

Abstract
Diffuse large B‐cell lymphoma (DLBCL), which is the most prevalent disease subtype of non‐Hodgkin lymphoma, is highly heterogeneous in terms of cytogenetic and molecular features. This study retrospectively investigated the clinical impact of G‐banding‐defined chromosomal abnormality on treatment outcomes of DLBCL in the era of rituximab‐containing immunochemotherapy. Of 181 patients who were diagnosed with DLBCL and treated with R‐CHOP or an R‐CHOP‐like regimen between January 2006 and April 2014, metaphase spreads were evaluable for G‐banding in 120. In these 120 patients, 40 were found to harbor a single chromosomal aberration type; 63 showed chromosomal abnormality variations (CAVs), which are defined by the presence of different types of chromosomal abnormalities in G‐banding, including 19 with two CAVs and 44 with ≥3 CAVs; and 17 had normal karyotypes. No specific chromosomal break point or numerical abnormality was associated with overall survival (OS) or progression‐free survival (PFS), but the presence of ≥3 CAVs was significantly associated with inferior OS rates (hazard ratio (HR): 2.222, 95% confidence interval (CI): 1.056–4.677, P = 0.031) and tended to be associated with shorter PFS (HR: 1.796, 95% CI: 0.965–3.344, P = 0.061). In addition, ≥3 CAVs more frequently accumulated in high‐risk patients, as defined by several conventional prognostic indices, such as the revised International Prognostic Index. In conclusion, our results suggest that the emergence of more CAVs, especially ≥3, based on chromosomal instability underlies the development of high‐risk disease features and a poor prognosis in DLBCL.
Keywords: Chromosomal abnormality, chromosomal abnormality variations, diffuse large B‐cell lymphoma, karyotypic evolution
Introduction
Non‐Hodgkin lymphoma (NHL) is a highly prevalent hematologic malignancy with a variety of disease subtypes with different histological findings, etiology, molecular features, and clinical manifestations. Diffuse large B‐cell lymphoma (DLBCL) is an aggressive and the most frequent subtype of NHL that is defined by histological characterization of diffuse and unstructured sheet architectures of medium‐ to large‐sized abnormal B‐cell lineage lymphoid cells 1. Recent progress in immunochemotherapy combining rituximab, an anti‐CD20 monoclonal antibody, and cytotoxic agents has greatly improved the treatment outcome of DLBCL, but approximately 30% of patients with DLBCL remain incurable 2, 3.
Chromosome abnormalities play critical roles in emergence of cancer‐initiating cells and in disease progression of most cancers. Especially in hematologic malignancies, identification of disease‐specific chromosomal abnormalities is essential for differential diagnosis of molecularly or biologically distinct disease subtypes with significantly different prognoses 4. In B‐cell NHLs, disease subtype‐specific chromosomal abnormalities are strongly associated with disease development, such as translocation t(14;18) involving BCL2 gene rearrangement in follicular lymphoma 5, 6, t(11;14) involving Cyclin D1 (CCND1) gene rearrangement in mantle cell lymphoma 7, or t(8;14) involving c‐MYC gene rearrangement in Burkitt lymphoma 8, 9. However, no specific chromosomal aberration has been shown to be diagnostically or prognostically relevant in DLBCL, although several abnormalities have been repeatedly identified. Double‐hit or triple‐hit B‐cell lymphomas harboring concomitant chromosomal rearrangements involving c‐MYC and BCL2 and/or BCL6 genes with unfavorable prognoses have previously been included in DLBCL, but these are considered to be an independent disease subtype in the latest WHO classification updated in 2016 10.
Tumors cells of DLBCL frequently possess random and complex chromosomal abnormalities and sometimes exhibit more than two chromosomal abnormality variations (CAVs), such as karyotypic evolution with additional chromosomal abnormalities or totally different patterns of chromosomal abnormalities. This suggests a contribution of karyotypic/genetic instability and additional acquisition of genetic changes to tumor progression. Considering that acquisition of additional karyotypic/genetic changes is vertically transmittable mechanisms for cancer adaptation and progression by constructing intratumor heterogeneity, which eventually leads to acquisition of therapeutic resistance 11, and in this study, we retrospectively investigated the clinical effects of particular chromosomal rearrangements and the number of CAVs on clinical outcomes of patients with DLBCL treated by rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisolone (R‐CHOP)‐based chemotherapy in a real‐world clinical setting.
Materials and Methods
Patients
We retrospectively analyzed the medical records of 465 patients with DLBCL diagnosed at three independent institutes in Kyoto, Japan, between January 2006 and April 2014. Among these patients, those with karyotypic analyses of biopsied specimens performed by G‐banding before the start of treatment by R‐CHOP or with an R‐CHOP‐like regimen were included in this study. The R‐CHOP‐like regimens included reduced R‐CHOP, R‐pirarubicin (THP)‐COP, and these chemotherapies combined with radiotherapy. This study was conducted in accordance with the ethical principles of the Declaration of Helsinki and was approved by the institutional review boards of all participating institutes.
Karyotypic analysis and counting of chromosomal abnormality variations (CAVs)
Classic karyotyping of metaphases by G‐banding was performed as described elsewhere 12. To avoid bias, interphase fluorescence in situ hybridization and molecular diagnostic tests were not considered for this analysis. Twenty metaphase spreads were normally analyzed for one biopsied specimen, and karyotypic aberration was determined in accordance with the International System for Human Cytogenetic Nomenclature (ISCN); however, the number of evaluable tumor‐derived metaphase cells for karyotypic analysis was <20 in some patients.
The number of CAVs was counted as follows: (i) 1, in a case with only one pattern of chromosomal abnormality identified throughout all analyzed metaphase cells, (ii) 2, in a case with metaphase cells with a major pattern of chromosomal aberration and a minor additional pattern of chromosomal aberration, (iii) also 2, in a case with metaphase cells with a major pattern of chromosomal aberration and a totally different pattern of chromosomal aberration, (iv) ≥3, in a case with metaphase cells with a major pattern of chromosomal aberration and more than two different patterns of additional chromosomal aberration, (v) also ≥3, in a case with more than three patterns of metaphase cells with totally different patterns of chromosomal aberration, and (vi) 0, in a case in which only a normal karyotype was identified. Constitutional karyotypes were not counted as abnormal. The counts for (ii) to (v) were used even if the findings were observed only in one metaphase spread.
To show examples of how we defined the number of CAVs, G‐banding data from two patients with DLBCL are shown in Figure 1A and B. As the first example, chromosomal feature of the patient #1 in Table S1 is shown in Figure 1A. The figure presents a major pattern of chromosomal aberration defined “a” found in 16 metaphase spreads, a minor pattern of additional chromosomal aberration defined “b” found in one metaphase spread, and other two metaphase spreads with 47 and 48 chromosomes. As the result, this case presents four CAVs with “a,” “b,” and the two unknown patterns. As the second example, chromosomal feature of the patient #29 in Table S1, a more complicated case, is shown in Figure 1B. The figure presents a major pattern of chromosomal aberration defined “a” found in eight metaphase spreads, two minor additional patterns of chromosomal aberration defined “b” and “c” found in six and one metaphase spreads respectively, two metaphase spreads defined “d” with no chromosomal aberration, and three other metaphase spreads with 46 chromosomes showing chromosomal aberrations, but further details are unknown. In this case, on the assumption that all the three metaphase spreads with unknown details shared totally identical pattern of chromosomal aberration, the number of CAVs would be 4 with “a,” “b,” “c,” and the unknown pattern. On the assumption that the three metaphase spreads with unknown details showed three different patterns of chromosomal aberration, the number of CAVs would be 6 with “a,” “b,” “c,” and three unknown patterns. In brief, the number of CAVs is 4 or more and 6 or less, as far as we can confirm from the available data. In contrast to the definition of clones by ISCN, requiring a chromosome gain or a structural rearrangement to be present in least two cells and a loss of chromosome to be present in at least three cells to be accepted as clonal 13, our definition of CAV includes single cell abnormalities such as the pattern “b” of the patient #1 or the pattern “c” of the patient #29.
Figure 1.

Two examples of the way how we defined the number of CAVs. White squares represent the original aberrations (a), and light and dark gray squares represent additional aberrations. b, c, and d represent minor clones with additional abnormalities and/or different chromosomal feature.
Statistics
Overall survival (OS) was defined as the time from start of treatment to death from any cause. Progression‐free survival (PFS) was defined as the time from start of treatment to the first sign of progression or death from any cause. PFS and OS rates were estimated using the Kaplan–Meier method. A log‐rank test and Cox proportional hazards regression analysis were used to evaluate differences between number of CAVs (≥3/0‐2) in OS and PFS. We also adjusted clinical background factors as confounders by Cox proportional hazard regression. Relationships of the number of CAVs with clinical background factors and prognostic indices: the International Prognostic Index (IPI) 14, revised IPI (R‐IPI) 15, National Comprehensive Cancer Network (NCCN)‐IPI 16, and Kyoto Prognostic Index (KPI), which we have recently developed 17, were evaluated by chi‐square test, except for that with age, which was examined by t‐test.
Results
Patients
Among 465 reviewed patients, karyotypic analyses by G‐banding were performed on biopsied tumor specimens before the start of R‐CHOP or R‐CHOP‐like regimens in 181. As shown in Table 1, the median age of the 181 patients was 70 years old, and the rates of male patients and patients with Eastern Cooperative Oncology Group performance status (PS) worse than 2 were 49.2% and 19.3%, respectively. The PFS and OS rates at 3 years of 181 patients were 68.7% and 79.7%, respectively. All three prognostic indices used in the study (R‐IPI, NCCN‐IPI, and KPI) largely successfully stratified the risks of patients, and survival of the KPI high‐risk group was the poorest in our analysis (Fig. S1).
Table 1.
Clinical background of the patients
| Item | Value |
|---|---|
| Age, median (range) | 70 (34–88) |
| Gender | |
| Male, n (%) | 89 (49.2) |
| Female, n (%) | 92 (50.8) |
| Performance status, n (%) | |
| 0–1 | 146 (80.7) |
| ≥2 | 35 (19.3) |
| Ann Arbor‐defined disease stage, n (%) | |
| Limited | 84 (46.4) |
| Advanced | 97 (53.6) |
| Serum LDH level, n (%) | |
| Normal range | 75 (41.4) |
| > x1–3 UNL | 86 (47.5) |
| ≥ x3 UNL | 20 (11.0) |
| 3‐year PFS (%) | 68.7 |
| 3‐year OS (%) | 79.7 |
| R‐IPI, n (%) | |
| Very Good | 76 (12.3) |
| Good | 24 (38.9) |
| Poor | 81 (48.8) |
| NCCN‐IPI, n (%) | |
| Low | 22 (10.8) |
| Low‐Int | 69 (35.5) |
| High‐Int | 59 (34.5) |
| High | 31 (19.2) |
| KPI, n (%) | |
| Low | 71 (36.0) |
| Low‐Int | 70 (38.9) |
| High‐Int | 15 (9.4) |
| High | 25 (15.8) |
PFS, progression‐free survival; OS, overall survival; R‐IPI, revised International Prognostic Index; NCCN‐IPI, National Comprehensive Cancer Network‐IPI; KPI, Kyoto Prognostic Index.
Results of chromosomal analysis
Among the 181 patients with biopsied specimens subjected to G‐banding, metaphase spreads were available for karyotypic analysis by G‐banding in 120 patients, and not available in 61 patients (Tables 2 and S1, Fig. S2). Neither OS nor PFS differed significantly between these groups of patients (Fig. S3). In the 120 patients with available metaphase spreads, 103 and 17 had abnormal and normal karyotypes, respectively. Regarding structural chromosomal abnormalities, 14q32 rearrangements were identified in 26 patients (21.7%), and other abnormalities detected at a rate of ≥5.0% included chromosomal rearrangements involving 3q27, 7q22, 8q24, 9p13, 11q13, and 18q21, which were identified in 16 (13.3%), 7 (5.8%), 8 (6.7%), 11 (9.2%), 6 (5.0%), and 20 (16.7%) cases, respectively. Numerous numerical chromosomal abnormalities were also detected, including both chromosomal gain and loss: The most frequent gains were +3 (N = 19, 15.8%), +7 (N = 18, 15.0%), and +18 (N = 16, 13.3%), and the most frequent losses were −13 (N = 27, 22.5%), −14 (N = 20, 16.7%), −4 (N = 20, 16.7%), −8 (N = 19, 15.8%), and −10 (N = 20, 16.7%) (Fig. 2). Marker chromosomes were also frequently observed. In the 103 patients with abnormal karyotypes, 40 harbored a single type chromosomal aberration only (i.e., one CAV), 19 had two CAVs, and 44 had ≥3 CAVs (Table S1).
Table 2.
Comparison between patients with ≥3 chromosomal abnormality variations (CAVs) and 0–2 CAVs in 120 patients with available metaphase spreads
| Subject | Total | CAV 0–2 | CAV ≥3 | P |
|---|---|---|---|---|
| Patient number | 120 | 76 | 44 | |
| Age, median (range) | 67.7 (34–85) | 67.8 | 67.6 | 0.917 |
| Gender (n) | ||||
| Male | 63 | 40 | 23 | 1.000 |
| Female | 57 | 36 | 21 | |
| Performance status (n) | ||||
| 0–1 | 92 | 65 | 27 | 0.005 |
| ≥2 | 28 | 11 | 17 | |
| Ann Arbor‐defined disease stage (n) | ||||
| Limited | 50 | 34 | 16 | 0.481 |
| Advanced | 70 | 42 | 28 | |
| Serum LDH level (n) | ||||
| Normal range | 50 | 35 | 15 | 0.378 |
| > x1–3 UNL | 53 | 32 | 21 | |
| ≥ x3 UNL | 17 | 9 | 8 | |
| Extranodal involvement (n) | ||||
| None | 59 | 39 | 20 | 0.668 |
| Present | 61 | 37 | 24 | |
| R‐IPI (n) | ||||
| Very Good/Good | 60 | 45 | 15 | 0.014 |
| Poor | 60 | 31 | 29 | |
| NCCN‐IPI (n) | ||||
| Low/Low‐Int/High‐Int | 95 | 64 | 31 | 0.120 |
| High | 25 | 12 | 13 | |
| KPI (n) | ||||
| Low/Low‐Int/High‐Int | 98 | 67 | 31 | 0.030 |
| High | 22 | 9 | 13 | |
The presence of ≥3 CAVs is shown to have significant association with PS, OS, R‐IPI, and KPI, but no association with other clinical backgrounds.
Figure 2.
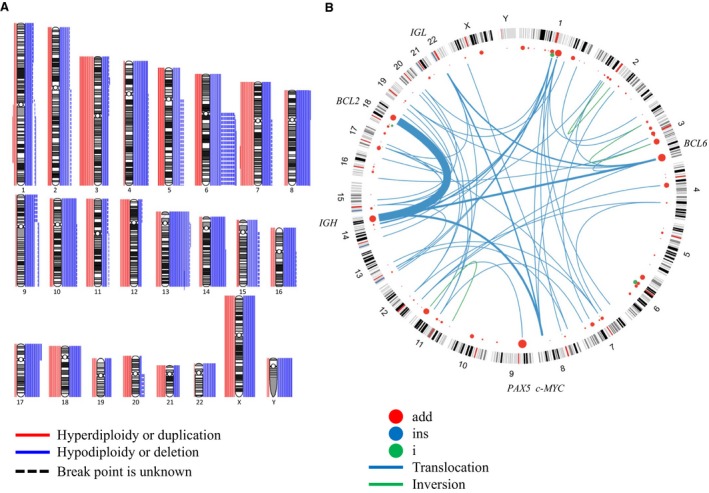
Numerical chromosomal abnormalities and chromosomal rearrangement break points/translocations. (A) Numerical abnormalities. Red lines on the left of each karyogram indicate gain or hyperdiploidy, and blue lines on the right indicate loss or hypodiploidy. In cases in which precise break points were not identified, dashed lines are shown. The number of lines for each chromosome shows the number of tumors with the abnormality. The frequent gains were +3 (N = 19), +7 (N = 18), and +18 (N = 16), and the frequent losses were −13 (N = 27), −14 (N = 20), −4 (N = 20), −8 (N = 19), and −10 (N = 20). (B) Structural abnormalities. Red points indicate break points of additional materials of unknown origins. The size of each point shows the number of tumors. Blue lines are chromosomal translocations. Each line weight shows the number of tumors. Abnormalities detected at a rate of >5.0% included chromosomal rearrangements involving 3q27 (N = 16), 7q22 (N = 7), 8q24 (N = 8), 9p13 (N = 11), 11q13 (N = 6), 14q32 (N = 29), and 18q21 (N = 20).
Prognostic impacts of sites of chromosomal rearrangements and number of CAVs
In investigations of the impact of sites of chromosomal rearrangements on survival outcomes, though the rearrangements involving 3q27, 7q22, 8q24, 9p13, 11q13, 14q32, or 18q21 tended to show elevated hazard ratio (HR) for OS, the statistical significance was not made clear (Fig. 3). In contrast, cases with ≥3 CAVs had significantly poorer OS compared to those with 0–2 CAVs (HR: 2.222, 95% confidence interval (CI): 1.056–4.677, P = 0.031) (Fig. 4A), and tended to have shorter PFS (HR: 1.796, 95% CI: 0.965–3.344, P = 0.061) (Fig. 4B). OS and PFS did not differ significantly between patients with one CAV and those without a CAV (data not shown). To adjust confounding, we next performed multivariate analysis including ≥3 CAVs with age and gender, which are obviously clear of influence by the number of CAVs. Presence of ≥3 CAVs showed significant impact to OS (HR: 2.142, 95% CI: 1.016–4.515, P = 0.045) and tended to show negative influence to PFS (HR: 1.768, 95% CI: 0.949–3.292, P = 0.072). We also performed multivariate analysis including ≥3 CAVs, age, gender, stage, and extranodal involvement, and the presence of ≥3 CAVs still showed relation to elevated hazard ratio for OS (HR: 2.066, 95% CI: 0.975–4.377, P = 0.058) and PFS (HR: 1.718, 95% CI: 0.921–3.207, P = 0.089). We did not regard the rest of prognostic factors utilized in R‐IPI, NCCN‐IPI, and KPI (poor performance status, elevated lactate dehydrogenase (LDH), and decreased albumin) as confounders, because it was unlikely that these factors affect the number of CAVs.
Figure 3.
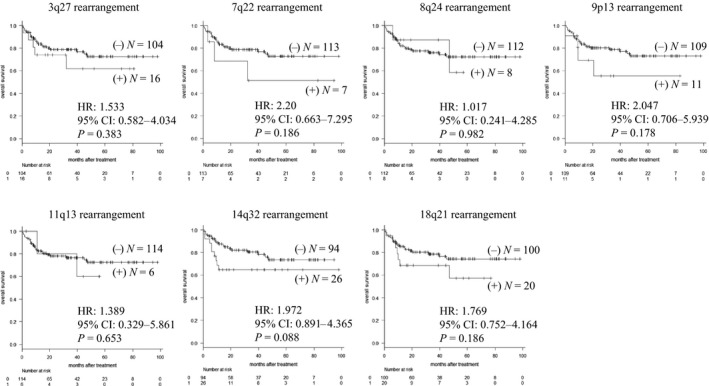
Overall survival of patients with and without chromosomal rearrangements involving 3q27, 7q22, 8q24, 9p13, 11q13, 14q32, and 18q21. HR, hazard ratio; CI, confidence interval. None of the sites of chromosomal rearrangement was significantly associated with OS.
Figure 4.
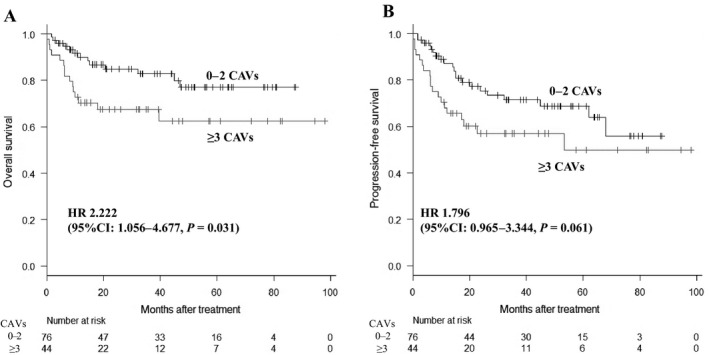
Overall survival (A) and progression‐free survival (B) of patients with 0–2 and ≥3 CAVs. HR, hazard ratio; CI, confidence interval. Cases with ≥3 CAVs had significantly poorer 3‐year OS compared to those with 0–2 CAVs (67.6% vs. 82.8%, P = 0.031) and tended to have shorter PFS.
Relationships among background factors, prognostic indexes, and number of CAVs
An evaluation of relationships between background factors and the number of CAVs showed that only PS ≥2 was significantly associated with ≥3 CAVs. Age, gender, serum LDH level, extranodal involvement, and Ann Arbor disease stage were not associated with the number of CAVs. In risk stratification using prognostic indices for DLBCL, patients with ≥3 CAVs were significantly more frequently found to be at high risk using the R‐IPI and KPI (Table 2). In each prognostic index‐defined risk group, the number of CAVs was not significantly associated with different survival outcomes of patients (data not shown).
Discussion
Acquisition of additional chromosomal abnormality (i.e., karyotypic evolution) is fueled by chromosomal instability due to loss of chromosome fidelity 18 and is frequently observed in hematologic malignancies in daily clinical practice. The prognostic impact of karyotypic evolution differs depending on the type of hematologic malignancy. For instance, it is associated with a poor prognosis in myelodysplastic syndrome 19, but does not significantly influence the prognosis of acute leukemias with core‐binding factor translocations or with t(9;11) 20. In DLBCL, the clinical impact of karyotypic evolution has not been fully evaluated, while the failure of anaphase accomplishment has been associated with a poor prognosis 21.
This study shows that chromosomal abnormality with ≥3 CAVs is related to a poor prognosis in DLBCL. An increased number of CAVs might be led by chromosomal instability and cause more advanced karyotypic evolution. The number of CAVs was also correlated with prognostic indices based on background factors and laboratory findings. Thus, our findings reveal that high‐risk patients defined by R‐IPI or KPI have distinct cytogenetic instability, compared to patients stratified as lower risk by these indices. These results suggest a need for identification of molecular mechanisms underlying chromosome instability in high‐risk patients. Our study showed the positive association between ≥3 CAVs and worse prognosis, while one or two CAVs did not show prognostic impact in DLBCL. Although the precise mechanism underlying the different prognostic impact between ≥3 CAVs and 1/2 CAVs remained to be verified, we suspect that ≥3 CAVs surrogates the boundary of chromosome instability which apparently makes the clinical manifestation more progressive with R‐CHOP(‐like) therapy.
In G‐banding analysis used in daily clinical practice, we found that the number of CAVs is often unclear, especially when there are many different CAVs to analyze. This problem was the major limitation in this study. However, even in this situation, it was possible to divide DLBCL into cases with 0, 1, 2, and ≥3 CAVs. We eventually focused on comparison of DLBCL with 0–2 and ≥3 CAVs because OS was most markedly different between these two groups. Another limitation was that metaphase spreads from G‐banding were not available in about one‐third of biopsied DLBCL specimens. Because our study suggests that increased number of CAVs due to chromosomal instability contribute to the development of high‐risk disease feature in DLBCL, our next research is focusing on the development of novel method which enables the detection of CAVs or chromosomal instability using nondividing cells and thereby provides a clue for therapeutic choice which is available in all patients with DLBCL in future.
Previous studies have evaluated the prognostic impacts of cytogenetic and molecular abnormalities in DLBCL. Although double‐hit or triple‐hit B‐cell lymphomas are well‐known with worse prognosis, previous studies concerning the prognostic impact of a c‐MYC rearrangement alone have been controversial. Some studies demonstrated that a c‐MYC rearrangement leads to poor prognosis even without BCL2 or BCL6 rearrangements 22, 23, while a c‐MYC rearrangement alone did not show significant prognostic impact in other studies 24, 25. The rearrangement of BCL2, BCL6, or PAX5 alone has not been associated with worse outcome in DLBCL patients 26, 27, while the prognostic impact of CCND1 rearrangement in DLBCL has been controversial 28. In this study, we found no significant association between the OS and chromosomal rearrangements involving 8q24, the site where c‐MYC locates, as well as rearrangements at other break points, including 3q27 involving BCL6, 9p13 involving PAX5, 11q13 involving CCND1, and 18q21 involving BCL2. While our data did not clarify the prognostic impact of the chromosomal rearrangements at specific break points, the number of CAVs showed significant association to the OS.
With recent advanced genomic technologies, such as gene expression profiling (GEP), microRNA (miRNA) profiling, genome‐wide copy number abnormalities, global methylation, mutation spectrum, and whole‐genome sequencing (WGS), more detailed molecular changes have been identified in DLBCL. The validity of GEP classification which reflects the cell‐of‐origin of tumor cells, that is, germinal center B‐cell‐like (GCB) type and activated B‐cell‐like (ABC) type 29, is supported by other study with high‐resolution genome‐wide copy number analysis 30. In addition, gene methylation profiles and miRNA signatures have been reported to be different between GCB‐DLBCL and ABC‐DLBCL 31, 32. WGS has identified widespread genomic mutations/rearrangements which are involved in lymphomagenesis of DLBCL 33. Concerning the genomic instability of DLBCL, targeted sequencing of 73 key DNA repair genes discovered somatic alterations in several novel and/or potentially functional important mutation targets in DLBCL, including CHEK2, PARP1, and DDB1, and several nonhomologous end‐joining (NHEJ) genes (DLRE1C, PRKDC, XRCC5, and XRCC6), as well as mismatch repair (MMR) genes (EXO1, MSH2, and MSH6) 34. Somatic mutations in DNA repair genes affected approximately half of DLBCL cases analyzed, and mutations in subsets of these genes, especially those belonging to the MMR and NHEJ pathways, indeed associated with different forms of genetic instability in tumors. However, those advanced genomic technologies have not been routinely used in daily clinical practice for DLBCL due to its technical difficulty and high cost. We, in this study, propose the relation of the number of CAVs to the prognosis of DLBCL which can be easily determined from routinely performed G‐banding. Future studies are necessary to explore the relationships among the number of CAVs, GEP classification, and gene mutation profiles for genetic instability in DLBCL.
Compared with GCB‐DLBCL, ABC‐DLBCL has been associated with an inferior prognosis even with rituximab‐containing immunochemotherapy, while recent studies have shown that strategies using lenalidomide, an immunomodulatory drug, or targeting B‐cell receptor signal improve the treatment outcome of ABC‐DLBCL 35, 36. Considering that difference in biological character between ABC‐DLBCL and GCB‐DLBCL is based on the distinct role of BCR signal pathway which is a completely different perspective from chromosomal instability, we expect patients with ≥3 CAVs to be found in both ABC and GCB‐DLBCL groups. However, it is possible that the clinical impact of the number of CAVs differs between ABC and GCB‐DLBCL, which is the remaining question we next have to work on.
In conclusion, this study suggests that more advanced cytogenetic evolution reflected by more CAVs is related to development of high‐risk disease and poor prognosis in DLBCL under R‐CHOP‐like strategy. The molecular basis for chromosomal instability requires further studies for identification of a high‐risk biomarker and development of novel diagnostic method for chromosomal instability and targeted therapeutics.
Conflict of Interest
The authors have declared no conflict of interest.
Supporting information
Figure S1. Overall survival of patients classified by R‐IPI, NCCN‐IPI, and KPI.
Figure S2. Patient cohort selection.
Figure S3. Overall survival (A) and progression‐free survival (B) of patients with and without available metaphase spreads.
Table S1. Clinical features, chromosomal abnormalities and number of chromosomal abnormality variations (CAVs) in 120 DLBCL patients with available metaphase spreads.
Acknowledgments
This work was supported in part by a Grant‐in‐Aid for Scientific Research from The Ministry of Education, Culture, Sports, Science and Technology of Japan (MEXT KAKENHI 16K09856) (MT); by a Grant‐in‐Aid for Young Scientists (B) (JSPS KAKENHI Grant Number JP16K21284) (YC); by the National Cancer Center Research and Development Fund (29‐A‐3); by a grant (Practical Research for Innovative Cancer Control) from the Japan Agency for Medical Research and Development (AMED) (17ck0106348 h0001); and by the Takeda Science Foundation and Astra Zeneca (JK).
References
- 1. Morton, L. M. , Wang S. S., Devesa S. S., Hartge P., Weisenburger D. D., and Linet M. S.. 2006. Lymphoma incidence patterns by WHO subtype in the United States. 1992‐2001. Blood 107:265–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Coiffier, B. , Lepage E., Briere J., Herbrecht R., Tilly H., Bouabdallah R., et al. 2002. CHOP chemotherapy plus rituximab compared with CHOP alone in elderly patients with diffuse large‐B‐cell lymphoma. N. Engl. J. Med. 346:235–242. [DOI] [PubMed] [Google Scholar]
- 3. Pfreundschuh, M. , Schubert J., Ziepert M., Schmits R., Mohren M., Lengfelder E., et al. 2008. Six versus eight cycles of bi‐weekly CHOP‐14 with or without rituximab in elderly patients with aggressive CD20 + B‐cell lymphomas: a randomised controlled trial (RICOVER‐60). Lancet Oncol. 9:105–116. [DOI] [PubMed] [Google Scholar]
- 4. Landau, D. A. , Carter S. L., Getz G., and Wu C. J.. 2014. Clonal evolution in hematological malignancies and therapeutic implications. Leukemia 28:34–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Rowley, J. D. 1988. Chromosome studies in the non‐Hodgkin's lymphomas: the role of the 14;18 translocation. J. Clin. Oncol. 6:919–925. [DOI] [PubMed] [Google Scholar]
- 6. Horsman, D. E. , Gascoyne R. D., Coupland R. W., Coldman A. J., and Adomat S. A.. 1995. Comparison of cytogenetic analysis, southern analysis, and polymerase chain reaction for the detection of t(14; 18) in follicular lymphoma. Am. J. Clin. Pathol. 103:472–478. [DOI] [PubMed] [Google Scholar]
- 7. Swerdlow, S. H. , Yang W. I., Zukerberg L. R., Harris N. L., Arnold A., and Williams M. E.. 1995. Expression of cyclin D1 protein in centrocytic/mantle cell lymphomas with and without rearrangement of the BCL1/cyclin D1 gene. Hum. Pathol. 26:999–1004. [DOI] [PubMed] [Google Scholar]
- 8. Dalla‐Favera, R. , Bregni M., Erikson J., Patterson D., Gallo R. C., and Croce C. M.. 1982. Human c‐myc onc gene is located on the region of chromosome 8 that is translocated in Burkitt lymphoma cells. Proc. Natl Acad. Sci. USA 79:7824–7827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Dave, S. S. , Fu K., Wright G. W., Lam L. T., Kluin P., Boerma E. J., et al. 2006. Molecular diagnosis of Burkitt's lymphoma. N. Engl. J. Med. 354:2431–2442. [DOI] [PubMed] [Google Scholar]
- 10. Sesques, P. , and Johnson N. A.. 2017. Approach to the diagnosis and treatment of high‐grade B‐cell lymphomas with MYC and BCL2 and/or BCL6 rearrangements. Blood 129:280–288. [DOI] [PubMed] [Google Scholar]
- 11. Bochtler, T. , Fröhling S., and Krämer A.. 2015. Role of chromosomal aberrations in clonal diversity and progression of acute myeloid leukemia. Leukemia 29:1243–1252. [DOI] [PubMed] [Google Scholar]
- 12. Huang, H. , and Chen J.. 2017. Chromosome Bandings. Methods Mol. Biol. 1541:59–66. [DOI] [PubMed] [Google Scholar]
- 13. Shaffer, L. G. , McGowan‐Jordan J., and Schmid M.. 2013. ISCN 2013: an international system for human cytogenetic nomenclature. P. 140 Karger Medical and Scientific Publishers, Basel. [Google Scholar]
- 14. International Non‐Hodgkin's Lymphoma Prognostic Factors Project . 1993. A predictive model for aggressive non‐Hodgkin's lymphoma. New Engl. J. Med. 329:987–994. [DOI] [PubMed] [Google Scholar]
- 15. Sehn, L. H. , Berry B., Chhanabhai M., Fitzgerald C., Gill K., Hokin P., et al. 2007. The revised International Prognostic Index (RIPI) is better predictor of outcome than the standard IPI for patients with diffuse large B‐cell lymphoma treated with R‐CHOP. Blood 109:1857–1861. [DOI] [PubMed] [Google Scholar]
- 16. Zou, Z. , Sehn L. H., Rademaker A. W., Gordon L. I., Lacasce A. S., Crosby‐Thompson A., et al. 2014. An enhanced International Prognostic Index (NCCN‐IPI) for patients with diffuse large B‐cell lymphoma treated in the rituximab era. Blood 123:837–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kobayashi, T. , Kuroda J., Yokota I., Tanba K., Fujino T., Kuwahara S., et al. 2016. The Kyoto Prognostic Index for patients with diffuse large B‐cell lymphoma in the rituximab era. Blood Cancer J. 6:e383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bakhoum, S. F. , and Compton D. A.. 2012. Chromosomal instability and cancer: a complex relationship with therapeutic potential. J. Clin. Invest. 122:1138–1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Heilig, C. E. , Löffler H., Mahlknecht U., Janssen J. W., Ho A. D., Jauch A., et al. 2010. Chromosomal instability correlates with poor outcome in patients with myelodysplastic syndromes irrespectively of the cytogenetic risk group. J. Cell Mol. Med. 14:895–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Byrd, J. C. , Mro′zek K., Dodge R. K., Carroll A. J., Edwards C. G., Arthur D. C., et al. 2002. Pretreatment cytogenetic abnormalities are predictive of induction success, cumulative incidence of relapse, and overall survival in adult patients with de novo acute myeloid leukemia: results from Cancer and Leukemia Group B (CALGB 8461). Blood 100:4325–4336. [DOI] [PubMed] [Google Scholar]
- 21. Bakhoum, S. M. , Danilova O. V., Kaur P., Levy N. B., and Compton D. A.. 2011. Chromosomal instability substantiates poor prognosis in patients with diffuse large B‐cell lymphoma. Clin. Cancer Res. 17:7704–7711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Landsburg, D. J. , Falkiewicz M. K., Petrich A. M., Chu B. A., Behdad A., Li S., et al. 2016. Sole rearrangement but not amplification of MYC is associated with a poor prognosis in patients with diffuse large B cell lymphoma and B cell lymphoma unclassifiable. Br. J. Haematol. 175:631–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Li, S. , Weiss V. L., Wang X. J., Desai P. A., Hu S., Yin C. C., et al. 2016. High‐grade B‐cell lymphoma with MYC rearrangement and without BCL2 and BCL6 rearrangements is associated with high P53 expression and a poor prognosis. Am. J. Surg. Pathol. 40:253–261. [DOI] [PubMed] [Google Scholar]
- 24. Cunningham, D. , Hawkes E. A., Jack A., Qian W., Smith P., Mouncey P., et al. 2013. Rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisolone in patients with newly diagnosed diffuse large B‐cell non‐Hodgkin lymphoma: a phase 3 comparison of dose intensification with 14‐day versus 21‐day cycles. Lancet 381:1817–1826. [DOI] [PubMed] [Google Scholar]
- 25. Lai, C. , Roschewski M., Melani C., Pittaluga S., Shovlin M., Steinberg S. M., et al. 2018. MYC gene rearrangement in diffuse large B‐cell lymphoma does not confer a worse prognosis following dose‐adjusted EPOCH‐R. Leuk. Lymphoma 59:505–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Dunleavy, K. 2015. Aggressive B cell Lymphoma: Optimal Therapy for MYC‐positive, Double‐Hit, and Triple‐Hit DLBCL. Curr. Treat. Options Oncol. 16:58. [DOI] [PubMed] [Google Scholar]
- 27. Je, E. M. , Yoo N. J., and Lee S. H.. 2014. PAX5 somatic mutation is rare in multiple myelomas and non‐Hodgkin lymphomas of Korean patients. Hematol. Oncol. 32:110–111. [DOI] [PubMed] [Google Scholar]
- 28. Jiang, C. , Shi X., and Fan C.. 2017. A cyclin D1‐positive diffuse large B‐cell lymphoma of germinal center B‐cell‐like subtype in the right tonsil: a rare case report. Medicine (Baltimore) 96:e6311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Alizadeh, A. A. , Eisen M. B., Davis R. E., Ma C., Lossos I. S., Rosenwald A., et al. 2000. Distinct types of diffuse large B‐cell lymphoma identified by gene expression profiling. Nature 403:503–511. [DOI] [PubMed] [Google Scholar]
- 30. Lenz, G. , Wright G. W., Emre N. C., Kohlhammer H., Dave S. S., Davis R. E., et al. 2008. Molecular subtypes of diffuse large B‐cell lymphoma arise by distinct genetic pathways. Proc. Natl Acad. Sci. USA 105:13520–13525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Shaknovich, R. , Geng H., Johnson N. A., Tsikitas L., Cerchietti L., Greally J. M., et al. 2010. DNA methylation signatures define molecular subtypes of diffuse large B‐cell lymphoma. Blood 116:e81–e89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Iqbal, J. , Shen Y., Huang X., Liu Y., Wake L., Liu C., et al. 2015. Global microRNA expression profiling uncovers molecular markers for classification and prognosis in aggressive B‐cell lymphoma. Blood 125:1137–1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Morin, R. D. , Mungall K., Pleasance E., Mungall A. J., Goya R., Huff R. D., et al. 2013. Mutational and structural analysis of diffuse large B‐cell lymphoma using whole‐genome sequencing. Blood 122:1256–1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. de Miranda, N. F. , Peng R., Georgiou K., Wu C., Falk S. E., Berglund M., et al. 2013. DNA repair genes are selectively mutated in diffuse large B cell lymphomas. J. Exp. Med. 210:1729–1742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Nowakowski, G. S. , LaPlant B., Macon W. R., Reeder C. B., Foran J. M., Nelson G. D., et al. 2015. Lenalidomide combined with R‐CHOP overcomes negative prognostic impact of non‐germinal center B‐cell phenotype in newly diagnosed diffuse large B‐Cell lymphoma: a phase II study. J. Clin. Oncol. 33:251–257. [DOI] [PubMed] [Google Scholar]
- 36. Zhang, L. H. , Kosek J., Wang M., Heise C., Schafer P. H., Chopra R., et al. 2013. Lenalidomide efficacy in activated B‐cell‐like subtype diffuse large B‐cell lymphoma is dependent upon IRF4 and cereblon expression. Br. J. Haematol. 160:487–502. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Overall survival of patients classified by R‐IPI, NCCN‐IPI, and KPI.
Figure S2. Patient cohort selection.
Figure S3. Overall survival (A) and progression‐free survival (B) of patients with and without available metaphase spreads.
Table S1. Clinical features, chromosomal abnormalities and number of chromosomal abnormality variations (CAVs) in 120 DLBCL patients with available metaphase spreads.