

Abstract
Key points
The uterine artery (UA) markedly vasodilates during pregnancy to direct blood flow to the developing fetus. Inadequate UA vasodilatation leads to intrauterine growth restriction and fetal death.
The large‐conductance voltage‐ and Ca2+‐activated K+ (BKCa) channel promotes UA vasodilatation during pregnancy.
We report that BKCa channel activation increases the UA diameter at late pregnancy stages in mice.
Additionally, a BKCa channel auxiliary subunit, γ1, participates in this process by increasing channel activation and inducing UA vasodilatation at late pregnancy stages.
Our results highlight the importance of the BKCa channel and its γ1‐subunit for UA functional changes during pregnancy.
Abstract
Insufficient vasodilatation of the uterine artery (UA) during pregnancy leads to poor utero‐placental perfusion, contributing to intrauterine growth restriction and fetal loss. Activity of the large‐conductance Ca2+‐activated K+ (BKCa) channel increases in the UA during pregnancy, and its inhibition reduces uterine blood flow, highlighting a role of this channel in UA adaptation to pregnancy. The auxiliary γ1‐subunit increases BKCa activation in vascular smooth muscle, but its role in pregnancy‐associated UA remodelling is unknown. We explored whether the BKCa and its γ1‐subunit contribute to UA remodelling during pregnancy. Doppler imaging revealed that, compared to UAs from wild‐type (WT) mice, UAs from BKCa knockout (BKCa −/−) mice had lower resistance at pregnancy day 14 (P14) but not at P18. Lumen diameters were twofold larger in pressurized UAs from P18 WT mice than in those from non‐pregnant mice, but this difference was not seen in UAs from BKCa −/− mice. UAs from pregnant WT mice constricted 20–50% in response to the BKCa blocker iberiotoxin (IbTX), whereas UAs from non‐pregnant WT mice only constricted 15%. Patch‐clamp analysis of WT UA smooth muscle cells confirmed that BKCa activity increased over pregnancy, showing three distinct voltage sensitivities. The γ1‐subunit transcript increased 7‐ to 10‐fold during pregnancy. Furthermore, γ1‐subunit knockdown reduced IbTX sensitivity in UAs from pregnant mice, whereas γ1‐subunit overexpression increased IbTX sensitivity in UAs from non‐pregnant mice. Finally, at P18, γ1‐knockout (γ1−/−) mice had smaller UA diameters than WT mice, and IbTX‐mediated vasoconstriction was prevented in UAs from γ1−/− mice. Our results suggest that the γ1‐subunit increases BKCa activation in UAs during pregnancy.
Keywords: BKCa channel, auxiliary subunits, vascular smooth muscle, uterine artery, utero‐placental perfusion, pregnancy
Key points
The uterine artery (UA) markedly vasodilates during pregnancy to direct blood flow to the developing fetus. Inadequate UA vasodilatation leads to intrauterine growth restriction and fetal death.
The large‐conductance voltage‐ and Ca2+‐activated K+ (BKCa) channel promotes UA vasodilatation during pregnancy.
We report that BKCa channel activation increases the UA diameter at late pregnancy stages in mice.
Additionally, a BKCa channel auxiliary subunit, γ1, participates in this process by increasing channel activation and inducing UA vasodilatation at late pregnancy stages.
Our results highlight the importance of the BKCa channel and its γ1‐subunit for UA functional changes during pregnancy.
Introduction
Fetal growth and development require a progressive increase in maternal utero‐placental circulation. Insufficient utero‐placental perfusion contributes to adverse pregnancy outcomes such as intrauterine growth restriction (IUGR), pre‐eclampsia and fetal loss (Chiswick, 1985; Konje et al. 2003; Browne et al. 2011; Rada et al. 2012; Cotechini et al. 2014; Janot et al. 2014). A major mechanism for increasing blood flow to the placenta is vasodilatation and remodelling of the uterine artery (UA) (Ford, 1982; Palmer et al. 1992; Osol & Cipolla, 1993; Hilgers et al. 2003; Mu & Adamson, 2006). Thus, during pregnancy, the UA diameter significantly increases (Osol & Cipolla, 1993; Cipolla & Osol, 1994) and UA resistance decreases (Ford, 1982; Palmer et al. 1992; Mu & Adamson, 2006).
One key player in pregnancy‐dependent increases in uterine blood flow is the large‐conductance voltage‐ and Ca2+‐activated K+ (BKCa) channel. Blocking BKCa channels reduces blood flow in UAs from pregnant sheep (Rosenfeld et al. 2001), and BKCa channel currents increase in sheep UA smooth muscle cells (SMCs) during pregnancy, although the BKCa protein expression level remains constant over pregnancy (Rosenfeld et al. 2009; Hu et al. 2011). In addition, BKCa channels regulate vascular function in UAs from non‐pregnant women (Rosenfeld et al. 2008), suggesting an evolutionarily conserved role in regulating UA function.
BKCa channel activation can be regulated by numerous mechanisms, such as membrane microdomain localization (Brainard et al. 2005; Alioua et al. 2008; Lu et al. 2010), alternative splicing (Korovkina et al. 2001; Curley et al. 2004; Zhu et al. 2005), and association with modulatory subunits (Knaus et al. 1994; Tanaka et al. 1997; Brenner et al. 2000; Yan & Aldrich, 2012; Evanson et al. 2014). Auxiliary subunits are proposed to confer cell‐ or tissue‐specific functional diversity. In vascular SMCs, the β1‐subunit enhances voltage activation and apparent Ca2+ sensitivity of the BKCa channel (McManus et al. 1995; Tanaka et al. 1997; Brenner et al. 2000). Moreover, expression of the BKCa channel β1‐subunit is higher in UAs from pregnant sheep than in those from non‐pregnant animals (Rosenfeld et al. 2009; Hu et al. 2011). This finding suggests that the β1‐subunit participates in the increased activity of BKCa channels in UA SMCs during pregnancy and may explain why BKCa activity increases even though expression level does not change (Rosenfeld et al. 2009; Hu et al. 2011).
Recent studies have revealed that, in addition to β‐subunits, BKCa channels associate with γ‐subunits (Yan & Aldrich, 2010, 2012). The first described member of this family, the γ1‐subunit (also referred to as the leucine‐rich repeat‐containing protein 26), markedly increases the voltage sensitivity of the BKCa channel, activating it at resting membrane potentials, without a rise in intracellular Ca2+ (Yan & Aldrich, 2010). The γ1‐subunit is expressed in cerebral artery SMCs, where it modulates BKCa channel activity and reduces myogenic tone (Evanson et al. 2014). However, whether the γ1‐subunit regulates BKCa channel activity in other vascular beds has not been explored.
Here, we tested the hypothesis that the γ1‐subunit increases BKCa channel activity in UA SMCs during pregnancy. We observed that UAs from BKCa channel knockout (BKCa −/−) mice had lower in vivo resistance and smaller in vitro diameter during pregnancy than those from wild‐type (WT) mice. Pharmacological blockade and single‐channel analysis of BKCa currents in UA SMCs indicated that BKCa activity and voltage sensitivity increased over the course of mouse pregnancy. Moreover, the increased BKCa channel activity in UAs from pregnant mice was reduced by knockdown of the γ1‐subunit. Finally, γ1‐subunit knockout mice exhibited reduced UA diameter and BKCa channel activity during pregnancy. Our findings suggest that the γ1‐subunit contributes to the pregnancy‐dependent increase in BKCa channel activity in the UA.
Methods
Mice
All animal work complied with the Guidelines for the Care and Use of Laboratory Animals set forth by the NIH and protocols approved by the Animal Care and Use Committee at Washington University in St Louis School of Medicine. BKCa −/− mice (a gift from Dr Andrea L. Meredith at University of Maryland) have stable, germ line‐transmissible deletion of exon 1 of the mSlo1 gene (Meredith et al. 2004). Because BKCa −/− males exhibit a significantly lower mating efficiency than WT mice, the mouse colony was maintained by breeding heterozygous mice. Genotyping was performed by PCR of tail DNA with the following primers: 5′‐TTCATCATCTTGCTCTGGCGGACG‐3′, 5′‐CCATAGTCACCAATAGCCC‐3′, 5′‐ATAGCCTGAAGAACGAGATCAGC‐3′ and 5′‐CCTCAAGAAGGGGACTCTAAAC‐3′, and confirmed by Transnetyx, Inc. (Cordova, TN, USA). Eight‐ to‐twelve‐week‐old homozygous BKCa −/− females and their WT littermates were mated to WT males, resulting in BKCa +/− and WT offspring, respectively. Mice lacking the lrrc26 gene (γ1−/−) were kindly provided by Dr Christopher Lingle (Washington University in St Louis) (Yang et al. 2017), and the colony was maintained as homozygous mutants. Day 0 of pregnancy was determined by the presence of a copulatory plug after mating for 2–3 h. Mice were killed by gradually increasing the CO2 flow rate into the chamber and then performing cervical dislocation. Primary UAs from both uterine arcades were dissected from non‐pregnant (NP) mice and from pregnant mice on days 14 (P14, after placentation) and 18 (P18, before labour initiation). After the dams were killed, the numbers of live pups were recorded, and pups were killed by decapitation.
Blood flow and embryonic measurements
P14 and P18 BKCa −/− and WT mice were sedated by subcutaneous administration of 1.2–5 mg midazolam. Mice remained awake but docile. In vivo analysis of blood flow was conducted with an Acuson Sequoia c256 echocardiographic system (Acuson Corp., Mountain View, CA, USA) fitted with a 15 MHz linear array oscillator/receiver. A 40 MHz linear array probe was applied to the chest to measure maternal heart rate and other cardiac parameters. The imaging probe was coupled to a Vevo 2100 imaging system (VisualSonics, Toronto, ON, Canada), generating ∼180–200 two‐dimensional frames per second. Uterine and fetal sonography was performed by applying the probe to the lower abdomen. Two‐dimensional sonographic images were used to measure crown–rump length (fetal size), placental thickness at the point of umbilical cord insertion, and fetal heart rate. All values were averaged from three pups per dam. Pulse‐wave Doppler evaluation was used to measure flow velocity in UAs adjacent to the gestational sacs and umbilical arteries. UA resistance index ((PSV − EDV)/PSV, where PSV is peak systolic velocity and EDV is end‐diastolic velocity) and UA pulsatility index ((PSV − EDV)/time‐averaged maximum velocity) were calculated as indicators of vascular resistance downstream of the UA. All sonographic images were analysed by an investigator blinded to the mouse genotype. After at least 1 h of recovery from sedation, P14 mice were housed until P18, when another ultrasound was performed. After at least 1 h of recovery from sedation after P18 ultrasound, vascular function was assessed in some of the mice.
Immunohistochemistry
P18 UAs were dissected in ice‐cold Dulbecco's phosphate buffered saline (Gibco, Carlsbad, CA, USA) and immediately fixed in 4% paraformaldehyde (Affymetrix, Cleveland, OH, USA) for 24 h at 4°C, embedded in 2% agar (Sigma‐Aldrich, St Louis, MO, USA), and then embedded in paraffin. Sections (5 μm) were adhered to slides, deparaffinized with xylenes (Sigma‐Aldrich), and rehydrated. Slides were treated with a citrate‐based solution (Vector Laboratories, Burlingame, CA, USA) for antigen retrieval and then stained with rabbit anti‐BKCa α (Abcam, Eugene, OR, USA, 1:100) or rabbit anti‐CD31 (Cell Signaling Technology, Danvers, MA, USA, 1:50) and wheat germ agglutinin (Alexa 555 conjugate, Invitrogen, Grand Island, NY, USA), mounted, and then imaged with a Nikon Diaphot fluorescence microscope.
Vascular function
UAs were dissected as described above, cannulated onto glass cannulas filled with Krebs solution (118 mm NaCl, 4.7 mm KCl, 2.5 mm CaCl2, 1.2 mm MgSO4, 1.2 mm KH2PO4, 25 mm NaHCO3, and 11 mm glucose), a common buffer used in physiological studies (Gutkowska et al. 1997; Knot & Nelson, 1998; Cheranov & Jaggar, 2004; Ketsawatsomkron et al. 2012), and secured with sutures in a Pressure Myograph System 110P (DMT‐USA, Ann Arbor, MI, USA). Oxygenated (95% O2 and 5% CO2) and warmed (37°C) Krebs buffer was continuously circulated through the chamber. Using a split‐screen microscope connected to a video camera, lumen and outer vascular diameters were continuously recorded with MyoVIEW software (DMT‐USA). Main UAs were pressurized to 60 mmHg (Hilgers et al. 2003) and allowed to equilibrate for a minimum of 45 min before study. To ensure viability, 50 mm KCl was added from a 2 m stock solution at the beginning and end of each experiment, and data were discarded if UAs showed <40% reduction of basal diameter in the presence of 50 mm KCl. Similar contractile responses were seen when Krebs was instead substituted with a high‐K+/low‐Na+ Krebs solution (R. A. Lorca, unpublished observations). Iberiotoxin (IbTX, Tocris, Bristol, UK) was sequentially applied at 1–100 nm in the presence of a low KCl concentration (15 mm), which slightly depolarizes SMCs, inducing an increase of intracellular Ca2+ and activating BKCa channels. The endothelium was removed before each experiment by passing air bubbles through the lumen and then flushing with Krebs buffer. As previously described (Ketsawatsomkron et al. 2012), endothelial removal was confirmed by detecting <30% vasorelaxation in the presence of 10 μm acetylcholine (Sigma‐Aldrich) in vessels pre‐constricted with 15 mm KCl. Lumen and external diameters of UAs pressurized at 60 mmHg in regular Krebs buffer but with no other stimulation were used to calculate wall thickness, cross‐sectional area and wall:lumen ratio.
Vascular SMC isolation
Single SMCs were enzymatically dissociated from UAs as described previously (Jackson et al. 1997). Briefly, isolated UAs were placed in ice‐cold Dulbecco's phosphate buffered saline, then cut into ≤2 mm‐long segments and incubated for 10 min at room temperature in dissociation solution (DS) containing (in mm): 145 NaCl, 4 KCl, 0.05 CaCl2, 1 MgCl2, 10 HEPES, 10 glucose (pH 7.4 with NaOH), and 0.1% bovine serum albumin (RPI, Mount Prospect, IL, USA). Then, the pieces were placed in DS containing (in mg ml−1): 1.5 papain (Worthington Biochemical, Lakewood, NJ, USA) and 1 dithiothreitol (Sigma‐Aldrich), and incubated at 37°C for 25 min. Next, the solution was replaced with DS containing (in mg ml−1): 1.5 collagenase type H (Sigma‐Aldrich), 1 trypsin inhibitor, and 0.5 elastase (both from Worthington Biochemical) and incubated at 37°C for 5 min. Pieces were incubated for 3 min at room temperature and then placed on ice for 5 min. After digestion, the pieces were gently triturated in ice‐cold DS until elongated vascular SMCs were observed. SMCs were placed on glass coverslips and allowed to attach for at least 40 min at 4°C before electrophysiological experiments were performed.
Electrophysiology
Excised‐patch recordings in the inside‐out configuration were performed on isolated SMCs at room temperature in a bath solution containing (in mm): 140 KCl, 20 KOH, 10 HEPES, 5 (H)EDTA, and 10 μm free‐Ca2+ (pH 7.2 with HCl). Free‐Ca2+ concentration was measured with a Ca2+‐sensitive electrode (Thermo Fisher Scientific, Rockford, IL, USA). The pipette solution contained (in mm): 140 KCl, 20 KOH, 2 MgCl2, and 10 HEPES (pH 7.4 with HCl). Single‐channel currents were recorded at a sampling rate of 100 kHz and filtered at 5 kHz by using an Axopatch 200B amplifier (Molecular Devices, Sunnyvale, CA, USA). pCLAMP 10 software (Molecular Devices) was used to evoke currents with 10 mV voltage steps (1000 ms duration) from −120 to +100 mV, from a holding potential of 0 mV. Mean open probability (P o) values were calculated with pCLAMP 10 software. Half‐maximal activation voltage (V 0.5) values were determined for each experiment by using GraphPad 6 software (San Diego, CA, USA) to fit membrane potential (V) to a Boltzmann function, P o = P o(max)/(1 + e–zF(V – V 0.5 /RT), where z is the effective charge, F is the Faraday constant, R is the ideal gas constant, and T is temperature.
RNA isolation and quantitative PCR
Total RNA from isolated UAs was extracted by using an Arcturus PicoPure RNA isolation kit (Thermo Fisher Scientific) and reverse transcribed with iScript reverse transcription supermix (Bio‐Rad, Hercules, CA, USA) according to the manufacturers’ instructions. The CFX96 Real‐time System with IQ‐SYBR Green Supermix (Bio‐Rad) was used for real‐time PCR. The primer sequences (Integrated DNA Technologies, Coralville, IA, USA) were: mouse BKCa α‐subunit (GenBank accession no: NM_010610.2), 5′‐AGATCGACATGGCTTTCAA‐3′ and 5′‐CAGGAGGGACTGTGAAGA‐3′; mouse BKCa β1‐subunit (NM_031169.4), 5′‐CTGGGAGTGGCAATGGTAGT‐3′ and 5′‐GCCCACAGCTGATACATTGA‐3′; mouse BKCa γ1‐subunit (NM_146117.2), 5′‐AAACTGAGACCCTGCTCTGC‐3′ and 5′‐GATGGCCAAACTAGCAAGGA‐3′; mouse succinate dehydrogenase complex flavoprotein subunit A (SDHA, NM_023281), 5′‐GGAACACTCCAAAAACAGACCT‐3′ and 5′‐CCACCACTGGGTATTGAGTAGAA‐3′. Thermal cycling conditions were as follows: 95°C for 3 min, followed by 40 cycles of amplification at 95°C for 10 s and 57°C for 30 s. All samples were analysed in triplicate; mRNA data are reported relative to SDHA.
Transfection of intact UAs
Isolated UAs were placed in ice‐cold Krebs buffer and reversibly permeabilized as described previously (Lesh et al. 1995). Briefly, UAs were placed for 20 min at 4°C in a solution containing (in mm): 120 KCl, 2 MgCl2, 10 EGTA, 5 Na2ATP, and 20 TES (pH 6.8 with KOH). After permeabilization, UAs were incubated for 90 min at 4°C in a similar buffer devoid of EGTA and containing 5 μg of non‐targeting (scrambled, Scr) control short hairpin RNA (shRNA) or γ1‐targeted shRNA in the pLKO vector (The Genome Institute, Washington University in St Louis, St Louis, MO, USA) for knockdown experiments. For overexpression experiments, UAs were incubated with 5 μg of either a pBudCE4.1 vector (Invitrogen) expressing the BKCa γ1‐subunit (GenBank accession number: NM_146117.2) or an empty pBudCE4.1 vector. In both cases, the MgCl2 concentration was then raised to 10 mm for 30 min, permeabilization was reversed by placing the UAs in a MOPS‐buffered solution containing (in mm): 140 NaCl, 5 KCl, 10 MgCl2, 5 glucose, and 2 MOPS (pH 7.1 with NaOH) at room temperature for 30 min, [Ca2+] was gradually increased from nominally Ca2+‐free to 0.01, 0.1 and 1.8 mm over a 45 min period, and then the UAs were incubated at 37°C in serum‐ and HEPES‐free Dulbecco's modified Eagle medium (DMEM; Invitrogen) for 48–60 h.
Statistical analyses
The numbers of pups per litter and data obtained from Doppler ultrasound and vascular reactivity experiments were subjected to two‐way ANOVA followed by Sidak's multiple comparison test (GraphPad 6 software). Patch‐clamp and quantitative PCR (qPCR) experiments were analysed by non‐parametric Kruskal–Wallis one‐way ANOVA (GraphPad 6 software). A P value < 0.05 was considered significant.
Results
Maternal and fetal characteristics
BKCa −/− dams had significantly fewer pups per litter than WT dams (Table 1). There were no differences between genotypes in pup size measured as crown–rump length by ultrasound (Table 1). However, placental thickness increased from P14 to P18 in BKCa −/− dams but not in WT dams (Table 1). WT dams showed significant decreases in UA resistance index (RI) from P14 to P18 (0.62 ± 0.03 vs. 0.47 ± 0.02, P < 0.01, Fig. 1 A and B), and in pulsatility index (PI) from P14 to P18 (1.09 ± 0.08 vs. 0.73 ± 0.04, P < 0.01, Fig. 1 A and C), suggesting greater downstream dilatation and utero‐placental blood flow. The decreases in UA RI and PI from P14 to P18 did not occur in the BKCa −/− dams (RI, 0.43 ± 0.03 in P14 and 0.48 ± 0.02 in P18; and PI, 0.67 ± 0.06 in P14 and 0.74 ± 0.04 in P18, Fig. 1 A–C). Both RI and PI were lower at P14 in BKCa −/− dams than in WT dams (P < 0.01, Fig. 1 A–C). The lower RI and PI in P14 BKCa −/− mice were associated with higher end‐diastolic velocities rather than lower peak systolic velocities (Fig. 1 D and E). Other maternal (WT vs. BKCa −/−) or fetal (WT vs. BKCa +/−) haemodynamic indices did not differ between genotypes (Table 1).
Table 1.
Maternal and fetal characteristics at P14 and P18
| WT dams | BKCa −/− dams | ||||||
|---|---|---|---|---|---|---|---|
| P14 (10) | P18 (11) | P P14 vs. P18 | P14 (5) | P18 (6) | P P14 vs. P18 | P WT vs. BKCa −/− | |
| Maternal heart rate (beats min−1) | 555 ± 8 | 564 ± 6 | NS | 555 ± 15 | 575 ± 10 | NS | NS |
| Relative wall thickness | 0.5 ± 0.02 | 0.49 ± 0.02 | NS | 0.53 ± 0.02 | 0.51 ± 0.03 | NS | NS |
| Fractional shortening (%) | 46± 1 | 45 ± 2 | NS | 50 ± 1 | 44 ± 2 | NS | NS |
| Ejection time (ms) | 43.4 ± 0.6 | 42.7 ± 1.2 | NS | 41.9 ± 1.7 | 43.3 ± 2.3 | NS | NS |
| UA PSV (mm s−1) | 413 ± 50 | 447 ± 43 | NS | 420 ± 39 | 516 ± 51 | NS | NS |
| UA EDV (mm s−1) | 151 ± 17 | 237 ± 22 | <0.05 | 237 ± 24 | 270 ± 27 | NS | <0.05 at P14 |
| UA TAmax velocity (mm s−1) | 238 ± 27 | 296 ± 28 | NS | 272 ± 23 | 336± 33 | NS | NS |
| WT pups | BKCa +/− pups | ||||||
|---|---|---|---|---|---|---|---|
| Number of pups per litter | 8.6 + 0.2 [5] | 8.6 + 0.3 [18] | NS | 5.3 + 1 [6] | 7 + 0.4 [7] | NS | <0.05 at P14 and P18 |
| Crown–rump length (mm) | 10.7 ± 0.3 | 16.8 ± 0.5 | <0.05 | 10.3 ± 0.4 | 16.2 ± 0.6 | <0.05 | NS |
| Placental thickness (mm) | 2.6 ± 0.2 | 2.8 ± 0.2 | NS | 2.4 ± 0.2 | 3.3 ± 0.1 | <0.05 | NS |
| Fetal heart rate (beats min−1) | 186.8 ± 5.7 | 243.3 ± 7.1 | <0.05 | 207.6 ± 12.0 | 230.3 ± 6.5 | NS | NS |
| UmbA TAmax velocity (mm s−1) | 126 ± 7 | 166 ± 10 | <0.05 | 124 ± 7 | 167 ± 6 | <0.05 | NS |
Data are means ± SEM. Number of dams are in parentheses, and in brackets where values were different. EDV, end‐diastolic velocity; PSV, peak systolic velocity; TAmax, time‐averaged maximum velocity; UmbA, umbilical artery. Significance columns (P) indicate differences between pregnancy stages or genotypes. NS, not significant. Two‐way ANOVA, Sidak.
Figure 1. Doppler measurements in UAs from pregnant WT and BKCa −/− mice.
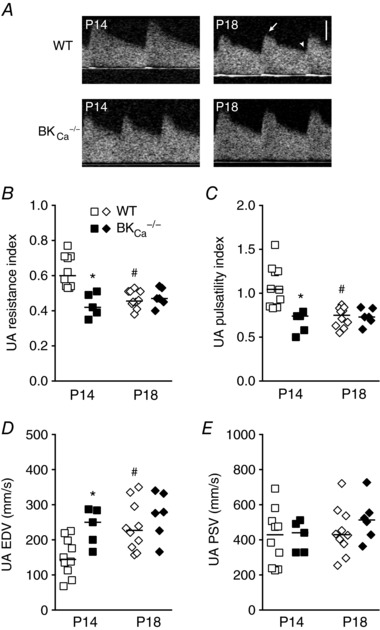
A, Doppler flow velocity waveforms obtained in UAs from WT and BKCa −/− mice at P14 and P18. Arrow and arrowhead indicate peak systolic velocity (PSV) and end‐diastolic velocity (EDV), respectively. Bar = 200 mm s−1. B–E, Doppler imaging measurements of resistance index (B), pulsatility index (C), EDV (D) and PSV (E) in UAs from WT (open symbols, n = 10–11) and BKCa −/− mice (filled symbols, n = 5–6) at P14 and P18. Symbols represent individual values; bars represent median values. * P < 0.05 compared to WT at same stage, # P < 0.05 compared to same genotype at P14.
UA diameter and BKCa channel activity during pregnancy
To better understand how the BKCa channel alters the vascular parameters observed in Fig. 1, we isolated UAs from WT and BKCa −/− dams and performed immunofluorescence to determine where BKCa was expressed. We observed BKCa protein in the SMC layer (Fig. 2 A) but not in the endothelial layer, which expressed the endothelial cell adhesion marker CD31 (Fig. 2 B). As expected, BKCa staining intensity was decreased in UAs from BKCa −/− mice (Fig. 2 C).
Figure 2. BKCa channel expression in UAs.
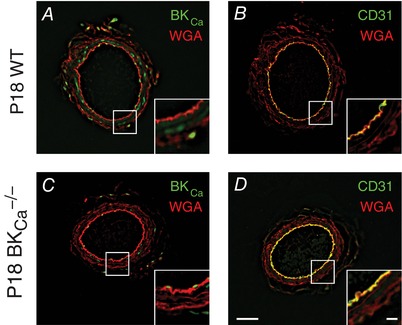
Immunohistochemistry showing expression of BKCa channel (green) in non‐pressurized P18 UAs from WT (A) and BKCa −/− (C) mice. CD31 (green) was used as an endothelial layer marker (B and D). Wheat germ agglutinin (WGA, red) was used to highlight vessel structure in all panels. Insets show high‐magnification views of boxed areas. Bars = 50 μm and 10 μm (insets).
To examine the role of SMC BKCa in control of UA diameter, we isolated UAs and removed the endothelium, which has a role in UA adaptation during pregnancy (Bird et al. 2000). In UAs from WT mice, diameter increased progressively between NP, P14 and P18 (281 ± 19.2 μm, 448.3 ± 24.8 μm and 584.1 ± 18.1 μm, P < 0.05, Fig. 3 A). In UAs from BKCa −/− mice, diameter increased between NP and P14 (266.5 ± 13.9 μm and 447.1 ± 6.9 μm, respectively, P < 0.05), but was not larger at P18 (433.2 ± 17.9 μm, Fig. 3 A). Moreover, mean diameter was significantly smaller in UAs from BKCa −/− mice than in those from WT mice at P18 (433.2 ± 17.9 μm vs. 584.1 ± 18.1 μm, P < 0.05, Fig. 3 A), indicating a role for BKCa channels in regulating UA smooth muscle basal tone during late pregnancy stages.
Figure 3. UA lumen diameter and BKCa activity in UAs from WT and BKCa −/− mice.
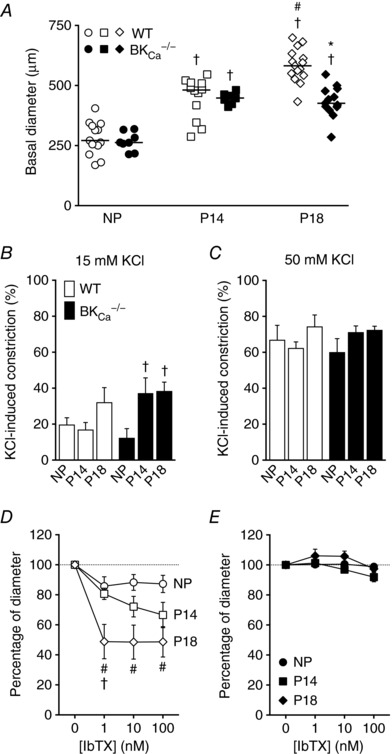
A, basal lumen diameter of pressurized UAs (60 mmHg) from NP, P14 and P18 WT (open symbols) and BKCa −/− (filled symbols) mice. Symbols are individual values; bars are median values. † P < 0.05 compared to NP of same genotype, # P < 0.05 compared to P14 of same genotype, * P < 0.05 compared to WT at same stage. B and C, vasoconstriction in response to 15 mm (B) or 50 mm KCl (C) in pressurized UAs isolated from NP, P14 or P18 WT (open columns, n = 7–9) and BKCa −/− mice (filled columns, n = 7–13). Columns are mean values ± SEM. † P < 0.05 compared to NP of the same genotype. D and E, constriction induced by 1–100 nm IbTX, measured as percentage of diameter after 15 mm KCl‐induced vasoconstriction of pressurized UAs from NP, P14 and P18 WT (D, n = 7–9) and BKCa −/− mice (E, n = 6–12). Symbols are mean ± SEM. # P < 0.05 compared to NP and † P < 0.05 compared to P14, at same IbTX concentration.
To determine whether BKCa contributed to UA architecture, we assessed wall thickness, cross‐sectional area and wall:lumen ratio. Consistent with previous findings (van der Heijden et al. 2005), UAs from P14 and P18 mice had thicker walls than those from NP mice (Table 2). However, we did not see this increase in wall thickness between NP and P14 or P18 in UAs from BKCa −/− mice (Table 3). In WT mice, the wall:lumen ratio was lower in UAs from P18 than in those from P14 or NP (Table 2), but this difference was not observed in UAs from BKCa −/− mice (Table 3). Despite these differences, wall:lumen ratio, cross‐sectional area and wall thickness did not differ significantly between UAs from WT and BKCa −/− mice at any stage (Tables 2 and 3).
Table 2.
UA vascular parameters in NP, P14 and P18 WT mice
| WT dams | |||
|---|---|---|---|
| NP (12) | P14 (12) | P18 (9) | |
| Wall thickness (μm) | 31 ± 2.5 | 42.4 ± 3.9† | 40.6 ± 2.7† |
| Cross‐sectional area (103 μm2) | 34.7 ± 4.8 | 64.8 ± 5.9† | 70.5 ± 5.2† |
| Wall:lumen ratio | 11.3 ± 1.6 | 10.2 ± 1.4 | 7.5 ± 0.5† |
Data are means ± SEM and number of vessels are in parentheses. † P < 0.05 compared to NP. 2‐way ANOVA, Sidak.
Table 3.
UA vascular parameters in NP, P14 and P18 BKCa −/− and γ1−/− mice
| BKCa −/− dams | γ1−/− dams | |||||
|---|---|---|---|---|---|---|
| NP (8) | P14 (9) | P18 (13) | NP (4) | P14 (5) | P18 (6) | |
| Wall thickness (μm) | 30.8 ± 1.8 | 38 ± 2.2 | 38.7 ± 1.9 | 30 ± 2.6 | 36.2 ± 1.2 | 41.5 ± 3.1 |
| Cross‐sectional area (103 μm2) | 28.8 ± 2.2 | 62.2 ± 5.2† | 57.7 ± 3.9† | 28.8 ± 2.7 | 53.6 ± 2.8 | 57.6 ± 5.6† |
| Wall:lumen ratio | 11.8 ± 1.1 | 8.5 ± 0.5 | 9.2 ± 0.8 | 11 ± 1.2 | 8.3 ± 0.2 | 10.6 ± 1.1 |
Data are means ± SEM and number of vessels are in parentheses. † P < 0.05 compared to NP of same genotype. 2‐way ANOVA, Sidak.
Because loss of BKCa affected UA diameter but had no effect on any structural measures, we wondered whether BKCa contributed to UA contraction in response to KCl‐mediated depolarization. We found that 15 mm KCl induced similar levels of constriction in UAs from NP, P14 and P18 WT mice (19.8 ± 3.8%, 17 + 3.9% and 32.2 ± 8.1% constriction, Fig. 3 B). In contrast, 15 mm KCl induced a larger constriction in UAs from P14 and P18 BKCa −/− mice than in those from NP BKCa −/− mice (37.2 ± 8.5% and 38.4 ± 4.9% vs. 12.4 ± 5.1% constriction, P < 0.05, Fig. 3 B), suggesting that BKCa channels attenuated the vasoconstriction associated with depolarization of UAs in pregnant mice. We observed no differences between genotypes or pregnancy stages at high depolarization levels obtained at a higher KCl concentration (50 mm, Fig. 3 C), indicating that the BKCa effect might be masked at higher KCl concentrations.
We next treated isolated UAs with 15 mm KCl, causing 20–40% constriction and ensuring activation of BKCa channels, and then treated them with the BKCa inhibitor IbTX, which produced a ∼15% reduction in diameter of pre‐constricted UAs from NP WT mice (Fig. 3 D). Increasing concentrations of IbTX induced a progressive decrease in diameter of UAs from P14 mice (19.3 ± 3.7%, 27.9 ± 6.8% and 33.4 ± 8.4%, with 1, 10 and 100 nm IbTX, respectively, Fig. 3 D). However, in UAs from P18 mice, all IbTX concentrations evoked a similar larger constriction (51.2 ± 11.4%, 51.5 ± 11.3% and 51.3 ± 10.1%, with 1, 10 and 100 nm IbTX, respectively, Fig. 3 D). These results suggest that UAs from P18 mice have higher IbTX sensitivity than those from NP and P14 mice. As expected, IbTX did not induce significant constrictions of UAs isolated from BKCa −/− mice, nor did pregnancy change the response (Fig. 3 E). These results indicate that increased BKCa channel activity contributes to the maintenance of a larger UA diameter during pregnancy.
BKCa channel currents in WT UA SMCs
To investigate the mechanism(s) by which BKCa activity was increased in UAs during pregnancy, we performed patch clamp analysis of single BKCa channels in UA SMCs isolated from NP, P14 or P18 WT mice (Fig. 4 A). In NP mice, the BKCa channel open probability (P o), in the presence of 10 μm Ca2+, increased with depolarizing voltages. When we fitted the data to the Boltzmann equation, we found a half‐maximal activation (V 0.5) of 4.7 ± 3 mV (Fig. 4 B). The goodness of fit (R 2) value for the curve fitting was 0.93 and the V 0.5 values of each patch were tightly clustered (Fig. 4 E). In contrast, when we performed similar experiments on UA SMCs isolated from P14 and P18 mice, the R 2 values were 0.81 and 0.76, respectively, indicating poor fits, and the V 0.5 values appeared to be in three clusters (Fig. 4 E). When we separately analysed the three clusters, which we termed ‘low’, ‘mid’ and ‘high’ voltage sensitivity, the R 2 values increased. Specifically, at P14, 82% of channels were low sensitivity (−5.8 ± 3.1 mV, R 2 = 0.94), 9% were mid sensitivity (−44.7 mV, R 2 = 0.98) and 9% were high sensitivity (−82.3 mV, R 2 = 0.95) (Fig. 4 C and E). At P18, 68% of channels were low sensitivity (−6.1 ± 3.9 mV, R 2 = 0.89), 28% were mid sensitivity (−39.2 ± 2.3 mV, R 2 = 0.97) and 4% were high sensitivity (−82.3 mV, R 2 = 0.99) (Fig. 4 D and E). We conclude that, at late stages of pregnancy, a subset of BKCa channels in UA SMCs have increased voltage sensitivity.
Figure 4. BKCa single‐channel voltage activation in UA SMCs from WT mice.
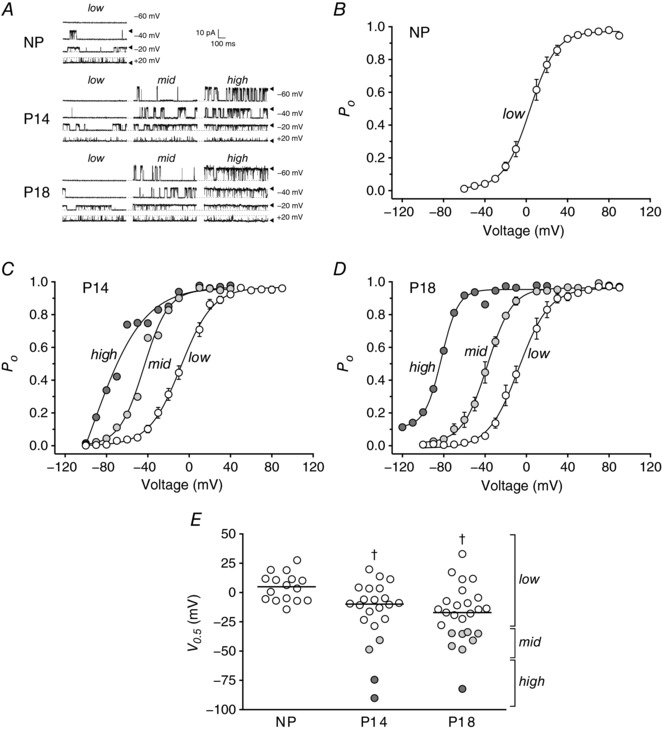
A, representative inside‐out patch‐clamp recordings from UA SMCs isolated from NP, P14 and P18 WT mice in the presence of 10 μm Ca2+ in the bath; membrane potential was held as indicated. Arrowheads and dashed lines indicate open and closed states of the channels, respectively. Single‐channel currents from NP, P14 and P18 showed different activation patterns (low, mid and high). B–D, voltage dependence of BKCa channel activation in UA SMCs isolated from NP (B), P14 (C) and P18 mice (D), expressed as open probability of the channel (P o), in the presence of 10 μm Ca2+. Single‐channel currents from NP, P14 and P18 showed different activation voltages, shown as different curves (low, mid and high; open, light grey and dark grey circles, respectively). Symbols are mean values ± SEM, except where n = 1 or 2. E, voltage of half‐maximal activation (V 0.5) obtained from voltage‐activation curves from NP (circles, N = 4 mice), P14 (squares, N = 9 mice) and P18 mice (triangles, N = 11 mice). Three sensitivities to voltage (low, mid and high) are depicted by open, light grey and dark grey circles, respectively. In P14, low sensitivity values were measured in 18 patches from 8 mice (mice 1, 2, 4, 5, 6, 7, 8, 9), mid sensitivity values were observed in 2 patches from 2 mice (mice 1 and 8) and high sensitivity values were measured in 2 patches from 2 mice (mice 1 and 3). In P18, low sensitivities were observed in 16 patches from 10 mice (mice 10, 11, 12, 13, 15, 16, 17, 18, 19, 20), mid sensitivity values were found in 7 patches from 6 mice (mice 14, 15, 16, 17, 18, 19) and high sensitivity was observed in 1 patch from 1 mouse (mouse 20). Symbols are individual patches; lines are median values. † P < 0.05 compared to NP.
Role of BKCa γ1‐subunit in UA vasodilatation during pregnancy
Because the BKCa γ1‐subunit increases the voltage sensitivity of the BKCa channel in SMCs from cerebral arteries (Evanson et al. 2014), we wondered whether the γ1‐subunit contributed to BKCa channel activity in UA SMCs during pregnancy. Thus, we analysed mRNA expression of BKCa α‐, β1‐ and γ1‐subunits in UAs from NP, P14 and P18 mice. mRNA expression levels of α‐ and β1‐subunits did not differ in UA SMCs from NP and pregnant animals nor change with advancing gestation (Fig. 5 A and B). However, the mRNA expression levels of the γ1‐subunit in UAs from P14 and P18 mice were 9.6‐ and 6.8‐fold greater, respectively, than in those from NP mice (Fig. 5 C).
Figure 5. mRNA expression of BKCa and its auxiliary β1‐ and γ1‐subunits in UA during pregnancy.
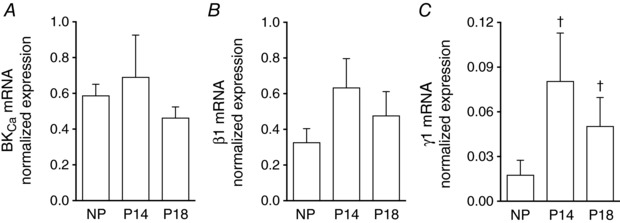
qPCR analysis of mRNA levels from BKCa α‐ (A, n = 6–10), β1‐ (B, n = 5–7) and γ1‐subunits (C, n = 6–8) in UAs isolated from NP, P14 and P18 WT mice. mRNA expression was normalized to expression of succinate dehydrogenase complex flavoprotein subunit A. Columns are mean values ± SEM. † P < 0.05 compared to NP.
To determine the contribution of the γ1‐subunit to UA diameter during pregnancy, we used four approaches. First, we delivered control scrambled (Scr) shRNA or γ1 shRNA into UAs from P14 and P18 WT mice to determine whether disruption of the γ1‐subunit decreased basal UA diameter. The γ1 shRNA lowered transcript levels of γ1 (46 ± 13% of Scr shRNA levels, P < 0.05) but had no effect on α‐ or β1‐subunit transcript levels (78 ± 15% and 97 ± 22%, respectively, of Scr shRNA levels). We found that basal UA diameter was smaller in the P14 and P18 UAs treated with either Scr or γ1 shRNAs than in non‐permeabilized UAs (compare Fig. 6 A–C to Fig. 3 A), indicating that shRNA delivery affected basal UA diameter non‐specifically. However, the basal diameters of UAs treated with γ1 or Scr shRNAs were equivalent (Fig. 6 A and C), indicating that γ1 did not contribute to basal diameter.
Figure 6. Functional knockdown or overexpression of BKCa channel γ1‐subunits in UAs isolated from pregnant or non‐pregnant mice.
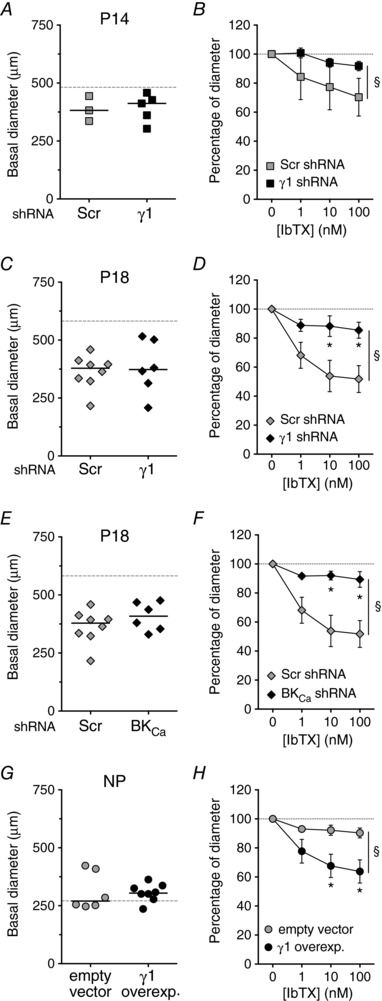
Basal lumen diameters and 1–100 nm IbTX responses of pressurized UAs from P14 (A and B), P18 (C–F) and NP (G and H) mice transfected with either Scr (grey symbols, A–F), empty vector (grey symbols, G and H), γ1‐subunit‐targeted shRNA (filled symbols, A–D), BKCa‐targeted shRNA (filled symbols, E and F) or γ1‐subunit overexpression construct (filled symbols, G and H). Grey dashed lines represent median diameter values of non‐permeabilized UAs, as shown in Fig. 3 A. Symbols in A, C, E and G are individual values, bars are median values. Symbols in B, D, F and H are mean ± SEM. * P < 0.05 compared to Scr shRNA or empty vector at same IbTX concentration, § P < 0.05 between curves. P14 (B, n = 3–5), P18 (D and F, n = 6–8) and NP (H, n = 8).
As a second approach to assess γ1 function, we knocked down γ1‐subunit expression and examined the effect of the BKCa inhibitor IbTX on UA diameter. If the γ1‐subunit was required for BKCa function, then, as we saw with UAs from BKCa −/− mice (Fig. 3 E), UAs should be less sensitive to IbTX and show a smaller decrease in diameter. Consistent with this, we found that both P14 and P18 UAs transfected with Scr shRNA constricted in response to IbTX, but those transfected with γ1 shRNA had an attenuated response to IbTX at P14 and P18 (Fig. 6 B and D). As expected, transfection of P18 UAs with BKCa shRNAs had no effect on basal diameter, but reduced the constriction in response to IbTX (Fig. 6 E and F).
As a third approach to assess γ1 function, we overexpressed the γ1‐subunit in NP UAs, reasoning that it should increase sensitivity to BKCa channel inhibition with IbTX. NP UAs in which the γ1‐subunit was overexpressed had equivalent basal diameter to those transfected with an empty vector (Fig. 6 G). However, NP UAs in which the γ1‐subunit was overexpressed had significantly larger IbTX‐induced contractions than those transfected with the empty vector (Fig. 6 H). In comparing Fig. 6 H to Fig. 3 D, we conclude that overexpression of the γ1‐subunit in NP UAs activated BKCa to a level similar to that measured in P14 UAs.
As a final approach to assess γ1 function, we examined UA function during pregnancy in γ1‐subunit knockout (γ1−/−) mice. UA basal diameter was similar between NP γ1−/− mice and WT mice (275 ± 16.8 μm and 281 ± 19.2 μm, Fig. 7 A). UA diameters increased during pregnancy and were similar between γ1−/− and WT mice at P14 (435.2 ± 9.1 μm and 448.3 ± 24.8 μm, Fig. 7 A). However, at P18, UA basal diameters were significantly smaller in γ1−/− mice than in WT mice (400.5 ± 24.9 μm vs. 584.1 ± 18.1 μm, P < 0.05, Fig. 7 A). Moreover, IbTX did not induce significant constrictions of UAs isolated from γ1−/− mice, nor did pregnancy change the response (Fig. 7 B). UA structural measurements, such as wall thickness, cross‐sectional area and wall:lumen ratio were similar between WT and γ1−/− mice (Tables 2 and 3). Finally, γ1−/− mice had significantly fewer pups per litter than WT mice (6.4 ± 0.8 vs. 8.4 ± 0.3, P < 0.05). In all of these assays, the effects of loss of γ1 were similar to the effects of loss of BKCa (compare to Fig. 3 A and E and BKCa −/− data in Tables 1 and 3). Together, these results suggest that the enhanced BKCa activity observed in UAs from pregnant mice was, in part, due to modulation by the γ1‐subunit.
Figure 7. UA lumen diameter and IbTX responses in UAs from γ1‐subunit knockout (γ1−/−) pregnant mice.
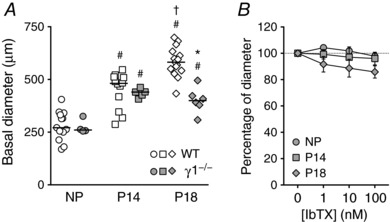
A, basal lumen diameter of pressurized UAs (60 mmHg) from NP, P14 and P18 WT (open symbols, same data as shown in Fig. 3 A) or γ1−/− mice (grey symbols). Symbols are individual values; bars are median values. † P < 0.05 compared to NP of same genotype, # P < 0.05 compared to P14 of same genotype, * P < 0.05 compared to WT at same stage. B, 1–100 nm IbTX‐induced constriction, measured as percentage of diameter after 15 mm KCl‐induced vasoconstriction of pressurized UAs from NP, P14 and P18 γ1−/− mice (grey symbols, n = 4–6). Symbols are mean ± SEM.
Discussion
The UA undergoes significant vasodilatation and remodelling during gestation, resulting in increased blood flow and supply of nutrients and oxygen to the developing fetus (Osol & Cipolla, 1993; Osol & Moore, 2014). Here, we present several lines of evidence that the BKCa channel and its auxiliary γ1‐subunit contribute to pregnancy‐dependent increases in UA diameter and blood flow in mice. First, in vitro analysis revealed that UA diameter steadily increased during pregnancy in WT mice. Second, both myography and electrophysiological analyses showed that BKCa channel activity increased in the UA during pregnancy. Finally, mice lacking either the BKCa channel α‐subunit or γ1‐subunit had reduced UA diameters at late pregnancy (P18).
Our results are consistent with the proposal that the BKCa channel attenuates vascular SMC contractions. In vascular SMCs, BKCa channel activity repolarizes the membrane potential after Ca2+‐mediated depolarization, inactivating voltage‐dependent Ca2+ channels and causing blood vessels to dilate (Brayden & Nelson, 1992; Nelson et al. 1995). Thus, the increased BKCa channel activity we observed in UAs from pregnant mice likely induces hyperpolarization of the SMC membrane, leading to UA vasodilatation. Future studies using sharp electrode recordings in intact UAs, or current‐clamp recordings or membrane potential‐sensitive dyes in isolated SMCs might elucidate the effect of pregnancy on the UA SMC membrane potential and the role of the BKCa channel in this regulation. Moreover, studies exploring Ca2+ dynamics in isolated UAs or individual SMCs at different stages of pregnancy could elucidate whether changes in Ca2+ sparks coupled to BKCa‐dependent spontaneous transient outward currents, as shown in other vascular beds (Jaggar et al. 2000), contribute to pregnancy‐dependent vasodilatation of UAs.
In SMCs, as in other cell types, BKCa channel voltage and Ca2+ sensitivity are modulated by the auxiliary β‐subunit. The β1‐subunit helps regulate vascular tone (Brenner et al. 2000; Leo et al. 2014), as evidenced by the fact that β1 knockout mice have altered myogenic tone and high blood pressure (Brenner et al. 2000; Pluger et al. 2000). In the UA, β1‐subunit protein expression is higher in pregnant sheep than in non‐pregnant animals (Rosenfeld et al. 2009; Hu et al. 2011) as a result of 17β‐oestradiol (Nagar et al. 2005; Hu et al. 2011) and epigenetic regulation of the β1‐subunit gene promoter (Chen et al. 2014), suggesting one mechanism to enhance activity of the BKCa channel.
BKCa is also regulated by the γ1‐subunit in other cell types. For example, in prostate cancer cells, the γ1‐subunit increases the voltage sensitivity of BKCa and allows its activation at resting membrane potential and low intracellular Ca2+ concentrations (Yan & Aldrich, 2010). Recently, Evanson et al. (2014) showed that the γ1‐subunit can increase both BKCa channel activity in vascular SMCs and BKCa‐induced vasodilatation of cerebral arteries (Evanson et al. 2014). Here, we found that knockdown of the γ1‐subunit partially reduced BKCa channel activity in UAs from P14 and P18 mice. Conversely, overexpression of the γ1‐subunit increased activity of the channel in UAs from NP mice. Confirming these findings, we found that UAs from γ1−/− P18 mice had smaller diameters and lower BKCa activity, measured as response to IbTX, than those from WT P18 mice. These observations suggest that the γ1‐subunit is, in part, responsible for the enhanced activation of BKCa channels and likely contributes to the resulting vasodilatation of the UA during pregnancy. Consistent with this idea, we found that the level of γ1 mRNA was higher in UAs from P14 and P18 mice than in those from NP mice. Unfortunately, we were unable to measure the level of γ1‐subunit protein or its association with BKCa during pregnancy because we lacked a sufficiently sensitive antibody. Future immunoblot and protein–protein interaction studies might reveal whether γ1‐subunit interaction with BKCa channel increases in UAs during pregnancy. Both oestrogen and progesterone increase the expression of the β1‐subunit in UAs (Nagar et al. 2005; Hu et al. 2011); thus, γ1 expression might also be hormonally regulated in UAs during pregnancy.
We speculate that the mid and high voltage sensitivities we observed in UA SMCs at P14 and P18 were due to the association of BKCa with the β1‐ and γ1‐subunits, respectively. The mid voltage sensitive channels were activated with a V 0.5 around −40 mV, which is comparable to that found in plasma membrane patches containing BKCa α‐ and β1‐subunits (Lippiat et al. 2003; Liu et al. 2014; Lorca et al. 2014). Although β1‐subunit mRNA levels in UAs did not significantly differ between NP, P14 and P18 mice, β1‐subunit protein levels may differ. For instance, the β1 protein level increases in UAs from pregnant sheep without significant change in mRNA level (Rosenfeld et al. 2009). The high voltage sensitive channels were activated with a V 0.5 around −80 mV, similar to that observed in the presence of the γ1‐subunit in other cell types (Yan & Aldrich, 2010, 2012). However, we note that the difference in γ1‐subunit mRNA level between UAs from pregnant and non‐pregnant mice is much bigger than the difference in numbers of low, mid and high voltage sensitivity channels. This may be because we extracted mRNA from whole UAs pooled from several mice but performed patch clamp on only a few cells within each UA.
Another limitation of our study is the apparent discrepancy between our in vivo and in vitro data. Given our finding that in vivo UA resistance was similar between BKCa −/− and WT mice at P18, we expected to find that in vitro UA diameters would be similar. Instead, UA diameter was smaller in BKCa −/− than in WT mice at P18. This difference could be due to the influence of the endothelium, which influences adaptation of the UA to pregnancy (Bird et al. 2000). The endothelium produces relaxing factors such as prostaglandins, nitric oxide and endothelial‐derived hyperpolarizing factor, all of which modulate BKCa channels in vascular SMCs (Tanaka et al. 2004). Thus, we specifically removed the endothelium for our in vitro studies to assess effects of BKCa on the smooth muscle layer. The fact that UA resistance did not increase as UA diameter decreased in BKCa −/− mice could indicate structural changes in arterioles, spiral arteries or placental vasculature, which could be assessed by microcomputed tomography. In this context, in mice, placentas become progressively thicker from P10.5 to around P14, then thickness remains constant until term (Mu et al. 2008). Our ultrasound studies confirmed that placental thickness in WT mice did not differ between P14 and P18. However, placentas in P18 BKCa −/− dams were thicker than those in P14 dams, suggesting differential placental growth. Given that performing Doppler imaging of UAs in NP mice is technically demanding, we were unable to measure UA vascular resistance in NP animals. Thus, we could not determine whether the lower UA resistance observed in BKCa −/− mice than in WT mice at P14 was specific to pregnancy or reflected an overall lower UA resistance in BKCa −/− mice. Another possibility is that compensation by other ion channels maintains a low UA resistance during pregnancy. Supporting this idea, a reduction in L‐type Ca2+ currents and increase in protein kinase A expression have been described in urinary bladder SMCs from global BKCa −/− mice, but not in mice with inducible SMC‐specific deletion of BKCa channels (Sprossmann et al. 2009). Thus, it is possible that compensatory mechanisms are also present in vascular SMCs in BKCa −/− mice.
Throughout pregnancy, both functional vasodilatation and structural remodelling result in changes in UA blood flow. Structural remodelling involves an increase in UA diameter (Cipolla & Osol, 1994; Hilgers et al. 2003; Mu & Adamson, 2006). In mice, the increase in UA diameter during pregnancy is accompanied by an increase in wall thickness and SMC hyperplasia (van der Heijden et al. 2005), and BKCa channels have been proposed to regulate cell proliferation (Bloch et al. 2007; Coiret et al. 2007). We observed a pregnancy‐dependent increase in the UA wall thickness in WT mice, but we did not find differences in wall thickness between NP and any pregnant stage in BKCa −/− mice, suggesting that this channel could play a role in the pregnancy‐induced hyperplasia of the UA SMCs and the resulting structural remodelling of these vessels.
Previous studies have shown that abnormal utero‐placental perfusion, due to reduced adaptation and/or dysfunction of the UA, contributes to the development of IUGR in humans (Konje et al. 2003) and rodents (Cotechini et al. 2014; Janot et al. 2014). In mice, bilateral ligation of the UA is a proposed model of IUGR (Janot et al. 2014), whereas in humans, women with lower uterine artery blood flow are at increased risk of developing IUGR and adverse pregnancy outcomes (Konje et al. 2003; Shwarzman et al. 2013). Here, we found that loss of BKCa did not affect pup size, measured as crown–rump length by ultrasound, a good predictor of fetal body weight and gestational age in mice (Mu et al. 2008). This is in contrast to an earlier report that BKCa −/− pups were smaller than their WT littermates (Meredith et al. 2004). However, in our study, all pups from WT or BKCa −/− dams were WT or BKCa +/−, respectively, indicating that loss of one copy of BKCa is insufficient to reduce pup size. Although fetal sizes were similar between the genotypes, the BKCa −/− dams had fewer pups than WT dams. A reduced number of pups, and hence lower UA resistance, could overcome the altered utero‐placental perfusion due to dysfunction in UA vasodilatation. Further studies might elucidate whether the decreased pup number reflects a defect in implantation or increased resorption in response to abnormal UA flow.
In this study, we described a key role of the BKCa channel in modulating UA diameter changes at late pregnancy in mice. We proposed that the γ1‐subunit participates in this process by increasing the activation of BKCa channels during pregnancy. Understanding the mechanisms underlying the functional and structural remodelling of the UA during pregnancy might contribute to the development of strategies to prevent dysfunctions of UA remodelling, which is essential for proper fetal development and maternal health.
Additional information
Competing interests
None declared.
Author contributions
R.A.L. and S.K.E. designed the study; R.A.L., M.W.‐P., W.E.F. and M.K.P. acquired and analysed the data; R.A.L., M.W.‐P. and S.K.E. interpreted the results and wrote the manuscript. All authors have approved the final version of the manuscript and agree to be accountable for all aspects of the work. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Funding
This work was supported by National Institutes of Health grant 5R01HD037831 and March of Dimes grant FY15‐147 to S.K.E., and American Heart Association Postdoctoral fellowship AHA/12POST10660000 to R.A.L.
Acknowledgements
We thank Dr Deborah J. Frank, Dr Kathryn G. Lamping and Dr Lorna G. Moore for critical reading and editing of the manuscript, Dr Molly J. Stout for scientific discussion, Dr Andrea L. Meredith for providing the BKCa −/− mice, Dr Christopher J. Lingle for providing the γ1−/− mice, Mrs Elise S. Bales for technical assistance with immunohistochemistry studies, the Cardiovascular Mouse Phenotyping Core at Washington University in St Louis for Doppler imaging assistance, and The Genome Institute at Washington University in St Louis for shRNA synthesis.
Biography
Ramón A. Lorca received his PhD in Physiology from the P. Catholic University of Chile in 2008. He did postdoctoral training at the University of Iowa and Washington University in St Louis. He is currently an instructor in the Department of Obstetrics and Gynecology at the University of Colorado Denver. His research interests focus on the regulation of vascular and uterine smooth muscle cell excitability during pregnancy.

Edited by: Laura Bennet & Kim Dora
This is an Editor's Choice article from the 15 March 2018 issue.
References
- Alioua A, Lu R, Kumar Y, Eghbali M, Kundu P, Toro L & Stefani E (2008). Slo1 caveolin‐binding motif, a mechanism of caveolin‐1‐Slo1 interaction regulating Slo1 surface expression. J Biol Chem 283, 4808–4817. [DOI] [PubMed] [Google Scholar]
- Bird IM, Sullivan JA, Di T, Cale JM, Zhang L, Zheng J & Magness RR (2000). Pregnancy‐dependent changes in cell signaling underlie changes in differential control of vasodilator production in uterine artery endothelial cells. Endocrinology 141, 1107–1117. [DOI] [PubMed] [Google Scholar]
- Bloch M, Ousingsawat J, Simon R, Schraml P, Gasser TC, Mihatsch MJ, Kunzelmann K & Bubendorf L (2007). KCNMA1 gene amplification promotes tumor cell proliferation in human prostate cancer. Oncogene 26, 2525–2534. [DOI] [PubMed] [Google Scholar]
- Brainard AM, Miller AJ, Martens JR & England SK (2005). Maxi‐K channels localize to caveolae in human myometrium: a role for an actin‐channel‐caveolin complex in the regulation of myometrial smooth muscle K+ current. Am J Physiol Cell Physiol 289, C49–C57. [DOI] [PubMed] [Google Scholar]
- Brayden JE & Nelson MT (1992). Regulation of arterial tone by activation of calcium‐dependent potassium channels. Science 256, 532–535. [DOI] [PubMed] [Google Scholar]
- Brenner R, Perez GJ, Bonev AD, Eckman DM, Kosek JC, Wiler SW, Patterson AJ, Nelson MT & Aldrich RW (2000). Vasoregulation by the β1 subunit of the calcium‐activated potassium channel. Nature 407, 870–876. [DOI] [PubMed] [Google Scholar]
- Browne VA, Toledo‐Jaldin L, Davila RD, Lopez LP, Yamashiro H, Cioffi‐Ragan D, Julian CG, Wilson MJ, Bigham AW, Shriver MD, Honigman B, Vargas E, Roach R & Moore LG (2011). High‐end arteriolar resistance limits uterine artery blood flow and restricts fetal growth in preeclampsia and gestational hypertension at high altitude. Am J Physiol Regul Integr Comp Physiol 300, R1221–R1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen M, Dasgupta C, Xiong F & Zhang L (2014). Epigenetic upregulation of large‐conductance Ca2+‐activated K+ channel expression in uterine vascular adaptation to pregnancy. Hypertension 64, 610–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheranov SY & Jaggar JH (2004). Mitochondrial modulation of Ca2+ sparks and transient KCa currents in smooth muscle cells of rat cerebral arteries. J Physiol 556, 755–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiswick ML (1985). Intrauterine growth retardation. Br Med J (Clin Res Ed) 291, 845–848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cipolla M & Osol G (1994). Hypertrophic and hyperplastic effects of pregnancy on the rat uterine arterial wall. Am J Obstet Gynecol 171, 805–811. [DOI] [PubMed] [Google Scholar]
- Coiret G, Borowiec AS, Mariot P, Ouadid‐Ahidouch H & Matifat F (2007). The antiestrogen tamoxifen activates BK channels and stimulates proliferation of MCF‐7 breast cancer cells. Mol Pharmacol 71, 843–851. [DOI] [PubMed] [Google Scholar]
- Cotechini T, Komisarenko M, Sperou A, Macdonald‐Goodfellow S, Adams MA & Graham CH (2014). Inflammation in rat pregnancy inhibits spiral artery remodeling leading to fetal growth restriction and features of preeclampsia. J Exp Med 211, 165–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curley M, Morrison JJ & Smith TJ (2004). Analysis of Maxi‐K alpha subunit splice variants in human myometrium. Reprod Biol Endocrinol 2, 67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evanson KW, Bannister JP, Leo MD & Jaggar JH (2014). LRRC26 is a functional BK channel auxiliary gamma subunit in arterial smooth muscle cells. Circ Res 115, 423–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ford SP (1982). Control of uterine and ovarian blood flow throughout the estrous cycle and pregnancy of ewes, sows and cows. J Anim Sci 55, Suppl. 2, 32–42. [PubMed] [Google Scholar]
- Gutkowska J, Jankowski M, Lambert C, Mukaddam‐Daher S, Zingg HH & McCann SM (1997). Oxytocin releases atrial natriuretic peptide by combining with oxytocin receptors in the heart. Proc Natl Acad Sci USA 94, 11704–11709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilgers RH, Bergaya S, Schiffers PM, Meneton P, Boulanger CM, Henrion D, Levy BI & De Mey JG (2003). Uterine artery structural and functional changes during pregnancy in tissue kallikrein‐deficient mice. Arterioscler Thromb Vasc Biol 23, 1826–1832. [DOI] [PubMed] [Google Scholar]
- Hu XQ, Xiao D, Zhu R, Huang X, Yang S, Wilson S & Zhang L (2011). Pregnancy upregulates large‐conductance Ca2+‐activated K+ channel activity and attenuates myogenic tone in uterine arteries. Hypertension 58, 1132–1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson WF, Huebner JM & Rusch NJ (1997). Enzymatic isolation and characterization of single vascular smooth muscle cells from cremasteric arterioles. Microcirculation 4, 35–50. [DOI] [PubMed] [Google Scholar]
- Jaggar JH, Porter VA, Lederer WJ & Nelson MT (2000). Calcium sparks in smooth muscle. Am J Physiol Cell Physiol 278, C235–C256. [DOI] [PubMed] [Google Scholar]
- Janot M, Cortes‐Dubly ML, Rodriguez S & Huynh‐Do U (2014). Bilateral uterine vessel ligation as a model of intrauterine growth restriction in mice. Reprod Biol Endocrinol 12, 62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ketsawatsomkron P, Lorca RA, Keen HL, Weatherford ET, Liu X, Pelham CJ, Grobe JL, Faraci FM, England SK & Sigmund CD (2012). PPARγ regulates resistance vessel tone through a mechanism involving RGS5‐mediated control of protein kinase C and BKCa channel activity. Circ Res 111, 1446–1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knaus HG, Folander K, Garcia‐Calvo M, Garcia ML, Kaczorowski GJ, Smith M & Swanson R (1994). Primary sequence and immunological characterization of β‐subunit of high conductance Ca2+‐activated K+ channel from smooth muscle. J Biol Chem 269, 17274–17278. [PubMed] [Google Scholar]
- Knot HJ & Nelson MT (1998). Regulation of arterial diameter and wall [Ca2+] in cerebral arteries of rat by membrane potential and intravascular pressure. J Physiol 508, 199–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konje JC, Howarth ES, Kaufmann P & Taylor DJ (2003). Longitudinal quantification of uterine artery blood volume flow changes during gestation in pregnancies complicated by intrauterine growth restriction. BJOG 110, 301–305. [PubMed] [Google Scholar]
- Korovkina VP, Fergus DJ, Holdiman AJ & England SK (2001). Characterization of a novel 132‐bp exon of the human maxi‐K channel. Am J Physiol Cell Physiol 281, C361–C367. [DOI] [PubMed] [Google Scholar]
- Leo MD, Bannister JP, Narayanan D, Nair A, Grubbs JE, Gabrick KS, Boop FA & Jaggar JH (2014). Dynamic regulation of β1 subunit trafficking controls vascular contractility. Proc Natl Acad Sci USA 111, 2361–2366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesh RE, Somlyo AP, Owens GK & Somlyo AV (1995). Reversible permeabilization. A novel technique for the intracellular introduction of antisense oligodeoxynucleotides into intact smooth muscle. Circ Res 77, 220–230. [DOI] [PubMed] [Google Scholar]
- Lippiat JD, Standen NB, Harrow ID, Phillips SC & Davies NW (2003). Properties of BKCa channels formed by bicistronic expression of hSloα and β1–4 subunits in HEK293 cells. J Membr Biol 192, 141–148. [DOI] [PubMed] [Google Scholar]
- Liu HW, Hou PP, Guo XY, Zhao ZW, Hu B, Li X, Wang LY, Ding JP & Wang S (2014). Structural basis for calcium and magnesium regulation of a large conductance calcium‐activated potassium channel with β1 subunits. J Biol Chem 289, 16914–16923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorca RA, Stamnes SJ, Pillai MK, Hsiao JJ, Wright ME & England SK (2014). N‐terminal isoforms of the large‐conductance Ca2+‐activated K+ channel are differentially modulated by the auxiliary β1‐subunit. J Biol Chem 289, 10095–10103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu T, Zhang DM, Wang XL, He T, Wang RX, Chai Q, Katusic ZS & Lee HC (2010). Regulation of coronary arterial BK channels by caveolae‐mediated angiotensin II signaling in diabetes mellitus. Circ Res 106, 1164–1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McManus OB, Helms LM, Pallanck L, Ganetzky B, Swanson R & Leonard RJ (1995). Functional role of the beta subunit of high conductance calcium‐activated potassium channels. Neuron 14, 645–650. [DOI] [PubMed] [Google Scholar]
- Meredith AL, Thorneloe KS, Werner ME, Nelson MT & Aldrich RW (2004). Overactive bladder and incontinence in the absence of the BK large conductance Ca2+‐activated K+ channel. J Biol Chem 279, 36746–36752. [DOI] [PubMed] [Google Scholar]
- Mu J & Adamson SL (2006). Developmental changes in hemodynamics of uterine artery, utero‐ and umbilicoplacental, and vitelline circulations in mouse throughout gestation. Am J Physiol Heart Circ Physiol 291, H1421–H1428. [DOI] [PubMed] [Google Scholar]
- Mu J, Slevin JC, Qu D, McCormick S & Adamson SL (2008). In vivo quantification of embryonic and placental growth during gestation in mice using micro‐ultrasound. Reprod Biol Endocrinol 6, 34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagar D, Liu XT & Rosenfeld CR (2005). Estrogen regulates β1‐subunit expression in Ca2+‐activated K+ channels in arteries from reproductive tissues. Am J Physiol Heart Circ Physiol 289, H1417–H1427. [DOI] [PubMed] [Google Scholar]
- Nelson MT, Cheng H, Rubart M, Santana LF, Bonev AD, Knot HJ & Lederer WJ (1995). Relaxation of arterial smooth muscle by calcium sparks. Science 270, 633–637. [DOI] [PubMed] [Google Scholar]
- Osol G & Cipolla M (1993). Pregnancy‐induced changes in the three‐dimensional mechanical properties of pressurized rat uteroplacental (radial) arteries. Am J Obstet Gynecol 168, 268–274. [DOI] [PubMed] [Google Scholar]
- Osol G & Moore LG (2014). Maternal uterine vascular remodeling during pregnancy. Microcirculation 21, 38–47. [DOI] [PubMed] [Google Scholar]
- Palmer SK, Zamudio S, Coffin C, Parker S, Stamm E & Moore LG (1992). Quantitative estimation of human uterine artery blood flow and pelvic blood flow redistribution in pregnancy. Obstet Gynecol 80, 1000–1006. [PubMed] [Google Scholar]
- Pluger S, Faulhaber J, Furstenau M, Lohn M, Waldschutz R, Gollasch M, Haller H, Luft FC, Ehmke H & Pongs O (2000). Mice with disrupted BK channel beta1 subunit gene feature abnormal Ca2+ spark/STOC coupling and elevated blood pressure. Circ Res 87, E53–60. [DOI] [PubMed] [Google Scholar]
- Rada CC, Pierce SL, Nuno DW, Zimmerman K, Lamping KG, Bowdler NC, Weiss RM & England SK (2012). Overexpression of the SK3 channel alters vascular remodeling during pregnancy, leading to fetal demise. Am J Physiol Endocrinol Metab 303, E825–E831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenfeld CR, Cornfield DN & Roy T (2001). Ca2+‐activated K+ channels modulate basal and E2β‐induced rises in uterine blood flow in ovine pregnancy. Am J Physiol Heart Circ Physiol 281, H422–H431. [DOI] [PubMed] [Google Scholar]
- Rosenfeld CR, Liu XT & DeSpain K (2009). Pregnancy modifies the large conductance Ca2+‐activated K+ channel and cGMP‐dependent signaling pathway in uterine vascular smooth muscle. Am J Physiol Heart Circ Physiol 296, H1878–H1887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenfeld CR, Word RA, DeSpain K & Liu XT (2008). Large conductance Ca2+‐activated K+ channels contribute to vascular function in nonpregnant human uterine arteries. Reprod Sci 15, 651–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shwarzman P, Waintraub AY, Frieger M, Bashiri A, Mazor M & Hershkovitz R (2013). Third‐trimester abnormal uterine artery Doppler findings are associated with adverse pregnancy outcomes. J Ultrasound Med 32, 2107–2113. [DOI] [PubMed] [Google Scholar]
- Sprossmann F, Pankert P, Sausbier U, Wirth A, Zhou XB, Madlung J, Zhao H, Bucurenciu I, Jakob A, Lamkemeyer T, Neuhuber W, Offermanns S, Shipston MJ, Korth M, Nordheim A, Ruth P & Sausbier M (2009). Inducible knockout mutagenesis reveals compensatory mechanisms elicited by constitutive BK channel deficiency in overactive murine bladder. FEBS J 276, 1680–1697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka Y, Koike K & Toro L (2004). MaxiK channel roles in blood vessel relaxations induced by endothelium‐derived relaxing factors and their molecular mechanisms. J Smooth Muscle Res 40, 125–153. [DOI] [PubMed] [Google Scholar]
- Tanaka Y, Meera P, Song M, Knaus HG & Toro L (1997). Molecular constituents of maxi KCa channels in human coronary smooth muscle: predominant α + β subunit complexes. J Physiol 502, 545–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Heijden OW, Essers YP, Spaanderman ME, De Mey JG, van Eys GJ & Peeters LL (2005). Uterine artery remodeling in pseudopregnancy is comparable to that in early pregnancy. Biol Reprod 73, 1289–1293. [DOI] [PubMed] [Google Scholar]
- Yan J & Aldrich RW (2010). LRRC26 auxiliary protein allows BK channel activation at resting voltage without calcium. Nature 466, 513–516. [DOI] [PubMed] [Google Scholar]
- Yan J & Aldrich RW (2012). BK potassium channel modulation by leucine‐rich repeat‐containing proteins. Proc Natl Acad Sci USA 109, 7917–7922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang C, Gonzalez‐Perez V, Mukaibo T, Melvin JE, Xia XM & Lingle CJ (2017). Knockout of the LRRC26 subunit reveals a primary role of LRRC26‐containing BK channels in secretory epithelial cells. Proc Natl Acad Sci USA 114, E3739–E3747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu N, Eghbali M, Helguera G, Song M, Stefani E & Toro L (2005). Alternative splicing of Slo channel gene programmed by estrogen, progesterone and pregnancy. FEBS Lett 579, 4856–4860. [DOI] [PubMed] [Google Scholar]