

Abstract

We explore the potential of DNA nanotechnology for developing novel optical voltage sensing nanodevices that convert a local change of electric potential into optical signals. As a proof-of-concept of the sensing mechanism, we assembled voltage responsive DNA origami structures labeled with a single pair of FRET dyes. The DNA structures were reversibly immobilized on a nanocapillary tip and underwent controlled structural changes upon application of an electric field. The applied field was monitored through a change in FRET efficiency. By exchanging the position of a single dye, we could tune the voltage sensitivity of our DNA origami structure, demonstrating the flexibility and versatility of our approach. The experimental studies were complemented by coarse-grained simulations that characterized voltage-dependent elastic deformation of the DNA nanostructures and the associated change in the distance between the FRET pair. Our work opens a novel pathway for determining the mechanical properties of DNA origami structures and highlights potential applications of dynamic DNA nanostructures as voltage sensors.
Keywords: DNA nanotechnology, single-molecule FRET, optical voltage measurements, nanocapillary, coarse-grained simulations
The design of nanometer scale biocompatible sensors has become a popular topic of research in recent years for purposes ranging from single-molecule sensing for disease diagnostics to understanding complex biological functionalities like neural cell responses.1−3 High spatial resolution is required for mapping of localized signals and thus a nanometer-sized footprint is essential for such a sensor.4 Optimum sensitivity is also key if the sensor is to be used as a reliable gauge of a biological process.5 In this study, we aimed to create a molecular device for optical voltage sensing. Future applications may include studying neuronal signaling and other biological processes resulting from a difference in potential across a membrane.
The application of light as a basis for voltage sensing offers a promising avenue to overcome the highly invasive nature of electrode-based techniques. Optical voltage-imaging requires the use of a reporter to translate the electrical signal into an optical readout signal. Quantitative measurements of the membrane voltage will thus rely on careful calibration of this optical probe and an understanding of the underlying voltage sensing mechanism. On the basis of the pioneering work of Cohen et al. in the 1970s,6 there have been enormous research efforts into the development of optical voltage sensing probes over the last four decades, which have yielded a number of different approaches, such as calcium imaging7 and voltage-sensitive dyes using for example electrochromic dyes9 or two-component FRET-based voltage sensors,10,11 whose fluorescence properties directly depend on the electric field across a membrane. However, these voltage sensing mechanisms can be hampered by some combination of limitations including low sensitivity, slow kinetics, or heavy capacitive loading.12,13
Here, we demonstrate the potential of DNA origami self-assembly as a tool to create a new generation of voltage sensors. DNA origami14−16 has emerged as a versatile and robust method to build nanoscale devices.17−19 Besides the programmability of the overall shape of the DNA-templated structure, the key strength of this technique lies in the ability to attach chemical functionalities such as fluorescent dyes,20 cholesterol moieties,21 or proteins22 with base-pair precision. This feature allows for easy and quick adaptation of designs to a particular experimental requirement. DNA-based force sensors that translate quantitatively a mechanical force to an optical signal have been developed previously.23−26 Shroff et al. used a single-stranded DNA oligomer labeled with a FRET pair to measure forces in the picoNewton (pN) range with magnetic tweezers.24 In 2016, Nickels et al. developed a DNA origami-based force clamp that allowed for the measurement of TATA-binding protein induced bending of a DNA duplex under tension.26
In this work, we develop a DNA origami based optical voltage sensing mechanism using FRET-based ratiometric signaling. The development of effective optical voltage sensors will encompass different functions. It includes not only the sensing and reporting mechanism, but biocompatibility and possible future cell membrane targeting.12 Because of the inherent flexibility of DNA nanotechnology, these functions can be optimized individually and ultimately be integrated into a single molecular device. Our previous work has demonstrated the ability of DNA origami to serve as a canvas for positioning fluorescent dyes with nanometer precision.27,28 The technique allows us to configure the distance between the dyes, a quantity which can be read out optically using a suitable donor–acceptor FRET pair. Complementary to this, we have also investigated the mechanical stability of DNA origami structures in an electric field29,30 and characterized the forces on DNA molecules in confined electric fields.31 We have shown that an increase in voltage can induce gradual distortions as well as distinct changes in conformation. Combining these unique features thus enables the design of dynamic voltage-responsive molecular rulers with optical readout. Here we present proof-of-principle simulations and experiments that demonstrate the successful use of a DNA origami structure labeled with a FRET dye pair, whose FRET signal is calibrated in response to the applied potential.
Results
Design of a Voltage-Responsive Fluorescent DNA Origami Structure
We created a voltage-responsive, fluorescent DNA origami structure that would undergo controlled structural changes upon applying an electric field. As shown in Figure 1, we used a two-layered DNA origami plate (170 bp × 24 helices × 2 helices) with a double-stranded leash (774 bp) and a central rectangular opening (42 bp × 6 helices) based on a previously published design.29 Further characterization of the structural details of this DNA origami plate is described in Materials and Methods and Supplementary Sections S1 and S2. The structure was labeled with a FRET dye pair whose FRET signal acts as the optical readout for the surrounding potential.32,33 We chose ATTO532 (donor) and ATTO647N (acceptor) as the FRET pair34,35 due to the large spectral overlap between donor emission and acceptor absorption and clear spectral separation between emissions. Both dyes also have similar quantum yields and brightness.
Figure 1.
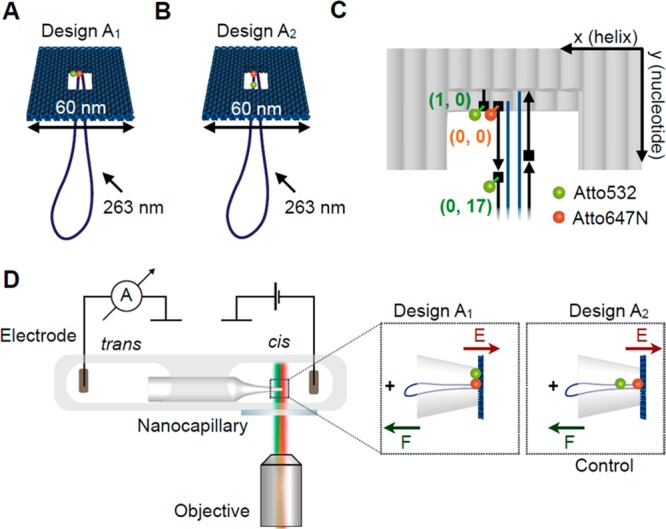
(A) Illustration of the DNA origami plate with a double-stranded leash protruding from its central aperture. The plate is labeled with a FRET pair located at the edge of the central opening. The donor dye (ATTO532, green) is located closer to the edge of the plate while the acceptor is close to the leash in the center (ATTO647N, red). (B) Illustration of the control DNA origami plate with leash labeled with the same FRET pair. The green donor dye is now located on the leash. (C) Exact positions of the dyes in the central part of the two-layered DNA origami platform. Each gray rod represents a double helix and the scaffold strand in dark blue and staple strands in black. The ATTO dyes are located in three locations. We denote the positions using a DNA origami coordinate system (x, y) with x (helix number) and y (nucleotide number). The acceptor dye (ATTO647N) is defined as origin (0, 0) and is attached to the 5′-end (illustrated as rectangle) of the staple strand marking the starting point of the leash with respect to the plate. One donor dye (ATTO532) can be positioned at the 5′-end of a staple one helix away (1, 0) at the edge of the plate. Another donor dye (ATTO532) can be positioned at the 5′-end of the staple strand adjacent to the acceptor strand in a distance of 17 nucleotides from the acceptor dye along the leash (0, 17). (D) Schematic of experimental design. The core of the experimental setup consists of a nanocapillary connecting two electrolyte reservoirs. A voltage can be applied across the nanocapillary for ionic current recordings. The microfluidic chip containing the nanocapillary is placed directly above a fluorescence microscopy objective for synchronous single-molecule fluorescence imaging. (Insets) The DNA origami plate is trapped onto the nanocapillary tip upon applying a positive voltage.
We strategically selected three different anchoring positions for accommodating the ATTO dyes on the DNA origami structures resulting in two different FRET pair arrangements. An overview of the two designs is shown in Figure 1A,B. As shown in Figure 1C, we employ a DNA coordinate system (x, y), where x is the helix number and y is the nucleotide number along the helix, to precisely define the dye positions and the theoretical interdye distances. We define the origin (0, 0) as the location of the ATTO647N, which is the acceptor common to either possible donor location. This position also marks the structural interface of the plate and the leash. One donor fluorescent label (ATTO532) is attached to the 5′-end of a staple strand using a 6-carbon linker terminating at the inner edge of the plate, one helix away from the acceptor (Figure 1C). Another donor fluorescent label (ATTO532) is in the same way attached to the 5′-end of the leash staple adjacent to the staple hosting the acceptor, which is displaced by 17 base pairs along the leash. This arrangement allowed us to assemble two different versions of the DNA origami plate, where a FRET pair is either located at the inner edge of the plate with the axis oriented perpendicular to the direction of the electric field (design A1, Figure 1A) or along the leash (control design A2, Figure 1B) with the axis aligned with the electric field vector. Because of previous evidence of an analogous two-layered plate deforming under increasing electric field strength,29 it was hypothesized that placing the dyes at different points on the main body of the plate as in design A1, will result in movement of the dyes as the helices change their relative positions, resulting in a change in FRET efficiency.
The estimated theoretical donor-to-acceptor distances are R1 = ∼3 nm (design A1) and R2 = ∼5.8 nm (design A2). These values were obtained from the distance between donor and acceptor attachment sites in an atomistic model of the plate constructed after a coarse-grained simulation of the object (see below) with a small 100 mV voltage applied across the nanocapillary.
Coarse-Grained Simulation of Voltage-Dependent Deformation
We carried out coarse-grained Brownian dynamics simulations to clarify whether changes in distances between the dyes were likely to occur when a range of voltages was applied to designs A1 and A2 immobilized on a capillary support. Bead-based models were constructed from the caDNAno design with potentials designed to capture properties that were experimentally determined (e.g., persistence length)36 and observed in all-atom simulations (e.g., potential of mean force between parallel DNA duplexes).37 The electrophoretic force due to the capillary was obtained from a continuum model. Materials and Methods and Supplementary Section 3 provide a complete description of the simulation setup and parameters.
Figure 2A illustrates the state of a typical coarse-grained simulation before and after the origami plate was captured by the nanocapillary under a 400 mV applied bias. Capture occurred within the first 10 μs of simulation. After capture the structure of the plate exhibited steady-state fluctuations for the remaining ∼30 μs, see Supplementary Movie 1. When the applied voltage was increased to 600 mV, the origami plate was seen to deform until it passed through the nanocapillary aperture, see Supplementary Movie 2. The configuration of the plate every 2 μs between 30 and 40 μs was used to initialize a finer-resolution coarse-grained simulation of the plate that allowed the distance between the FRET dyes to be estimated. The zoomed-in images show representative configurations of the plate in one such simulation, highlighting the positioning of the donor (green) and acceptor (red) dye for design A1. Despite undergoing considerable fluctuations, the distance between the dyes was systematically larger in the simulations of design A1 carried out at higher biases, Figure 2B and Supplementary Movie 3. Figure 2C plots the average change in the distance between the dyes with respect to the average distance at 100 mV as a function of applied voltage for both designs. A significant increase in distance is observed for design A1 between 150 and 400 mV, whereas there is a negligible distance change in the control design A2 where the dyes are positioned along the leash. These results suggest a possibility of observing a decreasing FRET signal for design A1 but not design A2, at increasing transmembrane voltages.
Figure 2.

Coarse-grained Brownian dynamics simulations of the voltage sensor. (A) (left) A typical simulation system. Electrostatic potential around the nanocapillary due to a 400 mV applied bias is superimposed with the initial configuration of the plate described using a low-resolution (five base pairs per bead) coarse-grained model. The configuration after 40 μs of simulation is shown to the right. Part of the nanocapillary is cut away for clarity. A high-resolution model of the plate (two beads per base pair) was used in subsequent simulations to measure the distance between the labeled nucleotides. The inset outlined in teal (left) shows the initial configuration of one such model under a 400 mV bias. The inset outlined in purple (right) shows the same model after a 500 ns simulation. Here, the side-by-side FRET pair of design A1 is shown, but similar simulations were used to estimate distances for both FRET pairs. (B) The distance between the labeled nucleotides during simulations of the high-resolution coarse-grained models at low (red) and high (dark red) applied biases for design A1. The circles indicate the states featured in panel A. (C) The distance between labeled nucleotides averaged over five simulations for each applied bias, relative to the distance obtained at 100 mV. The average distance between the nucleotides was obtained in each simulation, and the mean and standard error of the mean of the five distance values were calculated at each bias. The bars show the propagation of these errors for the difference of the distances. The lines show linear fits to the data.
Simultaneous Current/FRET Sensing
Having successfully simulated a voltage response in our origami design A1, a customized setup was constructed that enables simultaneous voltage control and dual-color fluorescence detection. The setup is equipped with a two-color alternating laser excitation (ALEX) scheme.38,39 This method allows for the quantitative measurement of FRET efficiency, independent of detection efficiency and the quantum yield of the dyes. From the alternation of two excitation wavelengths and detection of two emission bands, we obtain three relevant distinct photon streams IX(Y*). Here, X denotes the two emission windows D and A, which are D = 554–613 nm for donor emission and A = 658–742 nm for acceptor emission, respectively. Y* denotes the two excitation wavelengths D*= 532 nm and A* = 638 nm for donor (D*) and acceptor (A*) excitation, respectively. Direct excitation of the donor and acceptor results in the two fluorescence intensities ID(D*) and IA(A*) for donor and acceptor emission, respectively. Acceptor fluorescence sensitized by FRET from donor to acceptor is monitored in the third photon stream IA(D*). By determining correction factors, the FRET-induced signal can be disentangled from crosstalk terms such as donor leakage and direct acceptor excitation. However, the ALEX technique in this work was predominantly used to identify artifacts induced by acceptor photophysics and to determine the acceptor bleaching moment by observing the signal generated from direct excitation of the acceptor. ALEX is particularly useful for identifying incompletely labeled populations in automated analysis.38−40
For a detailed schematic of the setup, see Supplementary Figure S3. A microfluidic chip hosting the quartz nanocapillary was mounted on a glass slide that was directly placed on top of the fluorescence microscope objective (Figure 1D). In this way, the ionic current through the nanocapillary could be recorded while also imaging fluorescent structures in the vicinity of the nanocapillary tip. Because of the negative charge of the DNA backbone, DNA origami structures can be reversibly trapped on a glass nanocapillary tip upon applying a positive voltage. The applied electric field exerts forces on the arrested DNA origami structure inducing mechanical deformations. The FRET efficiency acted as the optical readout signal for monitoring local voltage-induced distortions of the DNA origami structure and thus reporting on the strength of the voltage applied.
Simultaneous Fluorescence and Ionic Current Measurements
Having characterized the bulk fluorescence properties of our fluorescently labeled DNA origami structures (Supplementary Section 5), we performed simultaneous ionic current measurements and time-resolved FRET-based fluorescence imaging at the single-molecule level. The DNA origami plates in the cis reservoir were repeatedly pulled onto the quartz nanocapillary tip by applying a positive voltage in the trans reservoir. Each successful attachment was indicated by a sudden drop in ionic current due to a reduced passage of ions, as expected.29 We also observed the characteristic second population of deeper blockage events at higher voltages (discussed further in Supplementary Section 6).29
Figure 3 demonstrates how the DNA origami trapping process is simultaneously observed with fluorescence and ionic current measurements. The DNA origami trapping event is identified by the sudden drop in ionic current at 10 s (change in conductance, ΔG ≈ 10%) that directly reflects the partial obstruction of the ion passage through the nanocapillary by the DNA origami plate. At 10 s, colocalized intensity increases appeared in all the three fluorescence channels (ID(D*), IA(D*), IA(A*), Figure 3A), which indicates attachment of the FRET pair labeled DNA origami structure on the capillary tip. The location of the fluorescence intensity coincides with the location of the nanocapillary tip determined from a brightfield image taken prior to starting measurements (Figure 3C).
Figure 3.
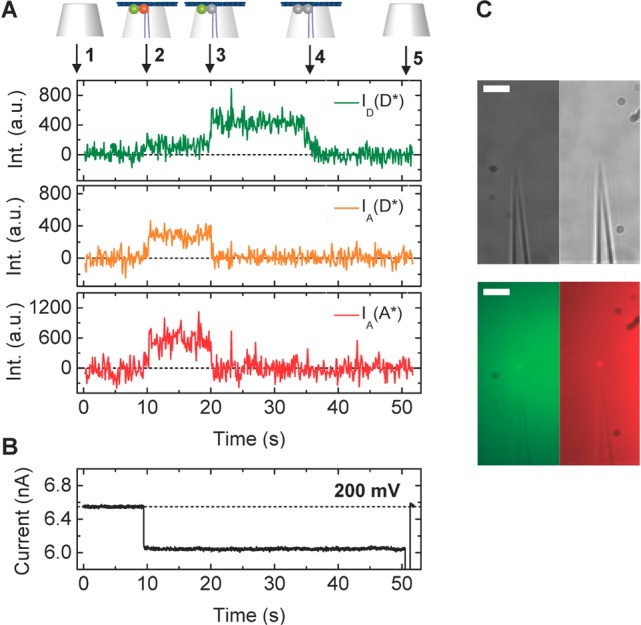
(A) Representative examples of fluorescence intensity traces correlated with voltage and ionic current recordings for DNA origami plates with design A1. (A) (top) Trace annotations: 1, Bare nanocapillary. 2, DNA origami trapping. 3, Acceptor bleaching. 4, Donor bleaching. 5, DNA origami ejection. (bottom) Fluorescence intensity ID(D*), IA(D*), IA(A*) traces. (B) Ionic current trace, please note that during these measurements the voltage was held constant at 200 mV until t = 50 s when the structure is removed by applying a brief voltage fluctuation from + to −1000 mV. (C) Brightfield (top) and fluorescence (bottom) images of a FRET pair labeled origami immobilized at the capillary tip in the donor (left) and acceptor (right) emission channels. Scale bar: 5 μm.
The fluorescent signals in ID(D*) and IA(A*) originate from direct excitation of the ATTO532 and ATTO647N dyes with the green and red laser, respectively, and confirm the presence of both labels independently. The fluorescence traces show a signature that provides compelling evidence of FRET. We start off at the fluorescence background level (1) and detect a simultaneous fluorescence increase in all the channels ID(D*), IA(D*), IA(A*) (2). While both dyes are active as monitored by ID(D*) and IA(A*), energy from the donor is transferred to the acceptor via FRET as apparent in IA(D*). As soon as the acceptor bleaches the FRET signal ceases as well (3). At the same time, the donor intensity rises instantly due to abrogated FRET by the acceptor (3). The sequence is completed by donor bleaching (4). An example of a representative FRET trace for design A2 is shown in Supplementary Figure S6.
Determining the Single-Molecule FRET Efficiency
From the traces, we determine the time-dependent proximity ratio Ei*(i), or uncorrected FRET efficiency. By taking the ratio of the FRET signal over the overall fluorescence intensity, we obtain
| 1 |
in each time frame i. In ALEX, the frames are counted independently in each channel. For each trace, we then obtain an overall single-molecule proximity ratio or uncorrected FRET efficiency Esm* by taking the average of all Ei*(i) values
| 2 |
within a window i to N while both donor and acceptor dyes are active, and where N is the total number of frames in this window. The start of the window i = 1 is defined by the insertion of the DNA origami platform (2) and the end i = N as the first bleaching event in at least one of the fluorescence channels ID(D*) and IA(A*) (3) (Figure 3A). In some cases, as supported by ionic current detection, simultaneous extinction of the fluorescence signal in all channels may indicate removal of the DNA origami platform from the nanocapillary tip rather than bleaching of both dyes at the same time. For the purpose of calculating the FRET proximity ratio, we do not make a distinction between the two possible origins of this event type and treat it as the end frame N of the window in either case.
Applying eqs 1 and 2 to calculate the proximity ratio Esm* of the example FRET traces, we extract E1* = 0.73 (Figure 3A) at a voltage of 200 mV.
These experiments at constant voltage demonstrate that the fluorescent intensity peaks appear simultaneously in all channels (ID(D*), IA(D*), IA(A*)), confirming the presence of both donor and acceptor dye as well as the proximity relative to each other. In addition, the drop in ionic current is synchronized with the increase in fluorescence intensity, as expected.
Voltage Dependence of FRET Proximity Ratio
In order
to investigate if our DNA structures showed a voltage-dependent FRET
change, origami structures were individually trapped onto the capillary
tip using a voltage from 100 to 400 mV. For each structure, their
fluorescence and current signal was measured and finally the structure
ejected. Data analysis, including consistency checks (see Materials and Methods for details), resulted in
around 50% of all DNA origami insertion events, as specified by a
drop in ionic current, with fluorescence traces qualifying for determination
of the FRET proximity ratio Esm*. The
total number of useable traces reached Ntot = 185 from eight capillaries for design A1 and Ntot = 241 from six capillaries
for design A2 over a voltage range of
100–400 mV, respectively. The intensity of the dyes was not
systematically effected by the presence of the electric field (Supplementary Figure S7) implying a negligible
voltage-induced photophysical effect on the dyes. From these traces
we determined the proximity ratio Esm*
as a function of the voltage applied over a range of 100–400
mV in 50 mV steps for the DNA origami plate with design A1 and A2, respectively. The
results are summarized in Figure 4A, B, where the proximity ratio is indicated relative
to the value in the absence of an applied voltage, that is, E*(x mV) – E*(0
mV). The experimental results were compared to the simulated change
in distances between the dyes with voltage (Figure 2). Specifically, we have converted the simulated
dye separations, r, into proximity ratios using the
expression 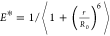 where R0 =
5.9 nm is the Förster radius.41 The
simulated FRET efficiency is approximate since the precise value for R0 depends on the local environment around the
dyes and because, lacking explicit representation of the dye linkers,
we used the labeling sites as a proxy for the dye positions. For the
majority of the values, the simulated proximity ratios agree well
with the experimental results within the spread of the data.
where R0 =
5.9 nm is the Förster radius.41 The
simulated FRET efficiency is approximate since the precise value for R0 depends on the local environment around the
dyes and because, lacking explicit representation of the dye linkers,
we used the labeling sites as a proxy for the dye positions. For the
majority of the values, the simulated proximity ratios agree well
with the experimental results within the spread of the data.
Figure 4.

Relative proximity ratio Esm* as a function of the voltage applied for design A1 (A) and for design A2 (B) from capillary experiments and coarse-grained simulations. Esm* is indicated relative to the value in the absence of an applied voltage i.e. E*(x mV) – E*(0 mV). For the unscaled proximity ratios, see Supplementary Figure S8. We used Ntot = 185 traces from 8 capillaries in design A1 and Ntot = 241 traces from 6 capillaries in design A2. Experimental zero voltage values were obtained by measuring the IA(D*) signal from origami structures immobilized on a coverslip using a BSA-biotin-neutravidin coating (outlined in Methods). The error bars correspond to the standard error of the mean. The simulated proximity ratio was calculated from the simulated distance between dye labeling sites, r using the expression relating FRET efficiency to distance E* = 1/⟨1 + (r/R0)6⟩ where R0 = 5.9 nm is the Förster radius.
We observe that the two designs behave differently for different applied voltages. For the DNA origami plate with design A1 where the donor dye is located on the edge of the inner plate aperture and the acceptor dye on the first leash staple, the relative proximity ratio Esm* shows a clear voltage dependent behavior. At low voltages (100–200 mV), Esm* remains stable. Above 200 mV, Esm* decreases in a linear fashion at ΔEsm*/ΔV ≈ 0.1/100 mV (Figure 4A). In contrast Esm* for design A2, where the donor and acceptor are positioned along the double-stranded leash, the relative proximity ratio Esm* stays constant over the entire voltage range 100–400 mV (Figure 4B). This trend was reproducible using capillaries with a smaller aperture, therefore with higher average resistances (102 ± 19 MΩ, SD) and a shifted origami trapping voltage range (200–800 mV, Supplementary Figure S9).
The data in Figure 4A, B are generated from multiple DNA origami trapping events where the DNA origami structure is exposed to a single, constant level of voltage. In contrast, Figure 5A, B show the proximity ratio for a single origami structure repeatedly exposed to a changing voltage. In the case of design A1, it can be seen that Esm* changes as expected for the change in voltage, that is, when ramping up the voltage from 100 to 400 mV, Esm* decreases (from ∼0.5 to ∼0.2), showing the great potential of our designs to convert a change in potential into an optical signal at the single-molecule level (Figure 5A). For additional examples see Supplementary Figure S10. In contrast to this, an isolated design A2 structure only fluctuates around a single Esm* value of ∼0.25 when exposed to a similar change in applied potential (Figure 5B). Our results prove that (i) DNA origami structures can be used as building blocks for optical voltage sensing and (ii) even the response of a single structure is in principle enough to detect a voltage change. This is a promising first step toward developing novel membrane potential sensors for complex environments as for example cellular membranes.
Figure 5.

Example trace (top) of the change in proximity ratio Esm* for a single trapped origami structure (measured and averaged proximity ratio for each voltage step) with steps in voltage (2 s, ΔV = 100 mV) from 100 to 400 mV (bottom) for (A) design A1 and (B) design A2.
The most obvious interpretation of the decrease in the uncorrected FRET efficiency as the voltage is stepped up is a gradual increase in distance between the donor and acceptor dye. On the basis of the assumption that the DNA origami insertion is directed by the double-stranded leash,29 the voltage sensing mechanism can be intuitively understood as a rope pulling on the DNA origami plate. The pulling force will directly depend on the strength of the electric field applied,29 which in design A1 is oriented almost perpendicularly with respect to the imaginary line connecting the FRET pair. The acceptor, positioned on the leash, will therefore be increasingly drawn away from the donor dye, attached to the plate adjacent to the leash by separation of adjacent helices, as the voltage and hence the force is increased. This interpretation is corroborated by the simulations of the origami structure (see Supplementary Movie 3). Therefore, in the case of design A1, our results show that there is potential for a FRET-based voltage sensor and we have provided a calibration of FRET signal with voltage between 200 and 400 mV.
By giving some simple physical arguments, we can also explain the voltage-independent behavior of the FRET efficiency in control design A2. The donor–acceptor pair is separated by 17 bp (5.8 nm) and the voltage is assumed to exert a force on the double-stranded DNA leash. The electric force normalized by the voltage applied on a DNA molecule in a nanopore has been estimated as κ ≈ 0.04 pN/mV.29,42,43 Within our voltage range of 100–400 mV, this yields an upper force limit of roughly 16 pN per DNA molecule. Because the persistence length of dsDNA is lp ≈ 50 nm,44 a leash section of length 6 nm is roughly straight at room temperature as seen in the simulations of the origami plate as well. Because we are far from the overstretching transition, the interdye spacing will remain unchanged (discussed further in Supplementary Section 12).
Designs A1 and A2 are thus exploring two different regimes in our system. A1 can clearly be used to detect electric field changes by following FRET efficiency. To the best of our knowledge this is the first time that structural changes in a DNA structure are used for voltage sensing at the single-molecule level.
Conclusion
We have explored DNA origami self-assembly as a novel avenue for creating the next generation of optical voltage sensing nanodevices. We successfully used coarse-grained Brownian dynamics to simulate the process of trapping DNA origami plates, labeled with a strategically located donor–acceptor pair, on a nanocapillary tip by applying a range of positive voltages. The designed structure was found to be highly sensitive to voltage changes above 200 mV, giving rise to a difference in proximity ratio (uncorrected FRET efficiency) of ΔEsm*/ΔV ≈ 0.1/100 mV. This voltage-sensitive FRET signal of our origami design was corroborated by experiments using a custom-built experimental setup, which allows for simultaneous ionic current recordings and single-molecule FRET measurements with two-color alternating laser excitation. This has enabled us to successfully obtain a calibration curve that relates an input voltage to an output optical signal in terms of FRET efficiency. The next major step forward will be to integrate a FRET-based optical voltage sensing mechanism into DNA nanopores that self-insert into lipid membranes. Following our successful proof-of-principle study, we believe that DNA origami based optical voltage sensing devices have great potential to be applied for live-cell imaging of transmembrane potentials in the near future.45
Materials and Methods
Coarse-Grained Brownian Dynamics Simulations
An in-house developed GPU-accelerated code called atomic resolution Brownian dynamics (ARBD)46 was used to perform simulations of the DNA origami object. The DNA origami models were constructed by importing caDNAno 2.547 as a Python module and directly querying the data structures to locate nucleotides and crossovers. Low-resolution models (∼5 bp/bead) were constructed to simulate the capture process and to provide initial coordinates for high-resolution models (2 beads/bp) that were used to estimate the distances between FRET labeled nucleotides. A detailed description of the model construction protocol and the model parameters is found in Supplementary Section 3.
Design and Assembly of the DNA Origami Structure
The open source software caDNAno was used to design and visualize the structure.48 The two-layered plate with a double-stranded leash was based on a previously published design.29 DNA staples were synthesized and purified by Integrated DNA Technologies, Inc. For caDNAno files and staple sequences see Supplementary Section 13.
To assemble the DNA origami structures, the staple strands and scaffold were mixed to a final concentration of 100 nM and 10 nM respectively in 16 mM MgCl2, buffered with 1× TE (10 mM Tris-HCl and 1 mM EDTA) at pH ∼ 8. The folding mixture was then subjected to a 23 h thermal-annealing process: 65 °C for 15 min, 65 to 60 °C in 5 cycles (−1 °C per cycle, 5 min each), 59 °C for 30 min, 59 to 40 °C in 19 cycles (−1 °C per cycle, 60 min each), 40 to 37 °C in 3 cycles (−1 °C per cycle, 30 min each), 37 to 25 °C in 12 cycles (−1 °C per cycle, 5 min each), and finally held at 4 °C. Following thermal-annealing, the folded DNA origami structures were purified from the excess staple strands in the buffer solution by centrifugation using Amicon Ultra-0.5 mL centrifugal filters (Merck) with a 100 kDa cutoff.
Characterization via UV–vis and Fluorescence Spectroscopy, AFM Imaging and Agarose Gel Electrophoresis
The concentration of the DNA structures was inferred from the absorbance at a wavelength of 260 nm, measured using a low-volume UV–vis spectrophotometer (Thermo Scientific, NanoDrop 2000). Concentrations obtained were typically ∼10 nM for the DNA origami plates.
Steady-state donor and acceptor fluorescence emission spectra were obtained on a Cary Eclipse Fluorescence Spectrophotometer by excitation at 500 and 600 nm and detection in the wavelength range 530–600 and 635–700 nm, respectively.
Agarose gels were prepared at a gel percentage of 1% and run at a voltage of 70 V for ∼3 h at 4 °C in an 11 mM MgCl2 solution buffered with 0.5× TBE. The DNA structures were stained postelectrophoresis with GelRed dye (Biotium) for UV-transillumination. For reference, lanes with the p8064 scaffold and a 1 kb ladder containing linear fragments of different lengths were included in the gel (Supplementary Figure S1).
For AFM imaging, 5 μL of DNA origami sample was diluted to ∼2 nM and deposited on a freshly cleaved mica surface (Agar Scientific). After an incubation period of 90 s, the surface was rinsed with Milli-Q water (Merck Millipore) and dried with nitrogen. Imaging was performed in air using a MFP-3D AFM System (Asylum, Oxford Instruments) in tapping mode. The resulting images were flattened and analyzed using the software Gwyddion (see Supplementary Figure S2).
Optical Setup for Single-Molecule FRET Imaging
The optical part of the custom-built setup was an inverted fluorescence microscope with wide-field illumination and two-color alternating laser excitation (ALEX). For a detailed schematic and for filter information, refer to Supplementary Section 4. The excitation source consists of two diode-pumped solid-state (DPSS) lasers. The green laser is centered at 532 nm (Laser Quantum gem, power range: 50–500 mW). The red laser is centered at 638 nm (Coherent CUBE, 25 mW, maximum power 50 mW).
Images were recorded using an electron multiplying charge-coupled device (EMCCD) camera (Andor iXon3 860), operated with an EM gain of 250, temperature −60 °C and frame rate 20 fps. The EMCCD chip hosts a two-dimensional array of 128 × 128 light-sensitive pixels. The combination of objective and tube lens results in a lateral magnification of ∼100 ×, corresponding to ∼0.24 μm per pixel.
Two-Color Alternating Laser Excitation (ALEX)
The ALEX is controlled by a Multistream device (Cairn Research), which allows synchronization of digital cameras and excitation light sources. The EMCCD camera generates a pulsing output Transistor-Transistor Logic (TTL) signal. In frame transfer mode, the TTL signal switches periodically between 5 V (ON) during the exposure time and 0 V (OFF) during the latency period. This TTL signal provides the input signal to the Multistream with four possible output channels. The green and red lasers are connected to one output channel each. The first TTL signal is used to trigger the green laser. The next TTL signal, which corresponds to the subsequent image frame, is used to trigger the red laser, and so forth. This creates alternating laser excitation in precise synchronization with the frame rate of the EMCCD camera.
Preparation of Microfluidic Device with Quartz Nanocapillaries
Quartz nanocapillaries with an outer diameter of 0.5 mm and an inner diameter of 0.3 mm were pulled as described previously29,30,49 and embedded into a 16-channel microfluidic device. For the assembly of the microfluidic device, a polydimethyl-siloxane (PDMS) (Sylgard 184, Dow Corning) mixture was prepared (curing agent and silicon 1:10 weight ratio) and baked at 60 °C for 3 h in a 3D-printed master mold. In the PDMS chip, each of 16 channels links an outer reservoir (trans) to a large, shared reservoir in the center of the chip (cis). Before inserting the nanocapillaries, 1 mm holes were punched through each trans reservoir and at the front and rear of the large cis reservoir. The nanocapillaries were then incorporated such that their tips point toward the central chamber and form the only connection between the trans and cis reservoirs. The PDMS chip with the nanocapillaries was sealed with a thin microscopy glass coverslip, leaving the punched holes as the only access points to the reservoirs for injection of liquids and immersion of electrodes (Supplementary Figure S11).
Following treatment with plasma-cleaning (Femto-Diener Electronic) at maximum power (109 W) for 5 min, an electrolyte solution was injected into the assembled chip through the holes. All measurements were performed in 500 mM KCl, 5.5 mM MgCl2 buffered with 0.5× TBE (pH 8.6). Ionic current measurements were carried out using a pair of Ag/AgCl electrodes connected to a patch-clamp amplifier (HEKA Elektronik, Germany) in voltage clamp mode. The ground electrode was inserted into the cis reservoir and the reference electrode into a trans reservoir. The microfluidic chip was enclosed with a Faraday cage. Hosting multiple nanocapillaries in one chip offers the possibility to select the most suitable candidates and to utilize one chip for several repeats. Before starting an experiment, each nanocapillary was screened for a stable baseline current, low root-mean-square (RMS) current noise and a resistance in the range 24–45 MΩ, which was obtained by measuring an I–V-curve. On average, the nanocapillary tip is suspended 0.25 mm above the glass surface assuming a symmetrical taper shape and an outer diameter of 0.5 mm.
Once the nanocapillary tip of choice was in focus, 100 μL of sample solution was injected, containing ∼100 pM DNA origami structures in 500 mM KCl, 5.5 mM MgCl2 buffered with 0.5× TBE. Dithiothreitol (DTT, 2.5 mM) was added for reducing blinking of the fluorescent dyes.50 DTT was used instead of a more conventional combination of an enzymatic oxygen scavenging system and Trolox as the oxygen scavenging system contains large molecules that introduced undesired noise into the ionic current recordings.
To obtain the FRET values at 0 V, the structures were assembled with 4× biotin anchors attached to the 5′ end of staples attached to the plate (for staple positions see Supplementary Section 13). A surface layer of origami sensors could then be formed using a BSA-biotin-neutravidin coating. A chamber (Grace Bio-Laboratories FlexWell) on a microscopy glass coverslip was incubated overnight with a BSA-biotin solution (1 mg/mL in PBS). The chamber was washed with 1× PBS buffer before a solution of neutravidin (1 mg/mL in PBS) was added for 15 min. After rinsing with PBS, the DNA origami sample was added at pM concentration. The origami were left for ∼5 min until the adsorption density was sufficient, as monitored by the EMCCD camera and then washed with PBS to remove unbound sample. The chamber was filled with the KCl electrolyte buffer (500 mM KCl, 5.5 mM MgCl2 buffered with 0.5× TBE) prior to imaging.
Criteria for Fluorescence Trace Selection
Before initiating a measurement, we ensured that the ionic current was at the bare nanocapillary level. In most cases, trapping of a DNA origami structure occurred within a few seconds. Recording the fluorescence traces over a period of ∼50 s allowed us to observe bleaching of the dyes frequently while also obtaining a relatively large number of traces in each experiment. The relative laser excitation powers were tuned such that acceptor bleaching predominantly preceded donor bleaching without exceeding a limit that leads to a largely “dark” DNA origami population. This enabled us to maximize the number of traces with a characteristic FRET signature. Within the voltage limits of 100–400 mV, the DNA origami trapping was usually stable for the duration of the trace recording. The average time period used for calculating the proximity ratio Esm* was 15.06 ± 14.72 s (SD), and could in exceptional cases even last over a minute until the DNA origami plate was deliberately ejected. The minimum duration was around 1 s. After 50 s, the DNA origami structure, if still present, was expelled from the nanocapillary, resetting the conditions for the next trace recording. A fluorescence trace was used for further analysis such as calculation of the proximity ratio Esm* if there was a signal both in ID(D*) and in IA(A*) accompanied by a synchronous drop in ionic current. Fluorescence transients with multilevel events such as multiple bleaching steps were excluded for the calculation of the proximity ratio Esm*. We extensively characterized DNA origami trapping events both by monitoring electric current response29 and simultaneous fluorescence and ionic current measurements.51 Here, we follow our established procedures and find that a negligible proportion of trapping events involved multiple origami structures. These events are identified during data analysis and then excluded. A more detailed discussion of our approach can be found in Supplementary Section 15.
Ionic Current Measurements and Simultaneous Fluorescence Imaging
Custom-written LabVIEW (LabVIEW 8.6, National Instruments) programs were applied to control the lasers and record single-molecule fluorescent images and ionic currents at the same time. The ionic current was recorded using a patch-clamp amplifier (HEKA Elektronik, Germany) at a bandwidth of 100 kHz and with an internal Bessel filter set to 10 kHz. The signals were digitized with a NI-PCIe-6351 card (National Instruments, U.S.A.).
Data Analysis
The raw data acquired from the single-molecule fluorescence recordings were visualized and processed using iSMS (v.2.01), an open-source software package for wide-field, single-molecule FRET microscopy on immobilized molecules.52
Acknowledgments
E.H. acknowledges financial support from the Swiss National Science Foundation (SNF). S.O.’s Ph.D. is supported by the Deutsche Forschungsgesellschaft DFG (TI 329/10-1). P.T. is grateful for support by the DFG (TI 329/10-1), the nanosystems initiative Munich (NIM), and the center for integrated protein science Munich (CIPSM). E.H., C.F., and U.F.K. were supported by a grant from the EPSRC Graphted EP/K016636/1 and ERC Consolidator Grant DesignerPores (647144). A.A. and C.M. acknowledge support from the National Science Foundation under Grants DMR-1507985 and PHY-1430124, National Institutes of Health Grant P41-GM104601 and the supercomputer time provided through XSEDE Allocation Grant MCA05S028 and the Blue Waters petascale supercomputer system (UIUC).
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.nanolett.7b05354.
Agarose gel and AFM images. Model description. Experimental setup. Bulk fluorescence emission spectra. Voltage-dependent properties of the DNA origami plates. Simultaneous fluorescence and ionic current measurements. Response of dye intensities to change in voltage. FRET response of origami designs to electric field. Estimated force required to stretch the dsDNA leash. Scaffold-staple layout and staple strand sequences. Diagram of PDMS-based microfluidic chip. Analysis of single and multiple trapping events (PDF)
Movie showing structure of plate exhibiting steady-state fluctuations (MPG)
Movie showing origami plate seemingly deforming until it passed through the nanocapillary aperture (AVI)
Movie showing distance between the dyes as systematically larger in the simulations of design A1 carried out at higher biases (MPG)
Author Present Address
⊥ (L.H.) Chemical Engineering and Biotechnology, Cambridge University, Philippa Fawcett Drive, Cambridge CB3 0AS, United Kingdom.
Author Contributions
# E.A.H. and C.F. contributed equally.
The authors declare no competing financial interest.
Supplementary Material
References
- Das S.; Carnicer-Lombarte A.; Fawcett J. W.; Bora U. Bio-Inspired Nano Tools for Neuroscience. Prog. Neurobiol. 2016, 142, 1–22. 10.1016/j.pneurobio.2016.04.008. [DOI] [PubMed] [Google Scholar]
- Hasanzadeh M.; Shadjou N. Electrochemical Nanobiosensing in Whole Blood: Recent Advances. TrAC, Trends Anal. Chem. 2016, 80, 167–176. 10.1016/j.trac.2015.07.018. [DOI] [Google Scholar]
- Jain K. K. Applications of Nanobiotechnology in Clinical Diagnostics. Clin. Chem. 2007, 53, 2002–2009. 10.1373/clinchem.2007.090795. [DOI] [PubMed] [Google Scholar]
- Cetin A. E.; Coskun A. F.; Galarreta B. C.; Huang M.; Herman D.; Ozcan A.; Altug H. Handheld High-Throughput Plasmonic Biosensor Using Computational on-Chip Imaging. Light: Sci. Appl. 2014, 3, e122. 10.1038/lsa.2014.3. [DOI] [Google Scholar]
- Sheehan P. E.; Whitman L. J. Detection Limits for Nanoscale Biosensors. Nano Lett. 2005, 5, 803–807. 10.1021/nl050298x. [DOI] [PubMed] [Google Scholar]
- Loew L. M.Design and Use of Organic Voltage Sensitive Dyes. In Membrane Potential Imaging in the Nervous System: Methods and Applications; Canepari M., Zecevic D., Eds.; Springer: New York, 2011; pp 13–23. [Google Scholar]
- Tsien R. Y. New Calcium Indicators and Buffers with High Selectivity against Magnesium and Protons: Design, Synthesis, and Properties of Prototype Structures. Biochemistry 1980, 19, 2396–2404. 10.1021/bi00552a018. [DOI] [PubMed] [Google Scholar]
- Platt J. R. Electrochromism, a Possible Change of Color Producible in Dyes by an Electric Field. J. Chem. Phys. 1961, 34, 862–863. 10.1063/1.1731686. [DOI] [Google Scholar]
- Gonzalez J. E.; Tsien R. Y. Improved Indicators of Cell Membrane Potential That Use Fluorescence Resonance Energy Transfer. Chem. Biol. 1997, 4, 269–77. 10.1016/S1074-5521(97)90070-3. [DOI] [PubMed] [Google Scholar]
- Cacciatore T. W.; Brodfuehrer P. D.; Gonzalez J. E.; Jiang T.; Adams S. R.; Tsien R. Y.; Kristan W. B.; Kleinfeld D. Identification of Neural Circuits by Imaging Coherent Electrical Activity with FRET-Based Dyes. Neuron 1999, 23, 449–459. 10.1016/S0896-6273(00)80799-0. [DOI] [PubMed] [Google Scholar]
- Peterka D. S.; Takahashi H.; Yuste R. Imaging Voltage in Neurons. Neuron 2011, 69, 9–21. 10.1016/j.neuron.2010.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrenberg B.; Montana V.; Wei M. D.; Wuskell J. P.; Loew L. M. Membrane Potential Can Be Determined in Individual Cells from the Nernstian Distribution of Cationic Dyes. Biophys. J. 1988, 53, 785–794. 10.1016/S0006-3495(88)83158-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothemund P. W. K. Folding DNA to Create Nanoscale Shapes and Patterns. Nature 2006, 440, 297–302. 10.1038/nature04586. [DOI] [PubMed] [Google Scholar]
- Douglas S. M.; Dietz H.; Liedl T.; Högberg B.; Graf F.; Shih W. M. Self-Assembly of DNA into Nanoscale Three-Dimensional Shapes. Nature 2009, 459, 414–418. 10.1038/nature08016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castro C. E.; Kilchherr F.; Kim D.-N.; Shiao E. L.; Wauer T.; Wortmann P.; Bathe M.; Dietz H. A Primer to Scaffolded DNA Origami. Nat. Methods 2011, 8, 221–229. 10.1038/nmeth.1570. [DOI] [PubMed] [Google Scholar]
- Chen Y. J.; Groves B.; Muscat R. A.; Seelig G. DNA Nanotechnology from the Test Tube to the Cell.. Nat. Nanotechnol. 2015, 10, 748–760. 10.1038/nnano.2015.195. [DOI] [PubMed] [Google Scholar]
- Pinheiro A. V.; Han D.; Shih W. M.; Yan H. Challenges and Opportunities for Structural DNA Nanotechnology. Nat. Nanotechnol. 2011, 6, 763–772. 10.1038/nnano.2011.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroener F.; Heerwig A.; Kaiser W.; Mertig M.; Rant U. Electrical Actuation of a DNA Origami Nanolever on an Electrode. J. Am. Chem. Soc. 2017, 139, 16510–16513. 10.1021/jacs.7b10862. [DOI] [PubMed] [Google Scholar]
- Stein I. H.; Schüller V.; Böhm P.; Tinnefeld P.; Liedl T. Single-Molecule FRET Ruler Based on Rigid DNA Origami Blocks. ChemPhysChem 2011, 12, 689–695. 10.1002/cphc.201000781. [DOI] [PubMed] [Google Scholar]
- Göpfrich K.; Li C.-Y.; Ricci M.; Bhamidimarri S. P.; Yoo J.; Gyenes B.; Ohmann A.; Winterhalter M.; Aksimentiev A.; Keyser U. F. Large-Conductance Transmembrane Porin Made from DNA Origami. ACS Nano 2016, 10, 8207–8214. 10.1021/acsnano.6b03759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell N. A. W.; Keyser U. F. Digitally Encoded DNA Nanostructures for Multiplexed, Single-Molecule Protein Sensing with Nanopores. Nat. Nanotechnol. 2016, 11, 645–651. 10.1038/nnano.2016.50. [DOI] [PubMed] [Google Scholar]
- Shen W.; Bruist M. F.; Goodman S. D.; Seeman N. C. A Protein-Driven DNA Device That Measures the Excess Binding Energy of Proteins That Distort DNA. Angew. Chem., Int. Ed. 2004, 43, 4750–4752. 10.1002/anie.200460302. [DOI] [PubMed] [Google Scholar]
- Shroff H.; Reinhard B. M.; Siu M.; Agarwal H.; Spakowitz A.; Liphardt J. Biocompatible Force Sensor with Optical Readout and Dimensions of 6 Nm 3. Nano Lett. 2005, 5, 1509–1514. 10.1021/nl050875h. [DOI] [PubMed] [Google Scholar]
- Tarsa P. B.; Brau R. R.; Barch M.; Ferrer J. M.; Freyzon Y.; Matsudaira P.; Lang M. J. Detecting Force-Induced Molecular Transitions with Fluorescence Resonant Energy Transfer. Angew. Chem., Int. Ed. 2007, 46, 1999–2001. 10.1002/anie.200604546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nickels P. C.; Wunsch B.; Holzmeister P.; Bae W.; Kneer L. M.; Grohmann D.; Tinnefeld P.; Liedl T. Molecular Force Spectroscopy with a DNA Origami-Based Nanoscopic Force Clamp. Science (Washington, DC, U. S.) 2016, 354, 305–307. 10.1126/science.aah5974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemmig E. A.; Creatore C.; Wünsch B.; Hecker L.; Mair P.; Parker M. A.; Emmott S.; Tinnefeld P.; Keyser U. F.; Chin A. W. Programming Light-Harvesting Efficiency Using DNA Origami. Nano Lett. 2016, 16, 2369–2374. 10.1021/acs.nanolett.5b05139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmied J. J.; Raab M.; Forthmann C.; Pibiri E.; Wünsch B.; Dammeyer T.; Tinnefeld P. DNA Origami–based Standards for Quantitative Fluorescence Microscopy. Nat. Protoc. 2014, 9, 1367–1391. 10.1038/nprot.2014.079. [DOI] [PubMed] [Google Scholar]
- Hernández-Ainsa S.; Misiunas K.; Thacker V. V.; Hemmig E. A.; Keyser U. F. Voltage-Dependent Properties of DNA Origami Nanopores. Nano Lett. 2014, 14, 1270–1274. 10.1021/nl404183t. [DOI] [PubMed] [Google Scholar]
- Li C.-Y.; Hemmig E. A.; Kong J.; Yoo J.; Hernández-Ainsa S.; Keyser U. F.; Aksimentiev A. Ionic Conductivity, Structural Deformation, and Programmable Anisotropy of DNA Origami in Electric Field. ACS Nano 2015, 9, 1420–1433. 10.1021/nn505825z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keyser U. F.; Koeleman B. N.; van Dorp S.; Krapf D.; Smeets R. M. M.; Lemay S. G.; Dekker N. H.; Dekker C. Direct Force Measurements on DNA in a Solid-State Nanopore. Nat. Phys. 2006, 2, 473–477. 10.1038/nphys344. [DOI] [Google Scholar]
- Roy R.; Hohng S.; Ha T. A Practical Guide to Single-Molecule FRET. Nat. Methods 2008, 5, 507–516. 10.1038/nmeth.1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gust A.; Zander A.; Gietl A.; Holzmeister P.; Schulz S.; Lalkens B.; Tinnefeld P.; Grohmann D. A Starting Point for Fluorescence-Based Single-Molecule Measurements in Biomolecular Research. Molecules 2014, 19, 15824–15865. 10.3390/molecules191015824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller B. K.; Reuter A.; Simmel F. C.; Lamb D. C. Single-Pair FRET Characterization of DNA Tweezers. Nano Lett. 2006, 6, 2814–2820. 10.1021/nl0619406. [DOI] [PubMed] [Google Scholar]
- List J.; Weber M.; Simmel F. C. Hydrophobic Actuation of a DNA Origami Bilayer Structure. Angew. Chem., Int. Ed. 2014, 53, 4236–4239. 10.1002/anie.201310259. [DOI] [PubMed] [Google Scholar]
- Knotts T. A.; Rathore N.; Schwartz D. C.; de Pablo J. J. A Coarse Grain Model for DNA. J. Chem. Phys. 2007, 126, 084901. 10.1063/1.2431804. [DOI] [PubMed] [Google Scholar]
- Yoo J.; Aksimentiev A. The Structure and Intermolecular Forces of DNA Condensates. Nucleic Acids Res. 2016, 44, 2036–2046. 10.1093/nar/gkw081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee N. K.; Kapanidis A. N.; Wang Y.; Michalet X.; Mukhopadhyay J.; Ebright R. H.; Weiss S. Accurate FRET Measurements within Single Diffusing Biomolecules Using Alternating-Laser Excitation. Biophys. J. 2005, 88, 2939–2953. 10.1529/biophysj.104.054114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapanidis A. N.; Laurence T. A.; Lee N. K.; Margeat E.; Kong X.; Weiss S. Alternating-Laser Excitation of Single Molecules. Acc. Chem. Res. 2005, 38, 523–533. 10.1021/ar0401348. [DOI] [PubMed] [Google Scholar]
- Kapanidis A. N.; Lee N. K.; Laurence T. A.; Doose S.; Margeat E.; Weiss S. Fluorescence-Aided Molecule Sorting: Analysis of Structure and Interactions by Alternating-Laser Excitation of Single Molecules. Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 8936–8941. 10.1073/pnas.0401690101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ATTO Gmbh. Fluorescent Labels and Dyes. Fluoresc. Labels Dye. Catal. 2013, 49, 1–45. [Google Scholar]
- Keyser U. F. Controlling Molecular Transport through Nanopores. J. R. Soc., Interface 2011, 8, 1369–1378. 10.1098/rsif.2011.0222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laohakunakorn N.; Ghosal S.; Otto O.; Misiunas K.; Keyser U. F. DNA Interactions in Crowded Nanopores. Nano Lett. 2013, 13, 2798–2802. 10.1021/nl401050m. [DOI] [PubMed] [Google Scholar]
- Smith S. B.; Cui Y.; Bustamante C. Overstretching B-DNA : The Elastic Response of Individual Double-Stranded and Single- Stranded DNA Molecules. Science (Washington, DC, U. S.) 1996, 271, 795–799. 10.1126/science.271.5250.795. [DOI] [PubMed] [Google Scholar]
- Lin M. Z.; Schnitzer M. J. Genetically Encoded Indicators of Neuronal Activity. Nat. Neurosci. 2016, 19, 1142–1153. 10.1038/nn.4359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comer J.; Aksimentiev A. Predicting the DNA Sequence Dependence of Nanopore Ion Current Using Atomic-Resolution Brownian Dynamics. J. Phys. Chem. C 2012, 116, 3376–3393. 10.1021/jp210641j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douglas S. M.; Marblestone A. H.; Teerapittayanon S.; Vazquez A.; Church G. M.; Shih W. M. Rapid Prototyping of 3D DNA-Origami Shapes with caDNAno. Nucleic Acids Res. 2009, 37, 5001–5006. 10.1093/nar/gkp436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D. N.; Kilchherr F.; Dietz H.; Bathe M. Quantitative Prediction of 3D Solution Shape and Flexibility of Nucleic Acid Nanostructures. Nucleic Acids Res. 2012, 40, 2862–2868. 10.1093/nar/gkr1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Göpfrich K.; Kulkarni C. V.; Pambos O. J.; Keyser U. F. Lipid Nanobilayers to Host Biological Nanopores for DNA Translocations. Langmuir 2013, 29, 355–364. 10.1021/la3041506. [DOI] [PubMed] [Google Scholar]
- Schmied J. J.; Raab M.; Forthmann C.; Pibiri E.; Wünsch B.; Dammeyer T.; Tinnefeld P. DNA Origami-Based Standards for Quantitative Fluorescence Microscopy. Nat. Protoc. 2014, 9, 1367–1391. 10.1038/nprot.2014.079. [DOI] [PubMed] [Google Scholar]
- Hernández-Ainsa S.; Bell N. A. W.; Thacker V. V.; Göpfrich K.; Misiunas K.; Fuentes-Perez M. E.; Moreno-Herrero F.; Keyser U. F. DNA Origami Nanopores for Controlling DNA Translocation. ACS Nano 2013, 7, 6024–6030. 10.1021/nn401759r. [DOI] [PubMed] [Google Scholar]
- Preus S.; Noer S. L.; Hildebrandt L. L.; Gudnason D.; Birkedal V. iSMS: Single-Molecule FRET Microscopy Software. Nat. Methods 2015, 12, 593–594. 10.1038/nmeth.3435. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.