

Abstract
Key points
Skeletal muscle contractile activity is associated with an enhanced reactive oxygen species (ROS) generation.
At very low , ROS generation by mitochondria can be elevated in intact cells.
An elevated intracellular oxidant activity may affect muscle force development and recovery from fatigue.
We treated intact single muscle fibres with a mitochondrial antioxidant and stimulated the fibres to contract at a low extracellular that is similar to the intracellular that is observed during moderate to intense exercise in vivo.
The mitochondrial antioxidant prevented a sustained decrease in the myofibrillar Ca2+ sensitivity and improved muscle submaximal force development after fatigue at low extracellular .
Abstract
Skeletal muscle can develop a prolonged low frequency‐stimulation force depression (PLFFD) following fatigue‐inducing contractions. Increased levels of reactive oxygen species (ROS) have been implicated in the development of PLFFD. During exercise the skeletal muscle intracellular decreases to relatively low levels, and can be further decreased when there is an impairment in O2 diffusion or availability, such as in certain chronic diseases and during exercise at high altitude. Since ROS generation by mitochondria is elevated at very low in cells, we tested the hypothesis that treatment of muscle fibres with a mitochondrial‐targeted antioxidant at a very low, near hypoxic, can attenuate PLFFD. We treated intact single fibres from mice with the mitochondrial‐specific antioxidant SS31, and measured force development and intracellular [Ca2+] 30 min after fatigue at an extracellular of ∼5 Torr. After 30 min following the end of the fatiguing contractions, fibres treated with SS31 showed significantly less impairment in force development compared to untreated fibres at submaximal frequencies of stimulation. The cytosolic peak [Ca2+] transients (peak [Ca2+]c) were equally decreased in both groups compared to pre‐fatigue values. The combined force and peak [Ca2+]c data demonstrated that myofibrillar Ca2+ sensitivity was diminished in the untreated fibres 30 min after fatigue compared to pre‐fatigue values, but Ca2+ sensitivity was unaltered in the SS31 treated fibres. These results demonstrate that at a very low , treatment of skeletal muscle fibres with a mitochondrial antioxidant prevents a decrease in the myofibrillar Ca2+ sensitivity, which alleviates the fatigue induced PLFFD.
Keywords: skeletal muscle, fatigue, hypoxia
Key points
Skeletal muscle contractile activity is associated with an enhanced reactive oxygen species (ROS) generation.
At very low , ROS generation by mitochondria can be elevated in intact cells.
An elevated intracellular oxidant activity may affect muscle force development and recovery from fatigue.
We treated intact single muscle fibres with a mitochondrial antioxidant and stimulated the fibres to contract at a low extracellular that is similar to the intracellular that is observed during moderate to intense exercise in vivo.
The mitochondrial antioxidant prevented a sustained decrease in the myofibrillar Ca2+ sensitivity and improved muscle submaximal force development after fatigue at low extracellular .
Introduction
Following a period of fatigue inducing contractions, skeletal muscle can demonstrate a long lasting impairment in submaximal force development that has been named “prolonged low frequency force depression” (PLFFD) (Edwards et al. 1977; Bruton et al. 2008; Balog, 2010; Cheng et al. 2015). PLFFD can last for up to 24 h or more in humans (Edwards et al. 1977; Place et al. 2015). Therefore, understanding the alterations that occur in skeletal muscle following fatigue may be of great relevance for the development of strategies to avoid a reduction in muscle function during and after strenuous physical activity. This is particularly important under conditions in which fatigue resistance is impaired by tissue hypoxia, such as during exercise at high altitude or for different chronic diseases states as in chronic obstructive pulmonary disease (COPD) (Gea et al. 2013) and chronic heart failure (CHF) (Rehn et al. 2012).
The mechanisms causing PLFFD remain unclear (Bruton et al. 2008; Balog, 2010; Place et al. 2015; Watanabe et al. 2015). One of the proposed mechanisms is that an increased generation of reactive oxygen species (ROS) resulting from a fatiguing contractile bout leads to oxidative modifications of proteins that will decrease the sarcoplasmic reticulum (SR) Ca2+ release and myofibrillar Ca2+ sensitivity during PLFFD (Bruton et al. 2008; Cheng et al. 2015; Watanabe et al. 2015). However, most of the studies examining PLFFD in isolated muscle models have been performed at supraphysiological extracellular (≥150 Torr) oxygen tensions () (Bruton et al. 2008; Cheng et al. 2015). Since ROS generation in various cellular compartments can be significantly affected by the (Clanton, 2007; Clanton et al. 2013), it is important to determine the role of ROS on the development of PLFFD under physiological, low conditions. During moderate to high intensity exercise the intracellular in human skeletal muscle fibres in vivo is quite low, decreasing from ∼30 Torr at rest to ∼3–5 Torr or less (Richardson et al. 2001, 2015; Wagner, 2012). Such physiological range of intracellular (i.e. 3–30 Torr) should not be restrictive to the rate of mitochondrial oxidative phosphorylation during exercise in healthy subjects (Wagner, 2012) due to the high affinity of cytochrome c oxidase for oxygen (Wilson et al. 1988). Conversely, the O2 availability can limit oxidative phosphorylation (i.e. hypoxia) (Connett et al. 1990) in highly trained subjects during maximal exercise (i.e. exercise induced hypoxia), or when exercise is performed at high altitude or in certain chronic disease patients (e.g. COPD and CHF) (Esposito et al. 2010; Poole & Jones, 2012; Cano et al. 2015; Hirai et al. 2015). In accordance with the idea that relatively low extracellular values are necessary to limit oxidative phosphorylation in muscle fibres, the critical extracellular estimated for rat skeletal myofibres working at their maximal oxygen consumption rate was estimated to be ∼4–6 Torr (Wüst et al. 2009). While controversial, it has been suggested that at very low , mitochondria may have an important role in ROS generation in intact contracting skeletal muscle as it has been demonstrated for other cell types (Guzy et al. 2005; Guzy & Schumacker, 2006; Clanton, 2007; Waypa et al. 2010; Clanton et al. 2013). Yet, the exact molecular mechanisms underlying ROS generation in skeletal muscle fibres, and potentially in skeletal muscle mitochondria, during contractions under near hypoxic are not clear.
The purpose of the present study was to test the hypothesis that under a very low extracellular , an exogenous mitochondrial‐targeted antioxidant can blunt or attenuate the development of PLFFD. We measured whether intact single muscle fibres treated with the mitochondrial‐targeted antioxidant SS31 preserves force development, free intracellular Ca2+ levels, and myofibrillar Ca2+ sensitivity 30 min after fatigue at an extracellular of 5 Torr. The results of the present study demonstrated that at near hypoxic conditions, treatment of skeletal muscle fibres with SS31 avoided a decrease in myofibrillar Ca2+ sensitivity and resulted in higher submaximal force development after fatigue.
Methods
Ethical approval
All experiments were approved by the University of California San Diego institutional animal care and use committee (IACUC) and complied with the National Institutes of Health (NIH) Guide for the Care and Use of Laboratory Animals in Research, all the regulations issued by the United States Department of Agriculture (USDA), and all regulations issued by the USDA implementing the Animal Welfare act. Male C57BL/6J mice (total of 14 mice) were obtained from The Jackson Laboratory (Bar Harbor, ME, USA). Animals were fed a standard diet and had access to food and water ad libitum. The animals (12–13 weeks old) were killed by an overdose of sodium pentobarbital administered by intraperitoneal injection, followed by cervical dislocation. Immediately after the animals were killed flexor digitorum brevis (FDB) muscles from both hindlimb feet were dissected.
Single fibre preparation, force and [Ca2+]c measurement
Isolated single muscle fibres with tendons intact from FDB muscles were mechanically dissected under dark field illumination at room temperature with dissecting solution (∼22°C; 136 mm NaCl, 5 mm KCl, 1.8 mm CaCl2, 0.5 mm MgCl2, 0.4 mm NaH2PO4, 11.9 mm NaHCO3 and 5.5 mm glucose). After dissection, each fibre was pressure injected with the Ca2+ sensitive fluorescent probe FURA‐2 (Invitrogen, Carlsbad, CA, USA) as previously described (Nogueira et al. 2013), and allowed to rest for 60 min. Platinum clips were attached to each tendon of the fibre. The fibre was mounted on a Small Intact Muscle Apparatus (model no. 801C, Aurora Scientific Inc., Aurora, ON, Canada) and placed on the stage of an inverted microscope. Tetanic contractions were evoked by parallel platinum electrodes (250 ms train duration, 0.5 ms biphasic pulses, 8 V) using a Grass S88X stimulator (Quincy, MA, USA). Force development was measured by a force transducer (model no. 403A, Aurora Scientific Inc.). The fibre length was adjusted to achieve the maximal isometric tetanic tension (L 0; set with 100 Hz stimulations), allowed to rest for 10 min, and perfused with Tyrode solution (121 mm NaCl, 5 mm KCl, 1.8 mm CaCl2, 0.5 mm MgCl2, 0.4 mm NaH2PO4, 24 mm NaHCO3, 5.5 mm glucose and 0.1 mm K2EGTA) which was continuously bubbled with 5% O2, 5% CO2 and N2 balance.
FURA‐2 fluorescence was measured combined with force measurements. FURA‐2 injected fibres were illuminated with two rapidly alternating (200 Hz) excitation wavelengths of 340 nm and 380 nm, and the ratio of fluorescence excitation (340 nm/380 nm; R) at an emission length of 510 nm was obtained. Fluorescence was converted to [Ca2+]c according to the following equation (Nogueira et al. 2013):
| (1) |
In this equation K D is the dissociation constant for Ca2+‐FURA‐2 and was set to 224 nm (Westerblad & Allen, 1991). β is the fluorescence ratio between high and no [Ca2+]c at 380 nm. R min and R max are the fluorescence ratios when Ca2+‐FURA‐2 binding is absent and saturated, respectively. R min was determined using an internal in vivo calibration described by Kabbara and Allen (Kabbara & Allen, 1999) with modifications (Nogueira et al. 2013). To determine R min, fibres were incubated for 30 min with a no‐calcium Ringer–EGTA solution (116.5 mm NaCl, 2 mm KCl, 2 mm NaH2PO4, 10 mm EGTA, at pH 7.3) supplemented with 30 mm caffeine. Fibres were then incubated with 100 μm BAPTA‐AM (the acetoxymethyl ester form of BAPTA), solubilized in DMSO (final [DMSO] = 1%), and incubated in Ringer–EGTA solution for 30 min, followed by washout with Ringer–EGTA solution for an additional 20 min. R min was determined as the smallest R value observed. To determine R max, another group of fibres were treated with 10 mm caffeine and 500 μm 2,5‐di‐tert‐butylhydroquinone (TBQ), a sarco(endo)plasmic reticulum Ca2+‐ATPase (SERCA) inhibitor, and stimulated at 120 Hz with 2 s train duration. To obtain the peak [Ca2+]c during contractions, the average FURA‐2 fluorescence ratio in the final 100 ms of stimulation was used.
The force–[Ca2+]c relationship was accessed in each fibre before and 30 min after fatigue by fitting the force relative to the maximum against the estimated peak tetanic [Ca2+]c for tetanic contractions evoked at 10–120 Hz with the following Hill equation (for details see Westerblad & Allen, 1993; Nogueira & Hogan, 2010).
| (2) |
where P is the relative force, P max is the force at saturating [Ca2+]c, Ca2+ 50 is the mid‐point of the force–[Ca2+]c relationship and N is a constant which describes the steepness of the function.
Since temperature can affect ROS generation, the fatigue protocol was performed at 28°C because it mimics the temperature of the FDB muscle in vivo (Bruton et al. 1998). In preliminary experiments, we observed a rapid loss of the FURA‐2 emission response to Ca2+ when fibres were kept at temperatures above 28°C and a very slow loss at 22°C. Due to the long‐lasting nature of the present experiments, the temperature was kept at 28°C only during the time period of the fatigue protocol and at 22°C in the remaining time of the experiment to avoid loss of the FURA‐2 fluorescent response to Ca2+.
SS31 treatment
Fibres were perfused with Tyrode solution in the absence (control) or presence of 1 μm SS31, a water soluble peptide that has antioxidant properties, but localizes primarily in the mitochondrial inner membrane (n = 7 fibres per group; each fibre was obtained from a different animal) (Zhao et al. 2004). The amount of SS31 used was based on an established dose–response curve for SS31 (Anderson et al. 2009). The average fibre diameter was not different between groups (35 ± 6 μm for control group and 36.8 ± 10 μm for the SS31 group).
Experimental protocol
The in the working chamber was measured by using a fibre optic oxygen sensor (Oxymicro, World precision Instruments, Sarasota, FL, USA), placed near the muscle fibre, throughout the experimental procedure, except when [Ca2+]c was measured to avoid interference from the light emitted by the O2 probe. Untreated and SS31 treated fibres were initially perfused with Tyrode solution continuously bubbled with a mixture of N2 and O2, so the final extracellular in the working chamber was either 40 Torr or 5 Torr. All gas mixtures contained 5% CO2 to maintain the solution's pH at ∼7.4. Each fibre was initially perfused with an extracellular of 40 Torr for 30 min, and stimulated to contract at different frequencies of pulses (1–150 Hz, 250 ms train duration, 0.5 ms biphasic pulses, 8 V) with 1 min intervals between contractions (to obtain a force–frequency curve) at 22°C. To minimize the contractile differences usually detected between the tested fibres, this work only used fibres in which the contraction force evoked at 30 Hz developed ∼40–60% of maximum tetanic force (52 ± 4% and 54 ± 5% of maximum force for untreated control and SS31 treated groups, respectively). After the first force–frequency curve at 40 Torr, the of the perfused solution was decreased to 5 Torr, which was kept until the end of the experimental procedure. In one group of fibres, 1 μm SS31 was added into the solution to scavenge mitochondrial ROS after the first force–frequency curve (22°C). Each fibre was equilibrated for 30 min with SS31, followed by a second force–frequency curve (pre‐fatigue force–frequency curve; 22°C). After a 10 min resting period, the temperature was raised to 28°C and each fibre performed a fatigue protocol. The fatigue protocol consisted of a series of repetitive contractions (stimulation trains) evoked by 100 Hz stimulations at a train frequency of 0.25 trains s−1 that was increased each minute to 0.33 and 0.5 trains s−1 as previously described (Gandra et al. 2012). The protocol was terminated when force was decreased to 30% of the initial, then the temperature was lowered to 22°C during recovery. To examine the fatigue‐induced PLFFD, a third force–frequency curve (22°C) was performed 30 min after the end of the fatigue protocol.
We have consistently observed for single fibres in our laboratory that the time to fatigue is significantly shorter when the extracellular is set to 5 Torr compared to 40 Torr (as exemplified in Fig. 1 A), which is similar to the resting mean capillary and intracellular in vivo (Richardson et al. 2006; Golub & Pittman, 2012). During a repetitive contractile period the oxygen consumption rate increases resulting in a decrease in the intracellular in a manner dependent on the extracellular as previously demonstrated for isolated fibres (Howlett et al. 2007). Therefore, it is very likely that during a fatigue protocol at 5 Torr the intracellular decreases to levels that limit oxidative phosphorylation in isolated fibres.
Figure 1. Single fibres treated with SS31 show no alterations in force generation and fatigue resistance at a low extracellular .
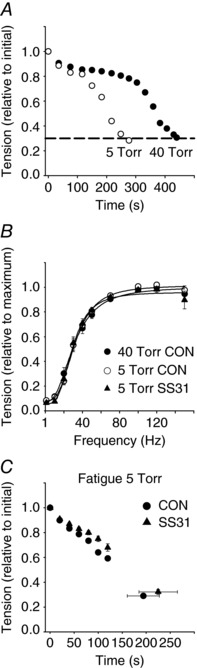
A, representative changes in tension generated by a single FDB fibre during fatiguing stimulations at an extracellular of 40 Torr (filled circles) and 5 Torr (open circles) under control conditions. B, force vs. stimulation frequency data from single fibres repeated after 60 min of constant perfusion at an extracellular of 40 Torr and 5 Torr, and 5 Torr in the presence of SS31 (40 Torr, filled circles; 5 Torr, open circles; and at 5 Torr plus SS31, filled triangles; n = 7). C, changes in tension generation during a fatiguing stimulation protocol at an extracellular of 5 Torr for control and SS31 treated fibres (control, filled circles; SS31, filled triangles; n = 7 fibres per group).
Statistics
Values are presented as mean ± SEM. Unpaired t test or two‐way ANOVA with Bonferroni's post‐test were used to determine statistically significant differences between untreated and SS31 treated groups as appropriate. Two‐way ANOVA repeated measures was used to determine with Bonferroni's post‐test intragroup differences. Analyses were performed using GraphPad PRISM version 4.00 software (GraphPad Software, La Jolla, CA, USA). The significance level was set at P < 0.05.
Results
Maximal and submaximal force development, as well as fatigue resistance, at very low extracellular were not altered by SS31
Neither SS31 incubation nor exposure to an extracellular of 5 Torr altered fibre maximal and submaximal force development evoked by different frequencies of stimulation under non‐fatiguing contractions (one contraction each 100 s; Fig. 1 B).
Treatment with SS31 did not affect fatigue resistance (determined by the time to fatigue) in single fibres stimulated at an extracellular of 5 Torr (Fig. 1 C). The time to observe a 70% fall in the initial tension during the fatigue protocol was 198 ± 29 s for control (untreated) fibres and 239 ± 38 s for SS31 treated fibres (P = 0.2661; n = 7; Fig. 1 C).
The mitochondrial antioxidant SS31 improved submaximal force development 30 min after fatigue at low extracellular
All fibres from the control and SS31 treated groups showed a complete recovery of the maximal tetanic force (evoked by high frequencies of pulse stimulation; i.e. 120 Hz) 30 min after the end of the fatigue protocol. Yet, fibres from both groups presented the expected PLFFD 30 min after fatigue (Fig. 2 A and B). Compared to pre‐fatigue values, the submaximal force development was significantly smaller 30 min after fatigue in control (between 20 and 70 Hz; Fig. 2 A) and in SS31 treated fibres (between 20 and 50 Hz; Fig. 2 B). However, fibres that were incubated with SS31 showed significantly greater force development during submaximal stimulation (between 30 and 50 Hz) 30 min after fatigue compared to control (e.g. at 40 Hz; 44 ± 4% vs. 60 ± 7% of maximal tetanic force for control and SS31, respectively, P < 0.01; Fig. 2 D).
Figure 2. Submaximal force generation is higher in SS31 treated single fibres 30 min after fatigue at low extracellular .
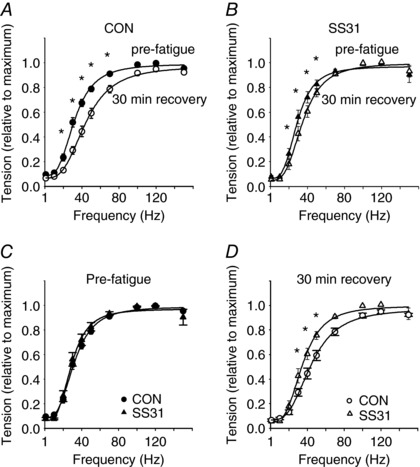
A, force vs. stimulation frequency data from control fibres before and after fatigue at 5 Torr (pre‐fatigue, filled circles; 30 min after fatigue, open circles; n = 7). B, force vs. stimulation frequency data from SS31 treated fibres before and after fatigue at 5 Torr (pre‐fatigue, filled triangles; 30 min after fatigue, open triangles; n = 7). C, pre‐fatigue force vs. stimulation frequency data from control fibres (filled circles; n = 7) and SS31 treated fibres (filled triangles; n = 7). D, force vs. stimulation frequency data obtained 30 min after fatigue from control fibres (filled circles; n = 7) and SS31 treated fibres (filled triangles; n = 7). * P < 0.05.
The mitochondrial antioxidant SS31 prevented the right‐shift in the force–Ca2+ relationship detected during PLFFD
The peak [Ca2+]c detected during submaximal and maximal contractions (between 10 and 120 Hz) was equally diminished (10–20% decrease) in the control and SS31 groups 30 min following the fatigue time point compared to pre‐fatigue values (Fig. 3 A). This is similar to the changes in peak [Ca2+]c reported in other studies examining PLFFD at supraphysiological (Cheng et al. 2015). To determine whether the effects detected on force when SS31 was present were due to changes in myofibrillar Ca2+ sensitivity, peak [Ca2+]c was plotted against peak force obtained from the force–frequency curves, before (unfatigued state) and 30 min after the fatigue protocol (PLFFD). In the control fibres, the force–peak [Ca2+]c relationship was right‐shifted during PLFFD (i.e. Ca2+ 50 was significantly increased from 618 ± 63 nm to 734 ± 90 nm, for pre‐fatigue and 30 min post‐fatigue, respectively; P < 0.05; Fig. 3 B). In contrast, fibres treated with SS31 did not show changes in the Ca2+ 50 (577 ± 36 nm vs. 560 ± 24 nm; pre‐fatigue and 30 min post‐fatigue, respectively; P = 0.273; Fig. 3 C). These data suggest that myofibrillar Ca2+ sensitivity was not different between pre‐fatigue compared to 30 min after fatigue when SS31 was present. This can also be noted in Fig. 3 D and E, which shows representative [Ca2+]c and force traces during submaximal stimulations in a control (Fig. 3 D) and an SS31 treated fibre (Fig. 3 E) before and 30 min after a fatigue protocol. The decrease in the [Ca2+]c 30 min after fatigue was similar for both fibres whereas the decline in the force developed was greater in the control fibre compared to SS31 treated fibre.
Figure 3. The prolonged decrease in myofibrillar Ca2+ sensitivity after fatigue at low was avoided by SS31.
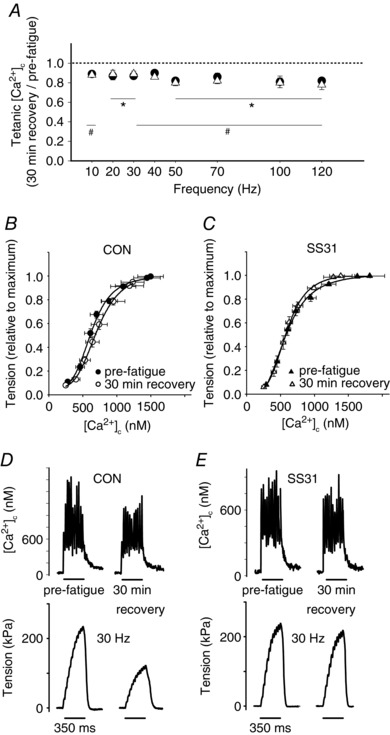
A, tetanic [Ca2+]c 30 min after fatigue relative to pre‐fatigue vs. stimulation frequency, data from control (filled circles; n = 7; * P < 0.05 vs. pre‐fatigue values) and SS31 treated fibres (open triangles; n = 7; # P < 0.05 vs. pre‐fatigue values). B, tension vs. [Ca2+]c data from control fibres (pre‐fatigue, filled circles; and 30 min after fatigue, open circles; at 5 Torr; n = 7). C, tension vs. [Ca2+]c data from SS31 treated fibres (pre‐fatigue, filled triangles; and 30 min after fatigue, open triangles; at 5 Torr; n = 7). D, representative [Ca2+]c and force generation records from a single control fibre during a submaximal stimulation (30 Hz) before fatigue and 30 min after fatigue at an extracellular of 5 Torr. E, [Ca2+]c and force generation records from a single fibre treated with SS31 during a submaximal stimulation (30 Hz) before fatigue and 30 min after fatigue at an extracellular of 5 Torr.
Discussion
The results of this study show that a mitochondrial antioxidant can mitigate the fatigue induced PLFFD in single skeletal muscle fibres at a very low extracellular . In isolated fibres at low extracellular , PLFFD resulted from a decrease in the peak tetanic Ca2+ concentration and in the myofibrillar Ca2+ sensitivity. When fibres were incubated with the mitochondrial‐specific antioxidant SS31, the prolonged decrease in myofibrillar Ca2+ sensitivity after fatigue was blunted. Thereby, SS31 partially prevents the fatigue‐induced prolonged fall in submaximal force development at low extracellular .
Extracellular condition in the single fibre model
We exposed single muscle fibres to a very low extracellular to replicate conditions in which O2 levels in skeletal muscle can be nearly restrictive for mitochondrial respiration during contractions. During exercise, skeletal muscle from COPD or CHF patients, or from climbers exposed to high altitude, may be subjected to hypoxic‐like states and to increased intracellular oxidative stress levels (Clanton, 2007). In isolated fibres NADPH oxidase was identified as the major source of ROS during contractions, but these experiments were performed at supraphysiological extracellular conditions (i.e. ambient ) (Michaelson et al. 2010; Sakellariou et al. 2013). However, it has been proposed that in physiological, near hypoxic conditions in skeletal muscle fibres during exercise, ROS formation by NADPH oxidase is inhibited due to its relatively high K m for O2 (Clanton, 2007; Brandes et al. 2014; Nisimoto et al. 2014). Since mitochondria are important sources of ROS when intact cells are exposed to low (Duranteau et al. 1998; Guzy et al. 2005; Guzy & Schumacker, 2006; Clanton, 2007; Waypa et al. 2010; Sabharwal et al. 2013), the goal of the present study was to determine if treatment with a mitochondrial‐targeted antioxidant can alter Ca2+ handling and force development in single fibres after fatiguing contractions at a low condition that closely mimics in vivo conditions that occur during contractions.
The critical intracellular reported for resting isolated fibres is ∼1.25 Torr (Richmond et al. 1997). As a result of contractile activity, the metabolic rate and mitochondrial oxygen consumption are elevated from resting levels and the critical during contractions will be higher than at rest (Golub & Pittman, 2012; Clanton et al. 2013). The critical extracellular during maximal work in rat plantaris muscle fibres was estimated to be between 4 and 6 Torr (Wüst et al. 2009). In our laboratory we have consistently observed that fatigue resistance for fibres from mice (with a diameter of ∼35 μm) contracting at an extracellular of ∼5 Torr is significantly reduced compared to an extracellular of 40 Torr, which is close to the in vivo interstitial (Golub & Pittman, 2012) (authors’ unpublished observations; see example in Fig. 1 A). This suggests that during a fatiguing contractile period at an extracellular of 5 Torr, oxidative phosphorylation is quite likely partially limited by oxygen diffusion from the extracellular environment to the mitochondria in isolated fibres. However, based on the model developed by our group (Stary & Hogan, 1999) the diameter of mouse fibres should be small enough to not restrict the extracellular oxygen diffusion to hinder mitochondrial respiration. Therefore, in this study the fatiguing contractile period was performed in physiological, near hypoxic, conditions, which should increase ROS generation from mitochondria (Guzy et al. 2005; Clanton, 2007; Clanton et al. 2013).
Treatment with a mitochondrial‐targeted antioxidant can improve PLFFD in isolated fibres at a low extracellular
The unaltered fatigue resistance observed with SS31 treatment was expected since treatments with antioxidants in general are not necessarily correlated with changes in fatigue resistance in isolated fibres (Zuo et al. 2011). Also, the treatment with antioxidants have been reported to have a greater effect on blunting PLFFD compared to increasing fatigue resistance (Kavdia, 2011). Enhanced levels of ROS generation resulting from muscle contractile activity has been implicated as one of the mechanisms causing a decrease in the peak tetanic [Ca2+]c during PLFFD, possibly due to a decrease in SR Ca2+ release, and a decrease in the sensitivity of the contractile sites to Ca2+ (Bruton et al. 2008; Cheng et al. 2015; Watanabe et al. 2015). Increased levels of O2 −• seems to impair the SR Ca2+ release in intact fibres (Bruton et al. 2008). In contrast, a prolonged exposure of unfatigued intact fibres to high amounts of H2O2 causes a decrease in the myofibrillar Ca2+ sensitivity, although the exposure to low concentrations of H2O2 can increase myofibrillar Ca2+ sensitivity (Andrade et al. 1998, 2001). Therefore, it has been proposed that different ROS (e.g. H2O2 or O2 −•) and the specific cellular sites where these oxidants are generated (e.g. mitochondrial matrix, cytosol) during contractile activity may have different effects on the myofibrillar Ca2+ sensitivity and the SR Ca2+ release in skeletal muscle fibres (Cheng et al. 2016). In this regard the antioxidant peptide SS31 can be used as an experimental probe to study the effects of mitochondrial ROS generation in cells (Min et al. 2011). SS31 accumulates specifically in the mitochondrial inner membrane and has the ability to scavenge H2O2 and OH•, thereby inhibiting intracellular oxidative stress such as lipid peroxidation (Zhao et al. 2004; Szeto, 2006). SS31 may also control mitochondrial ROS generation by stabilizing cardiolipin, a phospholipid specific to the mitochondrial inner membrane (Morin et al. 2003; Clanton et al. 2013; Szeto, 2014). Indeed, studies with sarcolemma‐permeabilized skeletal muscle fibres have shown that SS31 significantly diminishes H2O2 emission from mitochondria (Anderson et al. 2009; Siegel et al. 2013). We observed that treatment of isolated single myofibres with SS31 ameliorated the submaximal force development during PLFFD at very low extracellular conditions, which should mimic an oxygen‐restricted state (i.e. near hypoxia). During PLFFD, SS31 had no effects on the peak tetanic [Ca2+]c that had been diminished to a similar extent in control and SS31 treated fibres. The myofibrillar Ca2+ sensitivity was decreased 30 min after fatigue in the control untreated fibres, while SS31 treated myofibres did not demonstrate this reduction in Ca2+ sensitivity at the same time point. Similar to our results, in vitro SS31 treatment prevented a decrease in the myofibrillar Ca2+ sensitivity, and maintained force development within the control levels when ROS generation in mouse skeletal muscle was elevated by treatment with sphingomyelinase (Ferreira et al. 2012). In addition, SS31 prevented a decrease in myocardial force development in an ex vivo stunned heart model (Zhao et al. 2004). Thus, treatment with SS31 avoided a depression in the myofibrillar Ca2+ sensitivity during PLFFD at low extracellular , which may possibly be due to its effect in lowering mitochondrial ROS emission in skeletal muscle (Anderson et al. 2009; Powers et al. 2011).
The extracellular may impact the effects of SS31 on PLFFD
In contrast to our results, Cheng et al. reported that peak tetanic [Ca2+]c was not smaller during fatigue recovery compared to pre‐fatigue, when isolated fibres were treated with SS31 at a high, hyperoxic, extracellular . Therefore they concluded that PLFFD resulted from a decline in myofibrillar Ca2+ sensitivity (Cheng et al. 2015). It is possible that these different effects of SS31 on submaximal peak tetanic [Ca2+]c levels and myofibrillar Ca2+ sensitivity during PLFFD are due to the use of a low extracellular (∼5 Torr) in the present study instead of a supraphysiological (≥150 Torr) in the study of Cheng et al. Accordingly, it has been recently suggested that an increased ROS generation resulting from the use of supraphysiological in isolated muscle preparations in vitro may affect the development of PLFFD (Watanabe et al. 2015). Although controversial, in intact muscle cells, mitochondrial ROS formation can be increased in hypoxic as well as in hyperoxic conditions (Guzy et al. 2005; Guzy & Schumacker, 2006; Clanton, 2007; Murphy, 2009; Clanton et al. 2013). In isolated mitochondria ROS generation is directly proportional to the oxygen concentration (Clanton, 2007; Hoffman et al. 2007). Such a paradox may be explained by complex regulations and cell signalling mechanisms present in intact cells that are absent in isolated mitochondrial preparations (e.g. post‐translational protein modifications, accumulation of signalling molecules and allosteric regulations) (Hoffman et al. 2007; Clanton et al. 2013). However, it is likely that the rates of ROS formation within the distinct mitochondrial compartments are different at low and high . For example, in pulmonary artery smooth muscle cells, hypoxia prompts a decrease in ROS generation in the mitochondrial matrix and an increase in ROS production in the mitochondrial intermembrane space, which then diffuses to the cytosol (Waypa et al. 2010; Sabharwal et al. 2013). Notably, Cheng et al. (2015) have proposed that increases in the levels of superoxide radical in the mitochondria can directly affect Ca2+ release in adjacent SR, thereby linking ROS production in mitochondria to SR function in skeletal muscle (Eisner et al. 2013). Whereas, when the mitochondrial ROS is emitted to the cytosol, myofibrillar Ca2+ sensitivity should be decreased (Cheng et al. 2015). Thus, the surrounding the muscle fibre may be an important factor affecting the cellular location where reactive species levels may be increased during contractions, which will ultimately define the effects of treatment with SS31.
In conclusion, our data confirm that a decrease in myofibrillar Ca2+ sensitivity after fatigue is one of the mechanisms causing fatigue‐induced PLFFD at a low, but physiologically relevant, and that a mitochondrial‐targeted antioxidant treatment was able to prevent this decrease. The use of mitochondrial‐targeted antioxidants may constitute a potential strategy to improve muscle function after fatiguing contractions, particularly in conditions associated with a restrictive O2 delivery to skeletal muscle mitochondria which enhance mitochondrial ROS generation (e.g. COPD and high altitude). Since a certain level of oxidant activity is important for normal muscle function (e.g. glucose uptake, Sandström et al. 2006; force development, Andrade et al. 1998; and adaptation to exercise training, Kavdia, 2011), the use of antioxidants supplementation needs to be well deliberated to avoid blunting of ROS signalling. The use of mitochondrial specific antioxidants is a step forward for controlling oxidant activity in specific cellular compartments without drastically affecting overall cellular redox state, different cellular functions and muscle adaptation to exercise (Shill et al. 2016).
Additional information
Competing interests
None declared.
Author contributions
M.C.H, L.N. and P.G.G. participated in the conception and design of the study, interpretation of the data and drafting of the manuscript. Experimental data collection was performed at the Hogan Lab at University of California San Diego by P.G.G., A.A.S. and L.N. P.G.G., A.A.S., L.N., and M.C.H critically revised the manuscript. A.A.S. was a recipient of the American Physiological Society's Undergraduate Summer Research Fellowship Program. All authors approved the final version of the manuscript and agreed to be accountable for all aspects of the work. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Funding
This work was supported by the National Institute of Arthritis and Musculoskeletal and Skin Diseases of the National Institutes of Health under Award number AR‐069577 (to M.C.H.). The content of this study is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. L.N. and P.G.G are supported by funding from the Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq–Universal no. 424527/2016‐2; to L.N. and CNPq–Universal no. 423317/2016‐4; to P.G.G.). A.A.S. was a recipient of the American Physiological Society's Undergraduate Summer Research Fellowship Program.
Biographies
Paulo G. Gandra (on the right) is an assistant professor at the University of Campinas (Unicamp), Brazil. His research focuses on mechanisms of regulation of skeletal muscle metabolism.

Leonardo Nogueira, Ph.D. (on the left) is Associate Professor at UFRJ, Brazil and Assistant Research Scientist at UCSD. His research is focused on the role of nitric oxide production and metabolism on skeletal muscle function and development.
Michael C. Hogan, Ph.D. (in the middle) is Professor of Medicine at UCSD and his research is focused on the oxygen dependence of skeletal muscle function, bioenergetics of contracting skeletal muscle and the mechanisms of skeletal muscle fatigue.
Edited by: Scott Powers & Karyn Hamilton
References
- Anderson EJ, Lustig ME, Boyle KE, Woodlief TL, Kane DA, Lin C, Iii JWP, Kang L, Rabinovitch PS, Szeto HH, Houmard JA, Cortright RN, Wasserman DH & Neufer PD (2009). Mitochondrial H2O2 emission and cellular redox state link excess fat intake to insulin resistance in both rodents and humans. J Clin Invest 119, 573–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrade FH, Reid MB, Allen DG & Westerblad H (1998). Effect of hydrogen peroxide and dithiothreitol on contractile function of single skeletal muscle fibres from the mouse. J Physiol 509, 565–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrade FH, Reid MB & Westerblad H (2001). Contractile response of skeletal muscle to low peroxide concentrations: myofibrillar calcium sensitivity as a likely target for redox‐modulation. FASEB J 15, 309–311. [DOI] [PubMed] [Google Scholar]
- Balog EM (2010). Excitation‐contraction coupling and minor triadic proteins in low‐frequency fatigue. Exerc Sport Sci Rev 38, 135–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandes RP, Weissmann N & Schröder K (2014). Nox family NADPH oxidases: molecular mechanisms of activation. Free Radic Biol Med 76, 208–226. [DOI] [PubMed] [Google Scholar]
- Bruton JD, Lännergren J & Westerblad H (1998). Effects of CO2‐induced acidification on the fatigue resistance of single mouse muscle fibers at 28 degrees C. J Appl Physiol (1985) 85, 478–483. [DOI] [PubMed] [Google Scholar]
- Bruton JD, Place N, Yamada T, Silva JP, Andrade FH, Dahlstedt AJ, Zhang S‐J, Katz A, Larsson N‐G & Westerblad H (2008). Reactive oxygen species and fatigue‐induced prolonged low‐frequency force depression in skeletal muscle fibres of rats, mice and SOD2 overexpressing mice. J Physiol 586, 175–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cano I, Roca J & Wagner PD (2015). Effects of lung ventilation–perfusion and muscle metabolism–perfusion heterogeneities on maximal O2 transport and utilization. J Physiol 593, 1841–1856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng AJ, Bruton JD, Lanner JT & Westerblad H (2015). Antioxidant treatments do not improve force recovery after fatiguing stimulation of mouse skeletal muscle fibres. J Physiol 593, 457–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng AJ, Yamada T, Rassier DE, Andersson DC, Westerblad H & Lanner JT (2016). Reactive oxygen/nitrogen species and contractile function in skeletal muscle during fatigue and recovery. J Physiol 594, 5149–5160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clanton TL (2007). Hypoxia‐induced reactive oxygen species formation in skeletal muscle. J Appl Physiol (1985) 102, 2379–2388. [DOI] [PubMed] [Google Scholar]
- Clanton TL, Hogan MC & Gladden LB (2013). Regulation of cellular gas exchange, oxygen sensing, and metabolic control. Compr Physiol 3, 1135–1190. [DOI] [PubMed] [Google Scholar]
- Connett RJ, Honig CR, Gayeski TE & Brooks GA (1990). Defining hypoxia: a systems view of VO2, glycolysis, energetics, and intracellular PO2. J Appl Physiol (1985) 68, 833–842. [DOI] [PubMed] [Google Scholar]
- Duranteau J, Chandel NS, Kulisz A, Shao Z & Schumacker PT (1998). Intracellular signaling by reactive oxygen species during hypoxia in cardiomyocytes. J Biol Chem 273, 11619–11624. [DOI] [PubMed] [Google Scholar]
- Edwards RH, Hill DK, Jones DA & Merton PA (1977). Fatigue of long duration in human skeletal muscle after exercise. J Physiol 272, 769–778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisner V, Csordas G & Hajnoczky G (2013). Interactions between sarco‐endoplasmic reticulum and mitochondria in cardiac and skeletal muscle – pivotal roles in Ca2+ and reactive oxygen species signaling. J Cell Sci 126, 2965–2978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esposito F, Mathieu‐Costello O, Shabetai R, Wagner PD & Richardson RS (2010). Limited maximal exercise capacity in patients with chronic heart failure. J Am Coll Cardiol 55, 1945–1954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira LF, Moylan JS, Stasko S, Smith JD, Campbell KS & Reid MB (2012). Sphingomyelinase depresses force and calcium sensitivity of the contractile apparatus in mouse diaphragm muscle fibers. J Appl Physiol (1985) 112, 1538–1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gandra PG, Nogueira L & Hogan MC (2012). Mitochondrial activation at the onset of contractions in isolated myofibres during successive contractile periods. J Physiol 590, 3597–3609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gea J, Agusti A & Roca J (2013). Pathophysiology of muscle dysfunction in COPD. J Appl Physiol (1985) 114, 1222–1234. [DOI] [PubMed] [Google Scholar]
- Golub AS & Pittman RN (2012). Oxygen dependence of respiration in rat spinotrapezius muscle in situ. Am J Physiol Heart Circ Physiol 303, H47–H56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guzy RD, Hoyos B, Robin E, Chen H, Liu L, Mansfield KD, Simon MC, Hammerling U & Schumacker PT (2005). Mitochondrial complex III is required for hypoxia‐induced ROS production and cellular oxygen sensing. Cell Metab 1, 401–408. [DOI] [PubMed] [Google Scholar]
- Guzy RD & Schumacker PT (2006). Oxygen sensing by mitochondria at complex III: the paradox of increased reactive oxygen species during hypoxia. Exp Physiol 91, 807–819. [DOI] [PubMed] [Google Scholar]
- Hirai DM, Musch TI & Poole DC (2015). Exercise training in chronic heart failure: improving skeletal muscle O2 transport and utilization. Am J Physiol Heart Circ Physiol 309, H1419–H1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman DL, Salter JD & Brookes PS (2007). Response of mitochondrial reactive oxygen species generation to steady‐state oxygen tension: implications for hypoxic cell signaling. Am J Physiol Heart Circ Physiol 292, H101–H108. [DOI] [PubMed] [Google Scholar]
- Howlett RA, Kindig CA & Hogan MC (2007). Intracellular PO2 kinetics at different contraction frequencies in Xenopus single skeletal muscle fibers. J Appl Physiol (1985) 102, 1456–1461. [DOI] [PubMed] [Google Scholar]
- Kabbara AA & Allen DG (1999). Measurement of sarcoplasmic reticulum Ca2+ content in intact amphibian skeletal muscle fibres with 4‐chloro‐m‐cresol. Cell Calcium 25, 227–235. [DOI] [PubMed] [Google Scholar]
- Kavdia M (2011). Mathematical and computational models of oxidative and nitrosative stress. Crit Rev Biomed Eng 39, 461–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michaelson LP, Shi G, Ward CW & Rodney GG (2010). Mitochondrial redox potential during contraction in single intact muscle fibers. Muscle Nerve 42, 522–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min K, Smuder AJ, Kwon OS, Kavazis AN, Szeto HH & Powers SK (2011). Mitochondrial‐targeted antioxidants protect skeletal muscle against immobilization‐induced muscle atrophy. J Appl Physiol (1985) 111, 1459–1466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morin C, Zini R & Tillement JP (2003). Anoxia–reoxygenation‐induced cytochrome c and cardiolipin release from rat brain mitochondria. Biochem Biophys Res Commun 307, 477–482. [DOI] [PubMed] [Google Scholar]
- Murphy MP (2009). How mitochondria produce reactive oxygen species. Biochem J 417, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nisimoto Y, Diebold BA, Constentino‐Gomes D & Lambeth JD (2014). Nox4: a hydrogen peroxide‐generating oxygen sensor. Biochemistry 53, 5111–5120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nogueira L & Hogan MC (2010). Phenol increases intracellular [Ca2+] during twitch contractions in intact Xenopus skeletal myofibers. J Appl Physiol (1985) 109, 1384–1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nogueira L, Shiah AA, Gandra PG & Hogan MC (2013). Ca2+‐pumping impairment during repetitive fatiguing contractions in single myofibers: role of cross‐bridge cycling. Am J Physiol Regul Integr Comp Physiol 305, R118–R125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Place N, Ivarsson N, Venckunas T, Neyroud D, Brazaitis M, Cheng AJ, Ochala J, Kamandulis S, Girard S, Volungevičius G, Paužas H, Mekideche A, Kayser B, Martinez‐Redondo V, Ruas JL, Bruton J, Truffert A, Lanner JT, Skurvydas A & Westerblad H (2015). Ryanodine receptor fragmentation and sarcoplasmic reticulum Ca2+ leak after one session of high‐intensity interval exercise. Proc Natl Acad Sci USA 112, 15492–15497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poole DC & Jones AM (2012). Oxygen uptake kinetics. Compr Physiol 2, 933–996. [DOI] [PubMed] [Google Scholar]
- Powers SK, Hudson MB, Nelson WB, Talbert EE, Min K, Szeto HH, Kavazis AN & Smuder AJ (2011). Mitochondria‐targeted antioxidants protect against mechanical ventilation‐induced diaphragm weakness. Crit Care Med 39, 1749–1759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rehn TA, Munkvik M, Lunde PK, Sjaastad I & Sejersted OM (2012). Intrinsic skeletal muscle alterations in chronic heart failure patients: a disease‐specific myopathy or a result of deconditioning? Heart Fail Rev 17, 421–436. [DOI] [PubMed] [Google Scholar]
- Richardson RS, Duteil S, Wary C, Wray DW, Hoff J & Carlier PG (2006). Human skeletal muscle intracellular oxygenation: the impact of ambient oxygen availability. J Physiol 571, 415–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson RS, Newcomer SC & Noyszewski EA (2001). Skeletal muscle intracellular PO2 assessed by myoglobin desaturation: response to graded exercise. J Appl Physiol (1985) 91, 2679–2685. [DOI] [PubMed] [Google Scholar]
- Richardson RS, Wary C, Walter DW, Hoff J, Rossiter HB, Layec G & Carlier PG (2015). MRS evidence of adequate O2 supply in human skeletal muscle at the onset of exercise. Med Sci Sport Exerc 47, 2299–2307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richmond KN, Burnite S & Lynch RM (1997). Oxygen sensitivity of mitochondrial metabolic state in isolated skeletal and cardiac myocytes. Am J Physiol 273, C1613–C1622. [DOI] [PubMed] [Google Scholar]
- Sabharwal SS, Waypa GB, Marks JD & Schumacker PT (2013). Peroxiredoxin‐5 targeted to the mitochondrial intermembrane space attenuates hypoxia‐induced reactive oxygen species signalling. Biochem J 456, 337–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakellariou GK, Vasilaki A, Palomero J, Kayani A, Zibrik L, McArdle A & Jackson MJ (2013). Studies of mitochondrial and nonmitochondrial sources implicate nicotinamide adenine dinucleotide phosphate oxidase(s) in the increased skeletal muscle superoxide generation that occurs during contractile activity. Antioxid Redox Signal 18, 603–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandström ME, Zhang S‐J, Bruton J, Silva JP, Reid MB, Westerblad H & Katz A (2006). Role of reactive oxygen species in contraction‐mediated glucose transport in mouse skeletal muscle. J Physiol 575, 251–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shill DD, Southern WM, Willingham TB, Lansford KA, McCully KK & Jenkins NT (2016). Mitochondria‐specific antioxidant supplementation does not influence endurance exercise training‐induced adaptations in circulating angiogenic cells, skeletal muscle oxidative capacity or maximal oxygen uptake. J Physiol 594, 7005–7014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel MP, Kruse SE, Percival JM, Goh J, White CC, Hopkins HC, Kavanagh TJ, Szeto HH, Rabinovitch PS & Marcinek DJ (2013). Mitochondrial‐targeted peptide rapidly improves mitochondrial energetics and skeletal muscle performance in aged mice. Aging Cell 12, 763–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stary CM & Hogan MC (1999). Effect of varied extracellular PO2 on muscle performance in Xenopus single skeletal muscle fibers. J Appl Physiol (1985) 86, 1812–1816. [DOI] [PubMed] [Google Scholar]
- Szeto HH (2006). Mitochondria‐targeted peptide antioxidants: Novel neuroprotective agents. AAPS J 8, E521–E531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szeto HH (2014). First‐in‐class cardiolipin‐protective compound as a therapeutic agent to restore mitochondrial bioenergetics. Br J Pharmacol 171, 2029–2050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner PD (2012). Muscle intracellular oxygenation during exercise: Optimization for oxygen transport, metabolism, and adaptive change. Eur J Appl Physiol 112, 1–8. [DOI] [PubMed] [Google Scholar]
- Watanabe D, Kanzaki K, Kuratani M, Matsunaga S, Yanaka N & Wada M (2015). Contribution of impaired myofibril and ryanodine receptor function to prolonged low‐frequency force depression after in situ stimulation in rat skeletal muscle. J Muscle Res Cell Motil 36, 275–286. [DOI] [PubMed] [Google Scholar]
- Waypa GB, Marks JD, Guzy R, Mungai PT, Schriewer J, Dokic D & Schumacker PT (2010). Hypoxia triggers subcellular compartmental redox signaling in vascular smooth muscle cells. Circ Res 106, 526–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westerblad H & Allen DG (1991). Changes of myoplasmic calcium concentration during fatigue in single mouse muscle fibers. J Gen Physiol 98, 615–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westerblad H & Allen DG (1993). The influence of intracellular pH on contraction, relaxation and [Ca2+]i in intact single fibres from mouse muscle. J Physiol 466, 611–628. [PMC free article] [PubMed] [Google Scholar]
- Wilson DF, Rumsey WL, Green TJ & Vanderkooi JM (1988). The oxygen dependence of mitochondrial oxidative phosphorylation measured by a new optical method for measuring oxygen concentration. J Biol Chem 263, 2712–2718. [PubMed] [Google Scholar]
- Wüst RCI, Jaspers RT, van Heijst AF, Hopman MTE, Hoofd LJC, van der Laarse WJ & Degens H (2009). Region‐specific adaptations in determinants of rat skeletal muscle oxygenation to chronic hypoxia. Am J Physiol Heart Circ Physiol 297, H364–H374. [DOI] [PubMed] [Google Scholar]
- Zhao K, Zhao GM, Wu D, Soong Y, Birk AV, Schiller PW & Szeto HH (2004). Cell‐permeable peptide antioxidants targeted to inner mitochondrial membrane inhibit mitochondrial swelling, oxidative cell death, and reperfusion injury. J Biol Chem 279, 34682–34690. [DOI] [PubMed] [Google Scholar]
- Zuo L, Nogueira L & Hogan MC (2011). Reactive oxygen species formation during tetanic contractions in single isolated Xenopus myofibers. J Appl Physiol (1985) 111, 898–904. [DOI] [PMC free article] [PubMed] [Google Scholar]