Tsukamoto M, Okabe M, Kagami M, Nishimura K, et al. Pharmacological characterization and gene expression profiling of an L5/L6 spinal nerve ligation model for neuropathic pain in mice. Neuroscience. 2008;153:492–500. doi: 10.1016/j.neuroscience.2008.02.031. [DOI] [PubMed] [Google Scholar]
- 26.Mixcoatl-Zecuatl T, Quiñonez-Bastidas GN, Caram-Salas NL, Ambriz-Tututi M, Araiza-Saldaña CI, Rocha-González HI, et al. Synergistic antiallodynic interaction between gabapentin or carbamazepine and either benfotiamine or cyanocobalamin in neuropathic rats. Methods Find Exp Clin Pharmacol. 2008;30:431–41. doi: 10.1358/mf.2008.30.6.1232963. [DOI] [PubMed] [Google Scholar]
- 27.Cameron NE, Cotter MA, Ferguson K, Robertson S, Radcliffe MA. Effects of chronic alpha-adrenergic receptor blockade on peripheral nerve conduction, hypoxic resistance, polyols, Na+-K+-ATPase activity, and vascular supply in STZ-D rats. Diabetes. 1991;40:1652–8. doi: 10.2337/diab.40.12.1652. [DOI] [PubMed] [Google Scholar]
- 28.Coppey LJ, Davidson EP, Dunlap JA, Lund DD, Yorek MA. Slowing of motor nerve conduction velocity in streptozotocin-induced diabetic rats is preceded by impaired vasodilation in arterioles that overlie the sciatic nerve. Int J Exp Diabetes Res. 2000;1:131–43. doi: 10.1155/EDR.2000.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tsujino H, Kondo E, Fukuoka T, Dai Y, Tokunaga A, Miki K, et al. Activating transcription factor 3 (ATF3) induction by axotomy in sensory and motoneurons: A novel neuronal marker of nerve injury. Mol Cell Neurosci. 2000;15:170–82. doi: 10.1006/mcne.1999.0814. [DOI] [PubMed] [Google Scholar]
- 30.Kataoka K, Kanje M, Dahlin LB. Induction of activating transcription factor 3 after different sciatic nerve injuries in adult rats. Scand J Plast Reconstr Surg Hand Surg. 2007;41:158–66. doi: 10.1080/02844310701318288. [DOI] [PubMed] [Google Scholar]
- 31.Hanani M, Blum E, Liu S, Peng L, Liang S. Satellite glial cells in dorsal root ganglia are activated in streptozotocin-treated rodents. J Cell Mol Med. 2014;18:2367–71. doi: 10.1111/jcmm.12406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Donegan M, Kernisant M, Cua C, Jasmin L, Ohara PT. Satellite glial cell proliferation in the trigeminal ganglia after chronic constriction injury of the infraorbital nerve. Glia. 2013;61:2000–8. doi: 10.1002/glia.22571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Toth CC, Jedrzejewski NM, Ellis CL, Frey WH., 2nd Cannabinoid-mediated modulation of neuropathic pain and microglial accumulation in a model of murine type I diabetic peripheral neuropathic pain. Mol Pain. 2010;6:16. doi: 10.1186/1744-8069-6-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cheng KI, Wang HC, Chuang YT, Chou CW, Tu HP, Yu YC, et al. Persistent mechanical allodynia positively correlates with an increase in activated microglia and increased P-p38 mitogen-activated protein kinase activation in streptozotocin-induced diabetic rats. Eur J Pain. 2014;18:162–73. doi: 10.1002/j.1532-2149.2013.00356.x. [DOI] [PubMed] [Google Scholar]
- 35.Tsuda M, Ueno H, Kataoka A, Tozaki-Saitoh H, Inoue K. Activation of dorsal horn microglia contributes to diabetes-induced tactile allodynia via extracellular signal-regulated protein kinase signaling. Glia. 2008;56:378–86. doi: 10.1002/glia.20623. [DOI] [PubMed] [Google Scholar]
- 36.Wodarski R, Clark AK, Grist J, Marchand F, Malcangio M. Gabapentin reverses microglial activation in the spinal cord of streptozotocin-induced diabetic rats. Eur J Pain. 2009;13:807–11. doi: 10.1016/j.ejpain.2008.09.010. [DOI] [PubMed] [Google Scholar]
- 37.Watkins LR, Maier SF. Glia: a novel drug discovery target for clinical pain. Nat Rev Drug Discov. 2003;2:973–85. doi: 10.1038/nrd1251. [DOI] [PubMed] [Google Scholar]
- 38.Liao YH, Zhang GH, Jia D, Wang P, Qian NS, He F, et al. Spinal astrocytic activation contributes to mechanical allodynia in a mouse model of type 2 diabetes. Brain Res. 2011;1368:324–35. doi: 10.1016/j.brainres.2010.10.044. [DOI] [PubMed] [Google Scholar]
- 39.Ji RR, Strichartz G. Cell signaling and the genesis of neuropathic pain. Sci STKE. 2004;2004:reE14. doi: 10.1126/stke.2522004re14. [DOI] [PubMed] [Google Scholar]
- 40.Coull JA, Beggs S, Boudreau D, Boivin D, Tsuda M, Inoue K, et al. BDNF from microglia causes the shift in neuronal anion gradient underlying neuropathic pain. Nature. 2005;438:1017–21. doi: 10.1038/nature04223. [DOI] [PubMed] [Google Scholar]
- 41.LaBuda CJ, Little PJ. Pharmacological evaluation of the selective spinal nerve ligation model of neuropathic pain in the rat. J Neurosci Methods. 2005;144:175–81. doi: 10.1016/j.jneumeth.2004.11.008. [DOI] [PubMed] [Google Scholar]
- 42.Ma J, Yu H, Liu J, Chen Y, Wang Q, Xiang L. Metformin attenuates hyperalgesia and allodynia in rats with painful diabetic neuropathy induced by streptozotocin. Eur J Pharmacol. 2015;764:599–606. doi: 10.1016/j.ejphar.2015.06.010. [DOI] [PubMed] [Google Scholar]
- 43.Tanaka Y, Uchino H, Shimizu T, Yoshii H, Niwa M, Ohmura C, et al. Effect of metformin on advanced glycation endproduct formation and peripheral nerve function in streptozotocin-induced diabetic rats. Eur J Pharmacol. 1999;376:17–22. doi: 10.1016/s0014-2999(99)00342-8. [DOI] [PubMed] [Google Scholar]
- 44.Byrne FM, Cheetham S, Vickers S, Chapman V. Characterisation of pain responses in the high fat diet/streptozotocin model of diabetes and the analgesic effects of antidiabetic treatments. J Diabetes Res. 2015;2015:752481. doi: 10.1155/2015/752481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mao-Ying QL, Kavelaars A, Krukowski K, Huo XJ, Zhou W, Price TJ, et al. The anti-diabetic drug metformin protects against chemotherapy-induced peripheral neuropathy in a mouse model. PloS One. 2014;9:e100701. doi: 10.1371/journal.pone.0100701. [DOI] [PMC free article] [PubMed] [Google Scholar]