

Abstract
The behavior of proteins at biological and synthetic interfaces is often characterized by a strong history dependence caused by long relaxation times or irreversible transitions. In this work, we introduce the rate of adsorption as a means to systematically quantify the extent, and identify the underlying causes, of history dependence. We use multistep kinetic experiments in which the i′th step is an exposure of a Si(Ti)O2 surface to a flowing fibronectin or cytochrome c solution of concentration ci for a time ti (ci = 0 corresponds to a rinse) and measure the protein adsorption by optical waveguide light mode spectroscopy. The rate of adsorption is sensitive to the structure of the adsorbed layer, and we observe it to greatly increase, for a given adsorbed density, when going from a first to a subsequent adsorption step. This increase is most pronounced when the duration of the initial adsorption step is long. We attribute these observations to the gradual and irreversible formation of protein clusters or locally ordered structures and use them to explain previous underestimates of kinetic data by mesoscopic model descriptions. A thorough understanding of these complex postadsorption events, and their impact on history dependence, is essential for many physiological and biotechnological processes. Optical waveguide lightmode spectroscopy is a promising technique for their macroscopic quantification.
Keywords: optical waveguide lightmode spectroscopy, interfacial kinetics, surface diffusion, surface aggregation
A complete description of a system away from thermodynamic equilibrium must generally include information on its state at previous times. This “history dependence” may be caused by a slow relaxation toward equilibrium or the presence of certain essentially irreversible events. Systems of macromolecules adsorbed at a liquid-solid interface often exhibit a history dependence whose physical origin is the large number of energetically favorable contacts that must be broken for new states to be sampled. Synthetic neutral and charged polymers are notorious for possessing postadsorption relaxation rates sufficiently slow to inhibit true equilibration over reasonable experimental times (1–4). Proteins are biological polymers that, because of their strong internal cohesion, can have even slower relaxation rates than flexible synthetic polymers. They also differ fundamentally from synthetic polymers by their unique native conformation and their surface heterogeneity with respect to electrostatic (e.g., ref. 5) and electron donor-acceptor (6) potentials. These features engender certain postadsorption events like alteration in internal protein conformation (7–10) and surface aggregation (11–13) that may be essentially irreversible on an experimental time scale. Bioseparations, biocatalysis, and biosensing all involve protein adsorption. Thus, a predictive knowledge of its history dependence is clearly needed to design and control important biotechnological processes. Although the manifestation of history dependence in protein adsorption systems has been reported previously (14, 15), its extent has not been systematically quantified and a clear picture of the underlying mechanisms remains elusive.
History dependence can be analyzed by comparing systems of identical composition prepared along different compositional paths. In this work, we investigate history dependence in protein adsorption by considering systems of differing formation kinetics, that is, whose mass densities follow different time evolutions. We introduce the rate of adsorption as a measure of the degree of history dependence. In the absence of transport limitations, the rate of adsorption is a sensitive measure of the intermolecular and intramolecular structure of the adsorbed layer and may be used to ascertain the degree to which an adsorbed layer deviates from equilibrium, i.e., its history dependence.
To generate and analyze adsorbed protein systems of differing compositional histories, we use multistep kinetic experiments in which the i′th step is an exposure of the surface to a flowing protein solution of concentration ci for a time ti. Certain of these are rinses (ci = 0) during which a net desorption occurs. We use optical waveguide lightmode spectroscopy (OWLS) to continuously measure the mass of adsorbed protein during each experiment. We consider two proteins here, fibronectin and cytochrome c, and ours is a hydrophilic Si(Ti)O2 adsorbing surface. Kinetic data are collected with a time resolution as low as 2.9 s. A regeneration of the waveguiding surface allows for the direct comparison of independent experimental data sets.
One of our hopes is that this quantitative analysis of history dependence may provide a test of previously posed theoretical descriptions. For example, kinetic models accounting for history dependence through an irreversible change in internal protein conformation (16–18), although accurate to intermediate times, begin to deviate from experiment somewhat at longer times (19). Multistep adsorption analysis can serve as a systematic test of model assumptions and provide clues as to whether effects other than internal protein rearrangements might contribute to history dependence in a significant way. Another hope is that our findings will be useful in understanding multistep processes as they occur in practical applications such as molecular sensing and separation.
Materials and Methods
Our OWLS apparatus (Artificial Sensing Instruments, Zurich) consists of an aluminum flow cell of volume 70 μl whose bottom is a planar Si1−xTixO2 (where x = 0.25 ± 0.05) surface (20). A built-in temperature control device provides thermal stability (±0.05°C) during the measurements (all performed at 25°C). The experiment begins with a bare surface in equilibrium with a 10 mM Hepes buffer solution containing 100 mM NaCl (pH 7.4) flowing at 80 μl/min. This is then replaced by a solution of either human plasma fibronectin or horse heart cytochrome c (obtained from Sigma) dissolved in the Hepes buffer. The protein concentration, cb, is determined spectrophotometrically by using a coefficient for the absorption of a 1% protein solution at pathlength 1 cm, A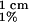 , of value 12.8 at 280 nm for fibronectin and 9.05 at 528 nm for cytochrome c. Using the Gouy-Chapman expression as done previously (21), we estimate the surface charge on the waveguide to be −12.0 μC/cm2.
, of value 12.8 at 280 nm for fibronectin and 9.05 at 528 nm for cytochrome c. Using the Gouy-Chapman expression as done previously (21), we estimate the surface charge on the waveguide to be −12.0 μC/cm2.
Protein molecules adsorbed at the waveguide-solution interface modify the mode spectrum of the waveguide. The adlayer evolution can be investigated by using an external polarized laser beam (λ = 632.8 nm), which is coupled into the waveguide via an incorporated diffraction grating (type 2400, Artificial Sensing Instruments). Mode eigenvalues N are determined by measuring the laser angle of incidence α at which the intensity of the guided wave is maximal. By repeatedly measuring the mode eigenvalues of either the transverse electric (TE) or transverse magnetic (TM) polarization, the mass of adsorbed protein per surface area, Γ, can be determined with a time resolution of 2.9 s. When measuring NTE alone, Γ is related to changes in NTE via (22)
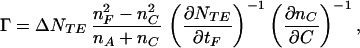 |
1 |
where n and t are refractive indices and thickness, respectively, of pure solvent (C), adsorbed layer (A), waveguiding film (F) or glass support (S). nC is measured experimentally in a refractometer (AR600, Leica Microsystems, Buffalo, NY), a value of 1.82 × 10−1 cm3/g (at 25°C and 632.8 nm) is used for the refractive index increment of the protein, ∂nC/∂c, and a value of 1.39 is used for nA (at 25°C and 632.8 nm). Accurate values for nF and tF are determined by first measuring the zero-order (m = 0) NTE and NTM modes of the bare surface before protein adsorption and then by solving (22)
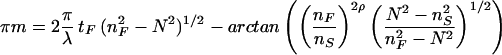 |
2 |
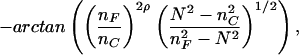 |
3 |
where ρ = 0 and 1 for the TE and TM modes, respectively, and nS = 1.52578 as given by the manufacturer. In Eq. 1 ∂NTE/∂tF is determined for each waveguide separately by measuring (once) both mode eigenvalues, NTE and NTM, simultaneously during protein adsorption. OWLS data published in the literature are usually collected by this form of double measurement, with a maximal time resolution of 23.9 s. The solution of the appropriate mode equations (20, 22) yields tA and nA of the protein adlayer, which, on transformation into Γ, can be compared with Eq. 1.
Desorption of reversibly bound molecules from the waveguiding film is studied by switching to flowing, protein-free buffer solution. Adsorption and readsorption steps are performed in a similar way, with the exception that in the latter case proteins adsorb to a partially covered surface from the beginning. A typical multistep experiment consists of: (i) adsorption of a protein during a specific time period followed by desorption until a steady state is reached; (ii) readsorption of the same protein with a subsequent desorption step; and (iii) either repetition of step ii or regeneration of the surface to start a new experiment.
To compare independent OWLS data sets in a quantitative way, it is vital to reuse a waveguiding surface several times. We therefore regenerate its chemical and physical properties through a cleaning procedure performed within the experimental setup. The surface is exposed to a flowing 1% Terg-A-Zyme (Alconox, New York) solution for 5 min and re-equilibrated in buffer afterward. (Terg-A-Zyme is an anionic detergent primarily consisting of sodium alkylarylsulfonate, phosphates, carbonates, and protease enzymes.) To test the efficacy of the regeneration step, we measured the contact angles of water on six of the waveguides used in this study by using a contact angle goniometer (model NRL 100, RameHart). We found advancing and receding angles to be identical and to have values before and after regeneration of, respectively, 17.9 ± 0.3° and 17.6 ± 0.7°. Without repositioning the waveguide in an OWLS experiment, the reproducibility between two independent adsorption runs with an intervening cleaning step is very high; we find the average standard deviation of the absolute rate curves to be less than 2% and that of the derivative curves (in the peak region) to be less than 10%.
Results
In Figs. 1 and 2, we show multistep kinetic adsorption curves for fibronectin at a bulk concentration cb = 50 μg/ml. In Fig. 1, protein molecules are desorbed from the waveguide after an initial adsorption time of 240 s. Fibronectin is readsorbed to the partially covered surface after a varying desorption period of 300 s, 900 s, or 1,800 s. The difference between the subsequent three readsorption curves is within the experimental uncertainty of our instrument. Therefore, in Fig. 1, we show only the run with a desorption period of 900 s and compare the trace of the readsorption curve with that of uninterrupted adsorption. These data clearly demonstrate that an early interruption of the adsorption process does not measurably influence the fibronectin deposition on the surface even for a desorption time period of up to 1,800 s.
Figure 1.

The adsorbed protein density versus time for a multistep OWLS experiment. After adsorption of fibronectin (cb = 50 μg/ml) for 240 s, reversibly bound protein molecules are removed from the waveguide surface during a desorption step (◊). The readsorption kinetics (+) are displayed twice, once shifted in time (dashed line), to compare its trace with the initial adsorption kinetics (◊) and the adsorption kinetics measured during a separate uninterrupted run on the same surface (continuous line). The protein solution is introduced at arrows a, and the buffer solution at arrows b. (Inset) The rate of adsorption as a function of deposited mass density for initial adsorption (◊), readsorption (+), and complete uninterrupted adsorption (continuous line).
Figure 2.

Adsorbed density of fibronectin (cb = 50 μg/ml) versus time as measured by OWLS for an initial adsorption of 1,900 s (◊), followed by desorption (◊) and readsorption (+). As in Fig. 1, the measured readsorption kinetics (+) are also displayed with a matching time shift (dashed line). The meaning of arrow a and b are as in Fig. 1. (Inset) Rate of adsorption as a function of deposited mass density for initial adsorption (◊) and readsorption (+).
To investigate the influence of a higher protein surface coverage on the readsorption kinetics, we allow fibronectin to attach to the waveguiding film over an extended time period (1,900 s) before desorption and readsorption (Fig. 2). Interestingly, the rate at which fibronectin readsorbs is now remarkably accelerated (compared with the rate measured at the same surface density during the initial step) and the previous saturation coverage is more rapidly achieved. The same observations can be drawn from similarly performed experiments at a protein bulk concentration of 100 μg/ml.
We next investigate whether this increase in deposition rate is related to fibronectin-specific features. For example, readsorption may occur onto empty pockets, formed by the desorption of molecules in close contact with those that are irreversibly bound, that could favor rebinding of molecules of similar structure. We thus consider readsorption of both fibronectin and cytochrome c on a surface already covered with fibronectin. Cytochrome c, much smaller and compact than fibronectin, is chosen for these rebinding experiments to rule out geometrical or biospecific effects. As shown in Fig. 3, we find that the readsorption kinetics of both proteins are accelerated and almost identical, but that fibronectin has, as expected, a greater saturation coverage than the smaller cytochrome c.
Figure 3.

Adsorbed protein density versus time as measured by OWLS for the adsorption and readsorption of fibronectin (+, cb = 100 μg/ml) followed by the readsorption of cytochrome c (○, cb = 100 μg/ml). Both readsorption curves are also displayed with a time shift (fibronectin: continuous line; cytochrome c: dashed line). The meaning of arrows a and b is the same as in Fig. 1. (Inset) Rate of readsorption as a function of deposited mass density for fibronectin (continuous line) and cytochrome c (dashed line).
We also perform multistep experiments of cytochrome c adsorption (cb = 100 μg/ml) at the same solid-liquid interface. We investigate the effect of previously adsorbed molecules on the readsorption kinetics by varying the duration of the initial adsorption step from 60 s (Fig. 4) to 2,400 s, 1,800 s, and finally 2,900 s (Fig. 5) and by comparing readsorption curves. We again observe that the readsorption kinetics are considerably accelerated, but in contrast to fibronectin, this is true even for short initial adsorption steps (Fig. 4). As with fibronectin, however, roughly the same saturation coverage is reached during initial and subsequent adsorption steps. Both observations have been reconfirmed by analogous experiments at a protein bulk concentration of 50 μg/ml.
Figure 4.

Adsorbed protein density versus time as measured by OWLS for the stepwise adsorption and readsorption of cytochrome c (◊, +, ○, cb = 100 μg/ml), with an initial adsorption step of 60 s (◊). Both readsorption curves are also displayed with a time shift (dashed and dash-dotted line for first and second readsorption, respectively) to compare them with curves obtained in an uninterrupted run on the same surface (continuous line). The meaning of arrows a and b is the same as in Fig. 1. (Inset) Rate of adsorption as a function of deposited mass density for initial adsorption (◊), first (+) and second readsorption (○).
Figure 5.

Adsorbed protein density versus time as measured by OWLS for stepwise adsorption and readsorption of cytochrome c (◊, +, ○, cb = 100 μg/ml), with an initial adsorption step of 2,900 s (◊). Both readsorption curves are also displayed with a time shift (first readsorption: continuous line; second readsorption: dashed line). The meaning of arrows a and b is the same as in Fig. 1. (Inset) Rate of adsorption as a function of deposited mass density for initial adsorption (◊), first (+) and second readsorption (○).
The rate and extent of desorption are also likely influenced by history-dependent features and, as discussed further below, provide a simple test of common theoretical assumptions. We investigate desorption by using multistep adsorption measurements via single-mode OWLS with a time resolution of 2.9 s (see Eq. 1). In Fig. 6, we show together the desorption curves from several cytochrome c (cb = 100 μg/ml) experiments. Because a structurally diverse protein “society” exists at the solid-liquid interface, one expects that the desorption kinetics are best described by a sum of exponential functions. We focus here on the initial period and thus measure the rate of desorption (kd) of the protein subpopulation with the greatest rate. In Fig. 7, we show the initial rates of desorption (graph a) and the percentages of reversibly bound protein (graph b) as a function of surface density for cytochrome c. We find both of these quantities to decrease in a roughly linear way with increasing surface density.
Figure 6.

Adsorbed protein density versus time as measured by OWLS, displaying the desorption kinetics of cytochrome c (cb = 100 μg/ml) after adsorption (◊), first (x) and second (+) readsorption steps. For the sake of clarity, only every fourth data point is shown for each desorption curve. All curves are shifted in time so that their starting points lie on the trace describing cytochrome c adsorption measured without interruption (dashed line).
Figure 7.

(a) The initial desorption rate, kd, versus surface density from which desorption began, Γmax, as determined from the cytochrome c desorption curves presented in Fig. 6. Data points belonging to desorption steps after adsorption (◊), first (x) and second (+) readsorption are distinguished. The uncertainty in the kd values is less than 8% and thus always smaller than the points. (b) The percentage of reversibly adsorbed protein (those desorbing during a 300-s rinse), R, versus Γmax. The meaning of the data point markers is the same as in a.
Discussion
Systems away from equilibrium generally exhibit history-dependent behavior. We observe this to be the case for adsorbed protein layers: the rate of adsorption (onto a partially saturated layer) can depend strongly on the layer's formation history. We suggest that the adsorption rate might serve as a convenient measure of the extent of the deviation from equilibrium or, equivalently, the history dependence. In the absence of transport limitations, the measured adsorption rate reflects the energetic and entropic interactions of a solution protein with previously adsorbed proteins and thus is a legitimate scalar measure of the adsorbed layer's structure. We observe that adsorption rates onto surfaces of identical composition can vary tremendously depending on their histories, indicating important differences in intermolecular and intramolecular adsorbed layer structures.
Two very different structural changes could cause the observed history dependence in adsorption rate. One of these is the formation of specific pockets by the assembly of irreversibly attached protein molecules around reversibly attached ones that favor readsorption of the same protein. Another is the irreversible formation of clusters or patch-wise aggregates leading to a more efficient molecular “packing” and a concomitant greater fraction of exposed surface. The accelerated kinetics also could be explained without invoking history dependence: patch-wise chemical or physical inhomogeneity of the waveguiding film could make certain surface regions more attractive than others for protein adsorption.
To determine which of these explanations is indeed causative, we use a mesoscopic model of the adsorption process in which the time evolution of adsorbed particle mass Γ follows the general kinetic equation
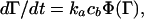 |
4 |
where ka is the intrinsic adsorption rate constant, cb is the bulk protein concentration, and Φ is the fraction of the surface available to additional adsorbing molecules. Eq. 4 is applicable during the “reaction” limited regime of the adsorption process (i.e., after the initial diffusion limited regime). Φ can be expressed as a power series of Γ, with coefficients that depend on the nature of the adsorption process (23, 24) and on the geometry of the adsorbing particles (25):
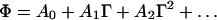 |
5 |
ka, on the other hand, depends on the potential energy (u) of interaction between an adsorbing protein and the surface and previously adsorbed proteins via a Boltzmann factor (19). Assuming u is an analytic function of Γ, we obtain
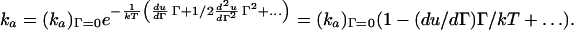 |
6 |
Inserting Eqs. 5 and 6 into Eq. 4, one predicts a linear relationship between dΓ/dt and Γ up to a certain surface coverage. This linearity is a feature we observe experimentally for both proteins as shown in the insets of Figs. 1–5. As cb is known and A0 = 1 for adsorption onto an empty surface, the intercept of the line extrapolated to Γ = 0 yields (ka)Γ=0 of the initial adsorption. The slope of the linear regions of the rate curves of both initial adsorption and readsorption vary proportionally to (ka)Γ=0cb by a nearly identical constant K.† We use the value of K calculated from the initial adsorption step to determine (ka)Γ=0 for the readsorption step and show the measured rate constants for both proteins in Table 1. We conclude that any difference between the intrinsic rate of initial adsorption and readsorption is much smaller than our measured difference in overall rate of initial adsorption and readsorption. This seems to rebut the possibility of inhomogeneity of the waveguiding film being responsible for the observed accelerated readsorption kinetics. [The difference in (ka)Γ=0 between the two fibronectin as well as the two cytochrome c studies is caused by the use of different Si1−xTixO2 waveguides.]
Table 1.
Adsorption rate constants of fibronectin and cytochrome c
| Protein | Δt (s) |
k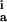 (10−5 cm/s) (10−5 cm/s) |
k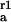 (10−5 cm/s) (10−5 cm/s) |
k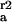 (10−5 cm/s) (10−5 cm/s) |
|---|---|---|---|---|
| Fibronectin | 240 | 4.5 ± 0.7 | 3.7 ± 0.8 | |
| Fibronectin | 1,900 | 6.4 ± 1.1 | 9.9 ± 1.2 | |
| Cytochrome c | 60 | 4.7 ± 0.6 | 3.2 ± 0.6 | 2.8 ± 0.7 |
| Cytochrome c | 2,900 | 1.9 ± 0.6 | 1.3 ± 0.5 | 1.2 ± 0.6 |
Rate constants are given for initial adsorption (k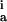 ) of duration Δt, first (k
) of duration Δt, first (k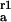 ) and second (k
) and second (k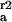 ) readsorption and are calculated as described in the text (see Discussion).
) readsorption and are calculated as described in the text (see Discussion).
An adsorbed layer evolving structurally so as to contain pockets of specificity can be ruled out as a major factor by comparing the kinetics of fibronectin and cytochrome c adsorbing to a surface already filled with irreversibly adsorbed fibronectin molecules (Fig. 3). Both proteins readsorb in a roughly equally accelerated fashion compared with continuous adsorption of fibronectin onto a bare Si1−xTixO2 surface. As shown in detail in the inset of Fig. 3, the Γ-dependent rate of mass deposition dΓ/dt is the same for both proteins within the experimental uncertainty.
We believe that the observed acceleration in adsorption rate is caused by the irreversible formation of protein clusters or locally ordered structures. The idea is that aggregation would allow for more of the surface to be exposed, leading to an increased rate of adsorption. As discussed above, this increase is caused by changes in Φ and not to changes in the intrinsic rate ka. Appreciable macromolecular surface mobility, a prerequisite to the formation of clusters, has been reported in experimental studies of serum albumin and DNA nucleotides (26–29). Evidence for protein clustering or ordered arrays has been reported for cytochrome P450 (13), cytochrome c (30), ferritin (11), and lysozyme (12) mainly by using techniques like transmission electron microscopy and scanning tunneling microscopy. Protein clustering also has been observed in Brownian dynamics simulations (31).
The fact that two proteins as different as fibronectin and cytochrome c exhibit history-dependent adsorption kinetics because of the irreversible formation of surface clusters suggests that the behavior may be universal. Although it is not the goal of this article to report an exhaustive study of protein/surface systems, we can report an exception to this behavior. Human serum albumin is a protein of size intermediate to fibronectin and cytochrome c that adsorbs somewhat weakly to Si(Ti)O2. We find this protein not to exhibit history-dependent adsorption kinetics (see Fig. 8).
Figure 8.

Adsorbed protein density versus time as measured by OWLS for the adsorption and readsorption of human serum albumin (⋄, +, cb = 100 μg/ml), with an initial adsorption step of 2,000 s (⋄). The readsorption curve is also displayed with a time shift (dashed line). The meaning of arrows a and b is the same as in Fig. 1.
History dependence in protein adsorption usually is attributed to the presence of irreversible, postadsorption transitions of either molecular orientation or intramolecular conformation. Indeed, these processes may be occurring in our experiments as well. Although it is thought that horse heart cytochrome c does not change its conformation considerably on adsorption (32), a broad molecular orientation distribution into hydrophilic glass adsorbed cytochrome c has been observed by total internal reflection fluorescence (30, 33). Human plasma fibronectin, on the other hand, is known to be very flexible and to exhibit either compact or very extended structures (depending on the type of surface) in an adsorbed state (34).
The presence of postadsorption transitions in orientation or conformation is insufficient, however, to explain the results reported here. To see why this is so, we consider the picture of partially reversible protein adsorption in which molecules initially attach in a reversible manner (with a structure similar to that in solution) and subsequently undergo an irreversible transition to a conformationally or orientationally altered structure possessing a greater degree of surface contact (16, 35). Within this picture, steric blocking of the transition prevents some of the molecules from becoming irreversibly bound; this explains observations of (i) a removal of only a certain fraction of adsorbed proteins during dilution of the bulk phase and (ii) an increase in maximal surface coverage with protein bulk concentration (36, 37). Based on this picture, one expects that the kinetics of a second adsorption step would be slower because of transitions of some of the proteins during the rinse, leading to a more efficiently blocked surface and a diminishment of the area available for subsequent protein adsorption. In contrast, our results demonstrate clearly that cytochrome c and fibronectin molecules readsorb more rapidly after a previous desorption step than when protein is adsorbed to the same surface without interruption (Figs. 2–5).
Simple new models of protein adsorption that account for history dependence can be tested by step-wise adsorption experiments as presented throughout this article. For example, a model of particles irreversibly adsorbing at random positions subject to steric exclusion rules, then randomly desorbing, then adsorbing again, etc. (24) predicts an accelerated second adsorption caused by a more compact particle arrangement after the desorption step. However, the acceleration observed in our experiments is much greater than that predicted by this simple approach. A model of particles adsorbing in a partially reversible manner, i.e., that adsorb reversibly and then spread to an irreversible state, all subject to steric exclusion rules (16–18), would predict a diminished rate for a readsorption step (because some proteins would spread whereas others desorb) and an increase in the fraction of reversibly bound protein with increased surface density (because latecomers have no space to spread). In fact, we observe just the opposite effect in our experiments. Nevertheless, a slow build-up of fibronectin clusters is consistent with the fact that the experiment can be modeled satisfactorily without considering clustering effects up to intermediate times (19). It is no surprise that the corresponding fit for cytochrome c is much less successful. It appears then that to be universally applicable, a mesoscopic model description must account for the effect of postadsorption cluster formation. A promising model possessing this feature has recently appeared (38, 39).
Common assumptions in mesoscopic adsorption models are initial intrinsic adsorption rates (ka)Γ=0, of uniform value throughout the surface and desorption rates, kd, independent of surface density. Our data suggest that the former but not the latter is valid for the systems studied here. It appears that an increased stability of adsorbed cytochrome c, as evidenced by a decreased kd, coincides with the gradual build-up of organized protein structures. These results are comparable to those found in displacement experiments with two types of polymer chains (40) and although there is no doubt about these proteins becoming more and more tightly bound to our surface with time, intramolecular structural rearrangements, changes in the state of surface hydration, or clustering effects are all possible explanations.
Conclusions
In this paper, we investigate the history dependence of fibronectin and cytochrome c adsorption onto Si(Ti)O2 by using OWLS. We observe that the rate of adsorption onto surfaces containing equal amounts of adsorbed protein can be greatly enhanced if the adsorbed layer is prepared by multiple adsorption steps, and we attribute this history dependence to the irreversible formation of clusters or ordered domains. Through its exquisite sensitivity to adsorbed layer structure, we propose that the rate of adsorption may be useful in determining the spatial and temporal characteristics of clustering needed to develop and test model descriptions.
Acknowledgments
We thank Prof. G. Mao for use of the contact angle goniometer and acknowledge the financial support of the National Science Foundation (CAREER Award CTS-9733310) and the National Institutes of Health (Grant No. R01-GM59487).
Abbreviations
- OWLS
optical waveguide lightmode spectroscopy
- TE
transverse electric
- TM
transverse magnetic
Footnotes
K (= A1 − A0(du/dΓ)/kT), may differ slightly between initial adsorption and readsorption because in the latter case, transitions involving proteins adsorbed during the first step lead to a nonunit A0. Nevertheless, this has only a very small effect on the rate curve's slope because (i) A0 is always expected to be close to unity and (ii) |du/dΓ/kT|≪|A1| based on the degree of electrostatic screening present in the systems considered here.
References
- 1.Douglas J F, Johnson H E, Granick S. Science. 1993;262:2010–2012. doi: 10.1126/science.262.5142.2010. [DOI] [PubMed] [Google Scholar]
- 2.Dijt J C, Cohen Stuart M A, Fleer G J. Macromolecules. 1994;27:3219–3228. [Google Scholar]
- 3.Rebar V A, Santore M M. Macromolecules. 1996;29:6262–6272. [Google Scholar]
- 4.Sukhishvili S A, Dhinojwala A, Granick S. Langmuir. 1999;15:8474–8482. [Google Scholar]
- 5.Koppenol W H, Margoliash E. J Biol Chem. 1982;257:4426–4437. [PubMed] [Google Scholar]
- 6.Calonder C, Talbot J, Ramsden J J. J Phys Chem B. 2001;105:725–729. [Google Scholar]
- 7.Soderquist M E, Walton A G. J Colloid Interface Sci. 1980;75:386–397. [Google Scholar]
- 8.Haynes C A, Norde W. J Colloid Interface Sci. 1995;169:313–328. [Google Scholar]
- 9.Green R J, Hopkinson I, Jones R A L. Langmuir. 1999;15:5102–5110. [Google Scholar]
- 10.Wertz C F, Santore M M. Langmuir. 1999;15:8884–8894. [Google Scholar]
- 11.Nygren H. Biophys J. 1993;65:1508–1512. doi: 10.1016/S0006-3495(93)81221-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Haggerty L, Lenhoff A M. Biophys J. 1993;64:886–895. doi: 10.1016/S0006-3495(93)81448-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ramsden J J, Bachmanova G I, Archakov A I. Phys Rev E. 1994;50:5072–5076. doi: 10.1103/physreve.50.5072. [DOI] [PubMed] [Google Scholar]
- 14.Mura-Galelli M J, Voegel J C, Behr S, Bres E F, Schaaf P. Proc Natl Acad Sci USA. 1991;88:5557–5561. doi: 10.1073/pnas.88.13.5557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nygren H, Stenberg M, Karlsson C. J Biomed Mater Res. 1992;26:77–91. doi: 10.1002/jbm.820260108. [DOI] [PubMed] [Google Scholar]
- 16.Van Tassel P R, Viot P, Tarjus G. J Chem Phys. 1997;106:761–770. [Google Scholar]
- 17.Brusatori M A, Van Tassel P R. J Colloid Interface Sci. 1999;219:333–338. doi: 10.1006/jcis.1999.6496. [DOI] [PubMed] [Google Scholar]
- 18.Minton A P. Biophys J. 1999;76:176–187. doi: 10.1016/S0006-3495(99)77187-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Calonder C, Van Tassel P R. Langmuir. 2001;17:4392–4395. [Google Scholar]
- 20.Ramsden J J. J Stat Phys. 1993;73:853–877. [Google Scholar]
- 21.Ramsden J J, Roush D J, Gill D S, Kurrat R, Willson R C. J Am Chem Soc. 1995;117:8511–8516. [Google Scholar]
- 22.Tiefenthaler K, Lukosz W. J Opt Soc Am B. 1989;6:209–220. [Google Scholar]
- 23.Schaaf P, Talbot J. J Chem Phys. 1989;91:4401–4409. [Google Scholar]
- 24.Van Tassel P R, Viot P, Tarjus G, Ramsden J J, Talbot J. J Chem Phys. 2000;112:1483–1488. [Google Scholar]
- 25.Ricci S M, Talbot J, Tarjus G, Viot P. J Chem Phys. 1992;97:5219–5228. [Google Scholar]
- 26.Michaeli I, Absolom D R, van Oss C J. J Colloid Interface Sci. 1980;77:586–587. [Google Scholar]
- 27.Burghardt T P, Axelrod D. Biophys J. 1981;33:455–467. doi: 10.1016/S0006-3495(81)84906-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tilton R D, Gast A P, Robertson C R. Biophys J. 1990;58:1321–1326. doi: 10.1016/S0006-3495(90)82473-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chan V, Graves D J, Fortina P, McKenzie S E. Langmuir. 1997;13:320–329. [Google Scholar]
- 30.Fraaije J G, Kleijn J M, van der Graaf M, Dijt J C. Biophys J. 1990;57:965–975. doi: 10.1016/S0006-3495(90)82616-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ravichandran S, Talbot J. Biophys J. 2000;78:110–120. doi: 10.1016/S0006-3495(00)76577-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Reynaud J A, Tavernier I, Yu L T, Cochet J M. Bioelectrochem Bioenerg. 1986;15:103–112. [Google Scholar]
- 33.Edmiston P L, Lee J E, Cheng S, Saavedra S S. J Am Chem Soc. 1997;119:560–570. [Google Scholar]
- 34.Erickson H P, Carrell N A. J Mol Biol. 1983;258:14539–14544. [PubMed] [Google Scholar]
- 35.Lundstrom I. Prog Colloid Polym Sci. 1985;70:76–82. [Google Scholar]
- 36.Kurrat R, Ramsden J J, Prenosil J E. J Chem Soc Faraday Trans. 1994;90:587–590. [Google Scholar]
- 37.Wahlgren M, Arnebrant T, Lundstrom I. J Colloid Interface Sci. 1995;175:506–514. [Google Scholar]
- 38.Minton A P. Biophys Chem. 2000;86:239–247. doi: 10.1016/s0301-4622(00)00151-4. [DOI] [PubMed] [Google Scholar]
- 39.Minton A P. Biophys J. 2001;80:1641–1648. doi: 10.1016/S0006-3495(01)76136-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Frantz P, Granick S. Phys Rev Lett. 1991;66:899–902. doi: 10.1103/PhysRevLett.66.899. [DOI] [PubMed] [Google Scholar]