

Abstract
Enzymatic installation of chlorine/bromine upon unactivated carbon centers provides a versatile, selective, and environmentally-friendly alternative to chemical halogenation. Iron(II)- and 2-(oxo)-glutarate (FeII/2OG)-dependent halogenases are powerful biocatalysts that are capable of cleaving aliphatic C-H bonds to install useful functional groups, including halogens. Using the structure of the Fe/2OG halogenase, WelO5, in complex with its small-molecule substrate, we identified a similar N-acyl amino acid hydroxylase, SadA, and reprogrammed it to halogenate its substrate, thereby generating a new chiral haloalkyl center. The work high-lights the potential of FeII/2OG enzymes as platforms for development of novel stereospecific catalysts for late-stage C-H functionalization.
Graphical abstract
Oxygen-activating FeII/2OG enzymes routinely chlorinate or brominate aliphatic carbon centers in reactions that, if successfully harnessed, could afford synthetically useful catalysts for halogenation of unactivated C-H bonds.1, 2 Eleven different FeII/2OG halogen-installing enzymes have been identified and biochemically characterized to date.3–13 Nine of these systems modify aliphatic centers in amino-acyl or fatty-acyl substrates and require a second carrier protein to deliver the chlorination/bromination target to the halogenase active site. The structural complexity and instability of the carrier-protein-linked substrates have been significant impediments to their use in synthetic applications. However, the recent discovery of two carrier-protein-independent FeII/2OG halogenases WelO5 and AmbO5,12, 14, 15 responsible for regio- and stereo-selective chlorination of aliphatic carbons in hapalindole-type indole alkaloids, provides new opportunities to overcome the barriers imposed by the general requirement for a carrier protein.
The FeII/2OG-dependent halogenases are part of a much larger superfamily of FeII/2OG oxygenases, of which the vast majority act as hydroxylases.2 In all superfamily members characterized to date, reaction of a 2OG-chelated FeII center with O2 proceeds through a hydrogen-abstracting FeIV=O (ferryl) intermediate that is produced in conjunction with breakdown of 2OG into succinate and CO2 (fig. S1). Ferryl accumulation has been observed in both the prototypical hydroxylases16, 17 as well as in the carrier-protein-dependent halogenases CytC318 and SyrB2.19 In all of these systems, the oxo unit of the ferryl intermediate removes a hydrogen atom (H•) from the primary substrate, yielding a substrate carbon radical (C•) and an FeIII-OH complex. In the hydroxylases, attack of the substrate C• upon the –OH ligand yields the hydroxylated product and an FeII complex.
FeII/2OG enzymes share a characteristic His-X-(Asp/Glu)-Xn-His metal-binding motif, in which the His and carboxylate residues form a triad of protein-derived iron ligands. The halogenases, however, universally deviate from this pattern and replace the carboxylate residue with Ala or Gly, an effective ligand deletion that opens a coordination site for the halide ion that is ultimately installed upon a substrate carbon during the reaction. During catalysis, the halogenases couple the substrate C• to the FeIII-coordinated halogen, enabled by substitution of the Asp/Glu carboxylate ligand found in the hydroxylases to accommodate coordination of –Cl or –Br.20 Given the relatively minimal modification required to promote alternative group transfer, universal translation of this activity to the more diverse set of substrates targeted within the larger superfamily of FeII/2OG enzymes could seemingly be accomplished by deletion of the carboxylate ligand. Several research groups have attempted to reprogram well-characterized FeII/2OG hydroxylases by single-site substitution of the Asp in the His-X-Asp motif. In the TauD and prolyl-4-hydroxlase targets tested thus far, this approach has not been successful. Loss of the carboxylate ligand additionaly yields an inability to form a productive FeII-2OG complex and failure to undergo substrate-triggered reaction with O2.21, 22
The difficulty in achieving halogenation by ligand deletion in a generic hydroxylation scaffold can also be rationalized by an additional requirement for a unique disposition of the halogenation substrate in the active site. A comprehensive investigation of SyrB2 reactivity showed that the halogenation target must reside distal to the oxo moiety of the ferryl intermediate when compared to substrates that are primarily hydroxylated.23, 24 Our recent x-ray structure of WelO5 in complex with its chlorination substrate, 12-epi-fischerindole U, revealed that the distinctive juxtaposition is accomplished by reconfiguring the FeII ligands at an intermediate stage of the reaction to move the oxygen-derived ligand away from substrate to suppress –OH transfer.25 The structure suggests the change occurs after addition of O2 to the WelO5•FeII•2OG•Cl/Br complex but prior to formation of the haloferryl intermediate. In reaction with substrate, the oxo unit of the WelO5 FeIV=O complex is oriented trans to the proximal His ligand and in the equatorial plane defined by the –Cl ligand and the succinate coproduct (fig. S1). This off-line oxo configuration is promoted by an unusual 2OG conformation and key second-sphere interactions, both distinct from those of canonical FeII/2OG hydroxylases. We show here that the structure of WelO5 can be used to identify an FeII/2OG hydroxylase platform that might be more easily converted to a halogenation catalyst by rational site-directed mutagenesis. In the appropriate structural context, reprogramming of outcome can be achieved by just a single substitution to enable halide coordination.
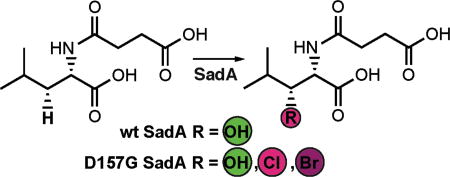
Query of the DALI 3-D structural comparison server26 with the coordinates of WelO5 identified the enzyme SadA from Burkholderia ambifaria (PDB accession code 3W21) as the match with the highest Z-score (fig. S2). Surprisingly, this hydroxylase, which shares only 19% sequence identity with WelO5, ranks above several carrier-protein-dependent halogenases. SadA was originally identified as part of an in vitro screen for catalysts that hydroxylate N-modified aliphatic and aromatic amino acids (Scheme 1).27 The enzyme has been characterized and optimized for use in the production of optically pure β-hydroxy amino acids.27, 28 Although the protein shares low overall sequence identity and modest alignment of topology (rmsd 2.6 Å over 193 Cα atoms) (fig. S3) with WelO5, the structures of the active sites of both proteins are strikingly similar (Fig. 1b).29 Note that SadA retains the FeII carboxylate ligand of the His-X-(Asp/Glu) motif, as is typical of FeII/2OG hydroxylases.
Scheme 1. Reactions catalyzed by SadA.

Figure 1.

A comparison of the active sites in halogenase WelO5 (a) and hydroxylase SadA (b). Gly 179 and Asp 157 in SadA were targeted for mutagenesis.
Reactions of wild-type (wt) SadA and its variants with N-succinyl-L-leucine substrate 1 were carried out under multiple turnover conditions and analyzed by liquid chromatography coupled to mass spectrometry (LC-MS; Fig. 2a). The wt enzyme catalyzed near complete consumption of the substrate and formation of a product with m/z ratio of 246.1, identical to the calculated mass of the anticipated β-hydroxy-N-succinyl-L-Leu product 2. The structure of 2 was verified by NMR analysis of the HPLC-purified product. Substitution of active site FeII carboxylate ligand Asp 157 by a Gly residue, giving D157G SadA, combined with inclusion of NaCl in the reaction mixture at concentrations in excess of 50 mM, yielded a new product 3 with a m/z ratio of 264.1 (Fig. 2a). This value corresponds to loss of a single hydrogen atom from N-succinyl-L-Leu and its replacement by chlorine. Note that production of 3 is not observed in the wild-type enzyme and occurs only after deletion of the carboxylate ligand in this scaffold. However, the modification is not sufficient to fully convert the outcome to exclusive halogenation and the peak for hydroxylated compound 2 is still present in significant amounts (70% of total product pool; Fig. 2b). The ligand substitution additionally results in FeII oxidation that is not linked to substrate conversion. Only 40% of D157G SadA succinate production is coupled to processing of 1, significantly less than the near 1:1 value measured in the wt reaction (fig. S4). Inclusion of NaBr in the reaction mixture yields analogous species 4 containing a single Br atom, a product with m/z = 308. The variant enzyme prefers the smaller chloride anion (Fig. 2b) resulting in greater production of 3 when both halides are present in equal proportions.
Figure 2.

Total ion chromatogram (TIC) showing production of chlorinated (3) and hydroxylated (2) N-succinyl-L-Leu with wt and variant SadA proteins under single turnover conditions (a). Asterisk denotes the peak for 2OG-derived co-product succinate. Reaction of D157G SadA with NaCl and/or NaBr yields chlorinated (3) and brominated (4) products (b). Single ion monitoring parameters and elution times are listed in Table S1.
The new activities of D157G SadA are 2OG-dependent (Fig. 3a) and product mass spectra are fully consistent with halogen installation (Fig. 3b–c). Chlorinated product, 3, exhibits the expected m/z + 2 peak at 1/3 intensity of the parent peak in the two ionized species that retain the halogen, associated with the naturallyoccurring 37Cl isotope (Fig. 3b, center and right). The dominant daughter ion (Fig. 3b, left) has lost the halide and the corresponding m/z + 2 peak is absent. The brominated product displays the anticipated 1:1 intensity ratio for its m/z + 2 peaks (Fig. 3c) in all three of the most prevalent fragments. The location of halogen installation in the HPLC-purified chlorination product 3 was verified by 1H-/13C-COSY and HSQC NMR spectroscopy. Comparison of the spectra to those of the hydroxylated product 2 confirms installation of the -Cl functional group at the C3 carbon of the Leu side chain, as evidenced by a downfield shift in the peaks associated with the protons at C2 and C3. Prior work established that the absolute stereochemical configuration of the wt SadA hydroxylation product 2 is L-threo-β-hydroxy-Leu,28 consistent with abstraction of the pro-(R) H-atom from C3. We anticipate an identical configuration for the halogenated compound generated by D157G SadA and observe a single product peak in our LC analyses of 3, implying formation of a single stereoisomer.
Figure 3.

Total ion chromatogram of D157G SadA reactions with and without 2OG (a). Observation of halogenated products 3 and 4 requires the cosubstrate. Mass spectra extracted at peak time points in (a) for chlorinated product 3 (b) and brominated product 4 (c). Intensities and m/z ratios are listed in Table S2.
To improve the chemoselectivity and efficiency of the engineered catalyst, with the ultimate goal of more complete conversion to the C3 halogenated version of 1, we inspected the SadA active site to identify additional sites that could contribute to control of outcome. We introduced a Ser residue that is implicated in stabilization of the uniquely disposed oxo/hydroxo ligand in WelO5,25 substituted for a native Gly at that position in SadA, in the background of the D157G variant (Fig. 1a). In WelO5, an S189A variant partially reverts the outcome to hydroxylation and we anticipated the opposite effect, a shift in favor of halogenation, in D157G/G179S SadA. Surprisingly, the ratio of –OH/–Cl modified products is unaltered in D157G/G179S (Fig. 2a). Diminished peak heights indicate a catalyst that is even less robust/ efficient than D157G SadA. These findings do not discount the importance of the Ser 179 equivalent in WelO5 halogenation. Instead, we hypothesize that wt SadA already employs an off-line intermediate stabilized by other means. Addition of ascorbate to D157G SadA also did not significantly improve activity, perhaps consistent with irreversible catalyst inactivation during turnover (fig. S5).
Recent efforts to exploit halogenases in catalyst-directed activation of C-H bonds have focused primarily on flavin-dependent tryptophan chlorinases30–32 and heme-based haloperoxidases.33, 34 Native and engineered versions of these proteins produce non-native halogenated compounds for use in stand-alone applications35–38 or as synthetic intermediates to novel aryl/amino/alkoxo compounds.39–42 With only a few exceptions,43, 44 the flavin- and heme-utilizing systems are limited to transformation of aromatic substrates. The ability to incorporate Fe/2OG halogenation platforms would significantly expand the chemical repertoire of such approaches to include routine halogenation of substrates with completely unactivated aliphatic carbon centers.
Supplementary Material
Acknowledgments
Funding Sources
A.J.M., X.L., and A.K.B. have filed an invention disclosure (U.S. Provisional No. 62/349,329) based on this work.
Funding sources include the University of Pittsburgh (X.L.), the National Institutes of Health (GM100011 to A.K.B.), and the Searle Scholars Program (A.K.B.).
We acknowledge the NMR facility and Prof. Xin Zhang in the Chemistry Department at the Pennsylvania State University for use of instrumentation. We also thank Lauren Rajakovich and Benjamin Allen for technical assistance, Cihad Sigindere for initial contribution of reagents, and J. Martin Bollinger Jr. for critical reading of the manuscript.
ABBREVIATIONS
- 2OG
2-(oxo)-glutarate
- wt
wild-type
- LC-MS
liquid chromatograpy mass spectrometry
- HPLC
high-performance liquid chromatography
- NMR
nuclear magnetic resonance
- COSY
correlation spectroscopy
- HSQC
heteronuclear single quantum coherence.
Footnotes
ASSOCIATED CONTENT
The Supporting Information is available free of charge on the ACS Publications website.
References
- 1.Vaillancourt FH, Yeh E, Vosburg DA, Garneau-Tsodikova S, Walsh CT. Chem Rev. 2006;106:3364–3378. doi: 10.1021/cr050313i. [DOI] [PubMed] [Google Scholar]
- 2.Bollinger JM, Jr, Chang W-c, Matthews ML, Martinie RJ, Boal AK, Krebs C. In: 2-oxoglutarate-dependent oxygenases. Hausinger RP, Schofield CJ, editors. The Royal Society of Chemistry; London: 2015. pp. 95–122. [Google Scholar]
- 3.Chang Z, Flatt P, Gerwick WH, Nguyen VA, Willis CL, Sherman DH. Gene. 2002;296:235–247. doi: 10.1016/s0378-1119(02)00860-0. [DOI] [PubMed] [Google Scholar]
- 4.Vaillancourt FH, Yeh E, Vosburg DA, O'Connor SE, Walsh CT. Nature. 2005;436:1191–1194. doi: 10.1038/nature03797. [DOI] [PubMed] [Google Scholar]
- 5.Vaillancourt FH, Yin J, Walsh CT. Proc Natl Acad Sci U S A. 2005;102:10111–10116. doi: 10.1073/pnas.0504412102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ueki M, Galonić DP, Vaillancourt FH, Garneau-Tsodikova S, Yeh E, Vosburg DA, Schroeder FC, Osada H, Walsh CT. Chem Biol. 2006;13:1183–1191. doi: 10.1016/j.chembiol.2006.09.012. [DOI] [PubMed] [Google Scholar]
- 7.Pratter SM, Ivkovic J, Birner-Gruenberger R, Breinbauer R, Zangger K, Straganz GD. Chembiochem. 2014;15:567–574. doi: 10.1002/cbic.201300345. [DOI] [PubMed] [Google Scholar]
- 8.Neumann CS, Walsh CT. J Am Chem Soc. 2008;130:14022–14023. doi: 10.1021/ja8064667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jiang W, Heemstra JR, Jr, Forseth RR, Neumann CS, Manaviazar S, Schroeder FC, Hale KJ, Walsh CT. Biochemistry. 2011;50:6063–6072. doi: 10.1021/bi200656k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Khare D, Wang B, Gu L, Razelun J, Sherman DH, Gerwick WH, Hakansson K, Smith JL. Proc Natl Acad Sci U S A. 2010;107:14099–14104. doi: 10.1073/pnas.1006738107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Galonić DP, Vaillancourt FH, Walsh CT. J Am Chem Soc. 2006;128:3900–3901. doi: 10.1021/ja060151n. [DOI] [PubMed] [Google Scholar]
- 12.Hillwig ML, Liu X. Nat Chem Biol. 2014;10:921–923. doi: 10.1038/nchembio.1625. [DOI] [PubMed] [Google Scholar]
- 13.Hillwig ML, Zhu Q, Ittiamornkul K, Liu X. Angew Chem Int Ed Engl. 2016;55:5780–5784. doi: 10.1002/anie.201601447. [DOI] [PubMed] [Google Scholar]
- 14.Hillwig ML, Fuhrman HA, Ittiamornkul K, Sevco TJ, Kwak DH, Liu X. Chembiochem. 2014;15:665–669. doi: 10.1002/cbic.201300794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hillwig ML, Zhu Q, Liu X. ACS Chem Biol. 2014;9:372–377. doi: 10.1021/cb400681n. [DOI] [PubMed] [Google Scholar]
- 16.Price JC, Barr EW, Glass TE, Krebs C, Bollinger JM., Jr J Am Chem Soc. 2003;125:13008–13009. doi: 10.1021/ja037400h. [DOI] [PubMed] [Google Scholar]
- 17.Price JC, Barr EW, Tirupati B, Bollinger JM, Jr, Krebs C. Biochemistry. 2003;42:7497–7508. doi: 10.1021/bi030011f. [DOI] [PubMed] [Google Scholar]
- 18.Galonić DP, Barr EW, Walsh CT, Bollinger JM, Jr, Krebs C. Nat Chem Biol. 2007;3:113–116. doi: 10.1038/nchembio856. [DOI] [PubMed] [Google Scholar]
- 19.Matthews ML, Krest CM, Barr EW, Vaillancourt FH, Walsh CT, Green MT, Krebs C, Bollinger JM., Jr Biochemistry. 2009;48:4331–4343. doi: 10.1021/bi900109z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Blasiak LC, Vaillancourt FH, Walsh CT, Drennan CL. Nature. 2006;440:368–371. doi: 10.1038/nature04544. [DOI] [PubMed] [Google Scholar]
- 21.Grzyska PK, Muller TA, Campbell MG, Hausinger RP. J Inorg Biochem. 2007;101:797–808. doi: 10.1016/j.jinorgbio.2007.01.011. [DOI] [PubMed] [Google Scholar]
- 22.Gorres KL, Pua KH, Raines RT. PLoS One. 2009;4:e7635. doi: 10.1371/journal.pone.0007635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Matthews ML, Neumann CS, Miles LA, Grove TL, Booker SJ, Krebs C, Walsh CT, Bollinger JM., Jr Proc Natl Acad Sci U S A. 2009;106:17723–17728. doi: 10.1073/pnas.0909649106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Martinie RJ, Livada J, Chang W-c, Green MT, Krebs C, Bollinger JM, Jr, Silakov A. J Am Chem Soc. 2015;137:6912–6919. doi: 10.1021/jacs.5b03370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mitchell AJ, Zhu Q, Maggiolo AO, Ananth NR, Hillwig ML, Liu X, Boal AK. Nat Chem Biol. 2016;12:636–640. doi: 10.1038/nchembio.2112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Holm L, Rosenström P. Nucleic Acids Res. 2010;38:W545–549. doi: 10.1093/nar/gkq366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hibi M, Kawashima T, Kasahara T, Sokolov PM, Smirnov SV, Kodera T, Sugiyama M, Shimizu S, Yokozeki K, Ogawa J. Lett Appl Microbiol. 2012;55:414–419. doi: 10.1111/j.1472-765X.2012.03308.x. [DOI] [PubMed] [Google Scholar]
- 28.Hibi M, Kasahara T, Kawashima T, Yajima H, Kozono S, Smirnov SV, Kodera T, Sugiyama M, Shimizu S, Yokozeki K, Ogawa J. Adv Synth Catal. 2015;357:767–774. [Google Scholar]
- 29.Qin HM, Miyakawa T, Jia MZ, Nakamura A, Ohtsuka J, Xue YL, Kawashima T, Kasahara T, Hibi M, Ogawa J, Tanokura M. PLoS One. 2013;8:e63996. doi: 10.1371/journal.pone.0063996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Brown S, O'Connor SE. Chembiochem. 2015;16:2129–2135. doi: 10.1002/cbic.201500338. [DOI] [PubMed] [Google Scholar]
- 31.Payne JT, Andorfer MC, Lewis JC. Methods Enzymol. 2016;575:93–126. doi: 10.1016/bs.mie.2016.03.024. [DOI] [PubMed] [Google Scholar]
- 32.van Pée KH, Milbredt D, Patallo EP, Weichold V, Gajewi M. Methods Enzymol. 2016;575:65–92. doi: 10.1016/bs.mie.2016.03.022. [DOI] [PubMed] [Google Scholar]
- 33.Yamada R, Higo T, Yoshikawa C, China H, Ogino H. J Biotechnol. 2014;192:248–254. doi: 10.1016/j.jbiotec.2014.10.030. [DOI] [PubMed] [Google Scholar]
- 34.Yamada R, Higo T, Yoshikawa C, China H, Yasuda M, Ogino H. Biotechnol Prog. 2015;31:917–924. doi: 10.1002/btpr.2117. [DOI] [PubMed] [Google Scholar]
- 35.Andorfer MC, Park HJ, Vergara-Coll J, Lewis JC. Chem Sci. 2016;7:3720–3729. doi: 10.1039/c5sc04680g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Payne JT, Poor CB, Lewis JC. Angew Chem Int Ed Engl. 2015;54:4226–4230. doi: 10.1002/anie.201411901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shepherd SA, Menon BRK, Fisk H, Struck AW, Levy C, Leys D, Micklefield J. Chembiochem. 2016;17:821–824. doi: 10.1002/cbic.201600051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shepherd SA, Karthikeyan C, Latham J, Struck AW, Thompson ML, Menon BRK, Styles MQ, Levy C, Leys D, Micklefield J. Chem Sci. 2015;6:3454–3460. doi: 10.1039/c5sc00913h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Deb Roy A, Gruschow S, Cairns N, Goss RJ. J Am Chem Soc. 2010;132:12243–12245. doi: 10.1021/ja1060406. [DOI] [PubMed] [Google Scholar]
- 40.Runguphan W, O'Connor SE. Org Lett. 2013;15:2850–2853. doi: 10.1021/ol401179k. [DOI] [PubMed] [Google Scholar]
- 41.Durak LJ, Payne JT, Lewis JC. ACS Catal. 2016;6:1451–1454. doi: 10.1021/acscatal.5b02558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Latham J, Henry JM, Sharif HH, Menon BR, Shepherd SA, Greaney MF, Micklefield J. Nat Commun. 2016;7:11873. doi: 10.1038/ncomms11873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chankhamjon P, Tsunematsu Y, Ishida-Ito M, Sasa Y, Meyer F, Boettger-Schmidt D, Urbansky B, Menzel KD, Scherlach K, Watanabe K, Hertweck C. Angew Chem Int Ed Engl. 2016;55:11955–11959. doi: 10.1002/anie.201604516. [DOI] [PubMed] [Google Scholar]
- 44.Podzelinska K, Latimer R, Bhattacharya A, Vining LC, Zechel DL, Jia ZC. J Mol Biol. 2010;397:316–331. doi: 10.1016/j.jmb.2010.01.020. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.