

Abstract
PIEZO1 is a large mechanosensitive ion channel protein. Diseases associated with PIEZO1 include autosomal recessive generalised lymphatic dysplasia of Fotiou (GLDF) and autosomal dominant dehydrated hereditary stomatocytosis with or without pseudohyperkalemia and/or perinatal oedema (DHS). The two disorders show overlapping features, fetal hydrops/perinatal oedema have been reported in both. Electrophysiological studies suggest opposite mechanisms of action: the mutations identified in GLDF patients cause a loss‐of‐function mechanism of disease and mutations in DHS patients cause gain of function. This raises the question: Is the pathogenic disease mechanism behind the fetal oedema the same in the two phenotypes? In this Symposium Review, we will discuss the two conditions and highlight key questions that remain to be answered. For instance, the perinatal oedema often resolves soon after birth and we are still at a loss to understand why. Are there any mechanisms which could compensate for the faulty PIEZO1 in these patients? Are there physiological changes at birth that are less reliant on the function of PIEZO1? Thus, there is a clear need for further studies into the two disorders, in order to fully understand the role of PIEZO1 in health and disease.
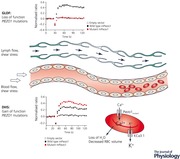
Keywords: PIEZO1, dehydrated hereditary stomatocytosis, generalised lymphatic dysplasia of fotiou, fetal hydrops
Introduction
Piezos are large mechanosensitive ion channel proteins. PIEZO1 is mainly expressed in non‐sensory tissues while PIEZO2 is predominantly found in sensory tissues (Wu et al. 2017). Many different roles are being assigned to PIEZO1, e.g. contributing to the regulation of urinary osmolarity (Martins et al. 2016), controlling blood pressure (Wang et al. 2016), or acting as a sensor of epithelial cell crowding and stretching (Gudipaty et al. 2017). PIEZO1 is also expressed in developing blood vessels and plays a key role in blood vessel formation (Li et al. 2014; Ranade et al. 2014). The Online Mendelian Inheritance of Man database (OMIM) lists two phenotypes associated with variants in the PIEZO1 gene. One is the autosomal dominant disorder, dehydrated hereditary stomatocytosis with or without pseudohyperkalemia and/or perinatal oedema (DHS; OMIM 194380) caused by gain‐of‐function mutations. The other is autosomal recessive generalised lymphatic dysplasia of Fotiou, with non‐immune fetal hydrops (GLDF; OMIM 616843) caused by biallelic, loss‐of‐function mutations. There are examples of allelic disorders in the literature where defects in a single gene are implicated in different phenotypes (Zhu et al. 2014). For example, biallelic loss‐of‐function mutations in SCN9A cause autosomal recessive congenital insensitivity to pain (Cox et al. 2006), whereas gain‐of‐function, heterozygous mutations in the same gene cause autosomal dominant primary erythromelalgia, where there is burning pain of the lower limbs (Cummins et al. 2004; Yang et al. 2004). However, the paradox here is that both GLDF and DHS have reported cases of perinatal oedema, so it appears that both loss‐of‐function and gain‐of‐function mutations in PIEZO1 can produce similar phenotypic features. In this Symposium Review, the two conditions caused by PIEZO1 mutations are presented, and overlapping features between the two phenotypes are discussed. We highlight key questions that remain to be answered to improve our current understanding of the role of PIEZO1 in health and disease.
DHS: dehydrated hereditary stomatocytosis – a gain‐of‐function phenotype
Dehydrated hereditary stomatocytosis with or without pseudohyperkalemia and/or perinatal oedema (DHS; OMIM 194380) is an autosomal dominant haemolytic anaemia. The condition is characterized by mild to moderate haemolysis with varying numbers of stomatocytes on peripheral blood smears. There can also be an elevated MCHC (mean corpuscular haemoglobin concentration) and decreased osmotic fragility. Sometimes high MCV (mean corpuscular volume) and reticulocyte counts are reported. Probably the most sensitive test for DHS is osmotic gradient ektacytometry, which measures red blood cell (RBC) membrane deformability as a function of osmolality. The ektacytometry profile of patients with DHS is shifted to the left indicating RBC dehydration (Fig. 1). Another indicator of DHS is stomacytic RBCs, where cells can have the appearance of ‘coffee beans’ (Fig. 2). Other frequent clinical findings in DHS patients associated with the haemolytic anaemia are splenomegaly (resulting from increased red cell trapping in the spleen), cholelithiasis (due to elevated bilirubin levels), infantile hepatitis, jaundice and iron overload, which can lead to hepatosiderosis or more widely distributed haemosiderosis. Some patients are asymptomatic, others have transient anaemia, which can go undetected for years. In patients with mild disease, stomatocytes are scarce on blood film, and often DHS patients only come to medical attention because of the unusual presentation of hepatosiderosis (Syfuss et al. 2006).
Figure 1. Ektacytometry curves in DHS.
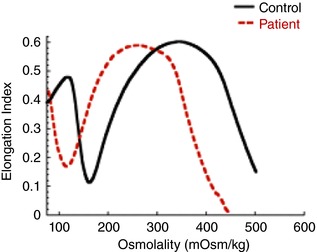
This idealised graph shows RBC membrane deformability (using the elongation index) as a function of osmolality. The ektacytometry osmotic deformability profile of a DHS patient (dashed red line) is shifted to the left of the healthy control (continuous black line) indicating high osmotic resistance and RBC dehydration.
Figure 2. Contrasting DHS and GLDF.
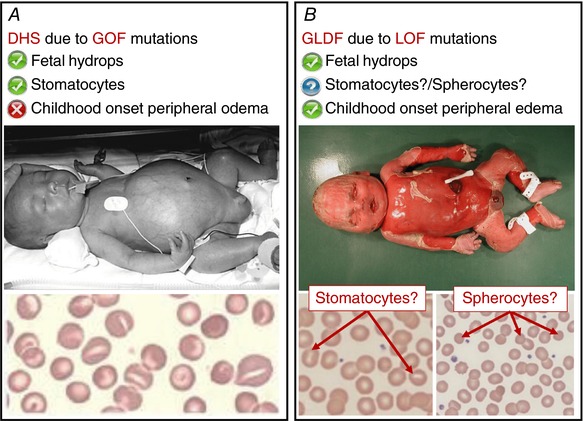
A, patients with DHS can present with fetal hydrops. The blood film of a DHS patient is characterised by mouth‐shaped (stomatocytic) red blood cells. The red blood cell dehydration seen in DHS patients is due to altered permeability of the RBC membrane which causes a loss of cation content due to the gain‐of‐function (GOF) mutations in PIEZO1. B, patients with GLDF can also present with fetal hydrops. In contrast to DHS, the GLDF patients often develop childhood onset peripheral oedema, indicating that in GLDF the hydrops could be due to an underlying lymphatic problem. Is the fetal hydrops due to the same underlying disease mechanism in the two conditions? Do all GLDF patients present with stomatocytes or spherocytes on their blood films? Not enough cases have been investigated to make a firm conclusion. Will the loss‐of‐function (LOF) mutations in GLDF patients lead to overhydrated red blood cells? Many questions remain unanswered, thus there is a clear need for more studies into the functional role of PIEZO1. Figure 2 A upper panel reproduced from Archives of Disease in Childhood: Fetal & Neonatal, Dehydrated hereditary stomatocytosis with transient perinatal ascites, A P Basu, P Carey, T Cynober, M Chetty, J Delaunay, G W Stewart, S Richmond, 88(5), F438‐9, Copyright 2003 with permission from BMJ Publishing Group Ltd. Figure 2 A lower panel reprinted from Blood Cells, Molecules, and Diseases, 56(1), Lydie Da Costa, Ludovic Suner, Julie Galimand, Amandine Bonnel, Tiffany Pascreau, Nathalie Couque, Odile Fenneteau, Narla Mohandas, Diagnostic tool for red blood cell membrane disorders: Assessment of a new generation ektacytometer, 9‐22, Copyright 2015 with permission from Elsevier.
Occasionally, pseudohyperkalemia is also observed in the blood samples of DHS patients. This is another indicator of defective cell membrane properties. It can also be a transient finding, as shown in a review of routine plasma potassium measurements over a 7‐year period in one patient, where values fluctuated between high‐normal and elevated (Syfuss et al. 2006).
Some individuals with DHS also present with perinatal oedema which can vary from ascites to severe hydrops, including pericardial or pleural effusions and/or subcutaneous oedema (including hands and feet). The perinatal oedema can be so severe that it is lethal, and in utero death due to generalised oedema has been reported. Some are stillborn or die soon after birth. The pregnancies are frequently complicated by polyhydramnios. If the baby survives the pregnancy, ventilation is often required at birth. The hydrops sometimes resolves in utero, at other times it resolves spontaneously within weeks or months after birth and never recurs. Some individuals present with chylothoraces upon the commencement of feeding. Entezami and colleagues treated a case of intra‐uterine anaemia but they were not able to cure the oedema, suggesting that the anaemia was not the cause of the oedema (Entezami et al. 1996). It has been suggested that the ascites can be of hepatic origin (Rees et al. 2004). A persistent cystic hygroma, reported in one DHS case, suggests the oedema could be lymphatic in origin (Ami et al. 2009). The mechanism by which perinatal oedema and ascites develop, and then spontaneously resolve in some fetal DHS cases, has not yet been elucidated. No consistent dysmorphic features are associated with DHS.
The age of onset varies widely. A family history is not essential for the diagnosis of DHS, but should there be one, there can be great inter‐familial and intra‐familial variability in expression. Because of the highly variable expression, DHS can be very difficult to diagnose, ranging from the absence of clinical symptoms to lethal perinatal oedema. For example, one report on intra‐familial variation described how one member only had haematological symptoms, while her cousin had perinatal oedema but no signs of haemolytic anaemia (Grootenboer et al. 2000). Several families have not been aware of their diagnosis until the occurrence of fetal oedema (Grootenboer et al. 2001).
The condition was first reported in 1971, where Miller and colleagues described a large family with over 50 affected members (Miller et al. 1971). Later, heterozygous missense mutations, segregating with the disease in DHS families (including the family described by Miller et al.), were identified in the PIEZO1 gene (Zarychanski et al. 2012). This finding was corroborated by additional studies of familial and sporadic cases (Albuisson et al. 2013; Andolfo et al. 2013). Interestingly, upon sequencing of the gene in the family described by Miller et al., three of the affected siblings were found to be homozygous for the variant, whereas the rest of the affected family members were heterozygous (Zarychanski et al. 2012). It was reported that the homozygotes had severe haemolytic anaemia with reticulocytosis, stomatocytosis and hyperbilirubinaemia, while the heterozygotes showed only cholelithiasis and intermittent jaundice. A number of cis‐missense variants have been identified, but which of those contribute to the DHS phenotype is unclear (Andolfo et al. 2013).
Human PIEZO1 is a highly polymorphic gene with many coding variants reported in the 1000 Genomes Project database, including variants predicted as deleterious. A recent case report suggests that the c.7505A>G variant (dbSNP: rs34830861) can cause a DHS phenotype (Del Orbe Barreto et al. 2016). Although the patient showed an abnormal curve shifted to the left on ektacytometry, and other blood findings supported the diagnosis of DHS, the rs34830861 SNP has been reported in 97 heterozygous cases in ExAC. Therefore, work to determine whether this mutation alters PIEZO1 channel activity is warranted to confirm that it is pathogenic. Until then, this finding should be interpreted with caution. Electrophysiological studies have been carried out for several DHS‐associated PIEZO1 mutations and whole‐cell patch clamp demonstrated that the tested mutations could be defined as gain of function (Albuisson et al. 2013; Bae et al. 2013). The experiments were done in transfected HEK293 cells, and showed that mutations in PIEZO1 slowed the channel inactivation rate. This could explain the red blood cell (RBC) dehydration seen in PIEZO1 mutation‐positive DHS patients. Mechanistically, the slower inactivation of mutant channels causes an excessive calcium (Ca2+) influx into the RBC, which promotes potassium (K+) efflux through KCa3.1 channels accompanied by osmotic efflux of water, with subsequent cell dehydration (Cahalan et al. 2015). A recent paper has shown that the RBC phenotype seen in DHS patients is not just related to delayed channel inactivation; a range of pathophysiological mechanisms are also contributing to the altered channel kinetics (Glogowska et al. 2017).
GLDF: generalised lymphatic dysplasia of Fotiou – a loss‐of‐function phenotype
Autosomal recessive generalised lymphatic dysplasia of Fotiou (GLDF; OMIM 616843) is characterised by a widespread lymphoedema that can affect all segments of the body. Like DHS, GLDF has a high incidence of non‐immune fetal hydrops, i.e. presenting with persistent bilateral pleural effusions, ascites and subcutaneous oedema, with either fetal death or a complete resolution of the hydrops postnatally. The pregnancies are frequently complicated by polyhydramnios. The babies are often hydropic at birth with generalised oedema and pleural effusions requiring ventilation for several weeks. After introduction of milk feeds, the pleural effusions become chylous.
Sometimes lymphoedema of the peripheries recurs in early childhood. This is mainly lower limb lymphoedema, but can also involve the hands, arms and face (which may be intermittent). Genital oedema, which may be intermittent, is frequent in males, sometimes with thickened scrotum, oedema of the foreskin, and hydroceles. Many patients have multiple episodes of cellulitis in their legs, and several of the patients have also had intermittent, severe facial swelling due to recurrent facial cellulitis. The facial cellulitis is a striking feature, and is rarely seen in other forms of primary lymphoedema. Four cases with severe, recurrent facial cellulitis and significant morbidity have been reported (Fotiou et al. 2015). There was high pyrexia and respiratory distress that led to admission to intensive care with ventilation. In addition to facial swelling, some of the patients had mild dysmorphic features (cupped simple ears, mild telecanthus due to epicanthic folds, and micrognathia). Some patients were also reported to have varicose veins, papillomatosis, verrucas and warts. Many of the affected children had systemic involvement with chylothoraces and/or pericardial effusions – but intestinal lymphangiectasia is rare (S. Mansour, personal observation).
It was noted that GLDF patients and unaffected carriers were also found to have mild changes (occasional stomatocytes and/or spherocytes) on their blood film, but with no history of haemolytic anaemia. It is questionable whether observations on the blood film are comparable to that of the DHS patients, as more work is needed in this area.
Through whole exome sequencing, Fotiou and colleagues identified biallelic, homozygous and compound heterozygous mutations in the PIEZO1 gene (Fotiou et al. 2015). The mutations segregated with the phenotype in the families. An independent study by Lukacs and colleagues confirmed this finding (Lukacs et al. 2015). A mix of missense, stop‐gain (nonsense), frameshift and splice site variants have been identified. Sanger sequencing of cDNA from carriers of a splice site mutation showed that intron 13 was not spliced out in the mutant protein. None of the variants, except for 2 found in cis, were found in the dbSNP, or 1000 Genomes Project databases or in 900 in‐house control samples (Fotiou et al. 2015).
Western blot analysis of RBCs from GLDF patients showed a complete absence or reduction of PIEZO1 protein expression compared to a healthy control (Fotiou et al. 2015). Analysis of RBCs from a patient with biallelic mutations in PIEZO1 showed decreased PIEZO1 function (Lukacs et al. 2015). This was mainly due to a reduced abundance of PIEZO1 channels in the RBC plasma membrane. The absence of symptoms in the parents carrying just one variant each argues against a dominant negative effect of the variants. Where the DHS‐associated PIEZO1 mutations have been shown to lead to dehydration of RBCs due to excessive calcium influx, the GLDF associated PIEZO1 mutations give rise to decreased PIEZO1 function with no calcium influx. As a consequence, reduced influx of calcium may cause fluid retention in the RBCs leading to ‘hyper‐hydration’ (Cahalan et al. 2015).
Piezo1 is expressed in embryonic endothelial cells and Piezo1 deficiency causes the disruption of vascular development in the mouse embryo. Loss of PIEZO1 results in reduced mechanosensitivity of the endothelial cells in terms of alignment and organization in the direction of flow in response to shear stress. Mechanistically, Piezo1 channel activity is stimulated by shear stress leading to calcium (Ca2+) entry in the endothelial cell, increased calpain activity, and modification of the actin cytoskeleton and the focal adhesions required for cell reorganization (Li et al. 2014; Ranade et al. 2014). Mechanotransduction is also a critical regulator of lymphatic vascular development, and shear stress triggers signalling pathways that promote lymphatic vessel maturation and lymphatic valve formation (Sabine et al. 2012; Cha et al. 2016). The loss of PIEZO1 function in GLDF patients, and consequent defects in mechanical stimulation and downstream signalling pathways, could disrupt embryonic lymphatic vascular development. This could result in the progression of hydrops or childhood onset peripheral oedema.
Discussion
In summary, we have presented two conditions caused by mutations in the PIEZO1 gene, autosomal recessive GLDF caused by loss of function, and autosomal dominant DHS where the disease is a gain of function. In disorders with phenotypic heterogeneity, where different mutations in the same gene give rise to different phenotypes, mutations usually have distinct effects on biochemical and cellular activities. Furthermore, disease specific mutations tend to cluster in key regions. For example, missense mutations in Dynamin2 (DNM2) have been described in association with four different phenotypes, depending on which domain of the DNM2 gene the mutation is located in (Brown et al. 2017). PIEZO1 missense variants are common in both GLDF and DHS, but a one domain–one disease pattern is not clear. The missense variants show a wide distribution across the gene, and the GLDF missense variants are interspersed among the DHS missense variants (Fotiou et al. 2015). Therefore, the clinical phenotype may depend on the nature of the conformational change produced by a disease specific mutation at the individual genomic position in PIEZO1. Interestingly, centronuclear myopathy (CNM), one of the DNM2‐associated phenotypes, is usually caused by mutations in the PH domain, but a few missense mutations have been identified interspersed with Charcot‐Marie‐Tooth (CMT) neuropathy‐associated missense mutations in the middle domain of the DNM2 gene. Chin and colleagues demonstrated that the mutations had different effects on protein function and that CMT was caused by loss‐of‐function mechanisms and CNM was caused by gain‐of‐function mechanisms (Chin et al. 2015).
The mutations in PIEZO1 have also been shown to have distinct effects. The gain‐of‐function mutations were reported to increase calcium influx into RBCs leading to their dehydration. The loss‐of‐function mutations were reported to yield no calcium influx, which may lead to the over‐hydration of RBCs. According to the literature, ektacytometry is the most effective method for diagnosing DHS, rather than direct observation of blood films alone. None of the patients of Fotiou et al. underwent ektacytometry, and it would be interesting to study this further, to see if the ektacytomery curves in the GLDF patients show a shift to the right, in contrast to the shift to the left observed in DHS patients. Could ektacytometry consequently be an equally suitable method for the diagnosis of GLDF?
Although caused by different disease mechanisms, the two disorders at first sight show some overlapping features. For example, fetal hydrops has been reported for both disorders (Fig. 2). Fetal hydrops is not the cause of disease, but rather an outcome of an underlying developmental problem (Bellini et al. 2015). Therefore, we need to understand better the fetal hydrops in both DHS and GLDF cases, to determine whether they have the same cause. The GLDF hydrops seems most likely to be lymphatic in origin, but is this also true in DHS? Or could it be hepatic in origin as previously suggested (Rees et al. 2004)? It is also unclear why the hydrops resolves at birth. Are there changes that occur then which could compensate for the defective function of PIEZO1?
Interestingly, in some GLDF patients, the fetal hydrops resolved soon after birth, but persistent peripheral oedema appeared later in childhood. This does not seem to be the case in DHS patients; at least this has not been reported. If, as suggested above, the hydrops is due to different disease mechanisms, the underlying fault is then not necessarily producing the same effect later in life. This could explain the development of peripheral oedema in one disorder and not the other. It is not clear whether the DHS patients have been examined for peripheral oedema later in life. Particularly those who presented with hydrops (if they survived). Perhaps the fetuses with perinatal oedema associated with the DHS phenotype, have a second, but as yet unidentified, hit or variant in PIEZO1 or a modifying gene.
The fact that the fetal hydrops in both conditions are able to fully resolve in the few weeks after birth suggests a physiological change at birth that is having a major effect on the oedema. If this could be understood, the potential for treatment of this, and other lymphatic related hydrops, is tangible. There is clearly a need for more work to be carried out, in order to fully understand the role of PIEZO1 in the two disorders presented here.
Additional information
Competing interests
The authors have no competing interests.
Author contribution
All authors contributed to the writing of this manuscript and the final version has been approved by all authors.
Funding
Our original work on autosomal recessive PIEZO1 mutations was supported by the British Heart Foundation and Newlife Foundation for Disabled Children.
Acknowledgements
Thanks to Melanie J. Ludlow for providing graphs for the abstract illustration.
Biographies
Silvia Martin‐Almedina is a postdoctoral research assistant at St George's, University of London. After being awarded a PhD in biochemistry and molecular biology from the University of Oviedo (Spain) she moved to Imperial College London where she became interested in vascular biology. Since 2014 she has been working at SGUL on the in‐vitro functional characterisation of primary lymphoedema genes.

Sahar Mansour is a consultant in clinical genetics, whose specialist interests include primary lymphoedema, dysmorphology, skeletal dysplasias and prenatal diagnosis. Professor Mansour is also an honorary professor in clinical genetics at St George's, University of London.
Pia Ostergaard is a reader in human genetics at St George's, University of London. Her research focus mainly lies within lymphovascular medicine and she works in close collaboration with the Lymphovascular Clinical Service at St George's Hospital. Her group has been researching the genetic causes of lymphoedema and has recently identified eight genes (including PIEZO1) that cause various types of primary lymphoedema.
This review was presented at the symposium ‘Piezo channel mechanisms in health and disease’, which took place at IUPS 38th World Congress, Rio de Janeiro, Brazil, 1–5 August 2017.
Edited by: Ole Petersen & David Beech
References
- Albuisson J, Murthy SE, Bandell M, Coste B, Louis‐dit‐Picard H, Mathur J, Feneant‐Thibault M, Tertian G, de Jaureguiberry J‐P, Syfuss P‐Y, Cahalan S, Garcon L, Toutain F, Rohrlich PS, Delaunay J, Picard V, Jeunemaitre X & Patapoutian A (2013). Dehydrated hereditary stomatocytosis linked to gain‐of‐function mutations in mechanically activated PIEZO1 ion channels. Nat Commun 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ami O, Picone O, Garcon L, Castel C, Guitton C, Delaunay J, Frydman R & Senat MV (2009). First‐trimester nuchal abnormalities secondary to dehydrated hereditary stomatocytosis. Prenat Diagn 29, 1071–1074. [DOI] [PubMed] [Google Scholar]
- Andolfo I, Alper SL, De Franceschi L, Auriemma C , Russo R, De Falco L, Vallefuoco F, Esposito MR, Vandorpe DH, Shmukler BE, Narayan R, Montanaro D, D'Armiento M, Vetro A, Limongelli I, Zuffardi O, Glader BE, Schrier SL, Brugnara C, Stewart GW, Delaunay J & Iolascon A (2013). Multiple clinical forms of dehydrated hereditary stomatocytosis arise from mutations in PIEZO1. Blood 121, 3925–3935. [DOI] [PubMed] [Google Scholar]
- Bae C, Gnanasambandam R, Nicolai C, Sachs F & Gottlieb PA (2013). Xerocytosis is caused by mutations that alter the kinetics of the mechanosensitive channel PIEZO1. Proc Natl Acad Sci USA 110, E1162–E1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellini C, Donarini G, Paladini D, Calevo MG, Bellini T, Ramenghi LA & Hennekam RC (2015). Etiology of non‐immune hydrops fetalis: an update. Am J Med Genet Part A 167A, 1082–1088. [DOI] [PubMed] [Google Scholar]
- Brown FC, Collett M, Tremblay CS, Rank G, De Camilli P, Booth CJ, Bitoun M, Robinson PJ, Kile BT, Jane SM & Curtis DJ (2017). Loss of Dynamin 2 GTPase function results in microcytic anaemia. Br J Haematol 178, 616–628. [DOI] [PubMed] [Google Scholar]
- Cahalan SM, Lukacs V, Ranade SS, Chien S, Bandell M & Patapoutian A (2015). Piezo1 links mechanical forces to red blood cell volume. Elife 4, e07370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cha B, Geng X, Mahamud MR, Fu J, Mukherjee A, Kim Y, Jho EH, Kim TH, Kahn ML, Xia L, Dixon JB, Chen H & Srinivasan RS (2016). Mechanotransduction activates canonical Wnt/beta‐catenin signaling to promote lymphatic vascular patterning and the development of lymphatic and lymphovenous valves. Genes Dev 30, 1454–1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin YH, Lee A, Kan HW, Laiman J, Chuang MC, Hsieh ST & Liu YW (2015). Dynamin‐2 mutations associated with centronuclear myopathy are hypermorphic and lead to T‐tubule fragmentation. Hum Mol Genet 24, 5542–5554. [DOI] [PubMed] [Google Scholar]
- Cox JJ, Reimann F, Nicholas AK, Thornton G, Roberts E, Springell K, Karbani G, Jafri H, Mannan J, Raashid Y, Al‐Gazali L, Hamamy H, Valente EM, Gorman S, Williams R, McHale DP, Wood JN, Gribble FM & Woods CG (2006). An SCN9A channelopathy causes congenital inability to experience pain. Nature 444, 894–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cummins TR, Dib‐Hajj SD & Waxman SG (2004). Electrophysiological properties of mutant Nav1.7 sodium channels in a painful inherited neuropathy. J Neurosci 24, 8232–8236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Orbe Barreto R, Arrizabalaga B, De la Hoz Rastrollo AB, Garcia‐Orad A, Gonzalez Vallejo I, Bento C, Villegas A & Garcia‐Ruiz JC (2016). Hereditary xerocytosis, a misleading anemia. Ann Hematol 95, 1545–1546. [DOI] [PubMed] [Google Scholar]
- Entezami M, Becker R, Menssen HD, Marcinkowski M & Versmold HT (1996). Xerocytosis with concomitant intrauterine ascites: First description and therapeutic approach. Blood 87, 5392–5393. [PubMed] [Google Scholar]
- Fotiou E, Martin‐Almedina S, Simpson MA, Lin S, Gordon K, Brice G, Atton G, Jeffery I, Rees DC, Mignot C, Vogt J, Homfray T, Snyder MP, Rockson SG, Jeffery S, Mortimer PS, Mansour S & Ostergaard P (2015). Novel mutations in PIEZO1 cause an autosomal recessive generalized lymphatic dysplasia with non‐immune hydrops fetalis. Nat Commun 6, 8085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glogowska E, Schneider ER, Maksimova Y, Schulz VP, Lezon‐Geyda K, Wu J, Radhakrishnan K, Keel SB, Mahoney D, Freidmann AM, Altura RA, Gracheva EO, Bagriantsev SN, Kalfa TA & Gallagher PG (2017). Novel mechanisms of PIEZO1 dysfunction in hereditary xerocytosis. Blood 130, 1845–1856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grootenboer S, Barro C, Cynober T, Schischmanoff PO, Ayoubi JM, Tchernia G, Delaunay J & Pons JC (2001). Dehydrated hereditary stomatocytosis: a cause of prenatal ascites. Prenat Diagn 21, 1114–1118. [PubMed] [Google Scholar]
- Grootenboer S, Schischmanoff PO, Laurendeau I, Cynober T, Tchernia G, Dommergues JP, Dhermy D, Bost M, Varet B, Snyder M, Ballas SK, Ducot B, Babron MC, Stewart GW, Gasparini P, Iolascon A & Delaunay J (2000). Pleiotropic syndrome of dehydrated hereditary stomatocytosis, pseudohyperkalemia, and perinatal edema maps to 16q23‐q24. Blood 96, 2599–2605. [PubMed] [Google Scholar]
- Gudipaty SA, Lindblom J, Loftus PD, Redd MJ, Edes K, Davey CF, Krishnegowda V & Rosenblatt J (2017). Mechanical stretch triggers rapid epithelial cell division through Piezo1. Nature 543, 118–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Hou B, Tumova S, Muraki K, Bruns A, Ludlow MJ, Sedo A, Hyman AJ, McKeown L, Young RS, Yuldasheva NY, Majeed Y, Wilson LA, Rode B, Bailey MA, Kim HR, Fu Z, Carter DAL, Bilton J, Imrie H, Ajuh P, Dear TN, Cubbon RM, Kearney MT, Prasad KR, Evans PC, Ainscough JFX & Beech DJ (2014). Piezol integration of vascular architecture with physiological force. Nature 515, 279–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukacs V, Mathur J, Mao R, Bayrak‐Toydemir P, Procter M, Cahalan SM, Kim HJ, Bandell M, Longo N, Day RW, Stevenson DA, Patapoutian A & Krock BL (2015). Impaired PIEZO1 function in patients with a novel autosomal recessive congenital lymphatic dysplasia. Nat Commun 6, 8329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martins JR, Penton D, Peyronnet R, Arhatte M, Moro C, Picard N, Kurt B, Patel A, Honore E & Demolombe S (2016). Piezo1‐dependent regulation of urinary osmolarity. Pflugers Arch 468, 1197–1206. [DOI] [PubMed] [Google Scholar]
- Miller DR, Rickles FR, Lichtman MA, La Celle PL, Bates J & Weed RI (1971). A new variant of hereditary hemolytic anemia with stomatocytosis and erythrocyte cation abnormality. Blood 38, 184–204. [PubMed] [Google Scholar]
- Ranade SS, Qiu Z, Woo S‐H, Hur SS, Murthy SE, Cahalan SM, Xu J, Mathur J, Bandell M, Coste B, Li Y‐SJ, Chien S & Patapoutian A (2014). Piezo1, a mechanically activated ion channel, is required for vascular development in mice. Proc Natl Acad Sci USA 111, 10347–10352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rees DC, Portmann B, Ball C, Mieli‐Vergani G, Nicolaou A, Chetty MC & Stewart GW (2004). Dehydrated hereditary stomatocytosis is associated with neonatal hepatitis. Br J Haematol 126, 272–276. [DOI] [PubMed] [Google Scholar]
- Sabine A, Agalarov Y, Maby‐El Hajjami H, Jaquet M, Hagerling R, Pollmann C, Bebber D, Pfenniger A, Miura N, Dormond O, Calmes JM, Adams RH, Makinen T, Kiefer F, Kwak BR & Petrova TV (2012). Mechanotransduction, PROX1, and FOXC2 cooperate to control connexin37 and calcineurin during lymphatic‐valve formation. Dev Cell 22, 430–445. [DOI] [PubMed] [Google Scholar]
- Syfuss PY, Ciupea A, Brahimi S, Cynober T, Stewart GW, Grandchamp B, Beaumont C, Tchernia G, Delaunay J & Wagner JC (2006). Mild dehydrated hereditary stomatocytosis revealed by marked hepatosiderosis. Clin Lab Haematol 28, 270–274. [DOI] [PubMed] [Google Scholar]
- Wang S, Chennupati R, Kaur H, Iring A, Wettschureck N & Offermanns S (2016). Endothelial cation channel PIEZO1 controls blood pressure by mediating flow‐induced ATP release. J Clin Invest 126, 4527–4536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Lewis AH & Grandl J (2017). Touch, tension, and transduction ‐ the function and regulation of Piezo ion channels. Trends Biochem Sci 42, 57–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Wang Y, Li S, Xu Z, Li H, Ma L, Fan J, Bu D, Liu B, Fan Z, Wu G, Jin J, Ding B, Zhu X & Shen Y (2004). Mutations in SCN9A, encoding a sodium channel alpha subunit, in patients with primary erythermalgia. J Med Genet 41, 171–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarychanski R, Schulz VP, Houston BL, Maksimova Y, Houston DS, Smith B, Rinehart J & Gallagher PG (2012). Mutations in the mechanotransduction protein PIEZO1 are associated with hereditary xerocytosis. Blood 120, 1908–1915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu X, Need AC, Petrovski S & Goldstein DB (2014). One gene, many neuropsychiatric disorders: lessons from Mendelian diseases. Nat Neurosci 17, 773–781. [DOI] [PubMed] [Google Scholar]