

Abstract
Glioblastoma is the most aggressive primary brain tumor in humans and is virtually incurable with conventional therapies. Chimeric antigen receptor (CAR) T cell therapy targeting the glioblastoma antigen EphA2 is an attractive approach to improve outcomes because EphA2 is expressed highly in glioblastoma but only at low levels in normal brain tissue. Building upon our previous findings in this area, we generated and evaluated a panel of EphA2-specific CARs. We demonstrate here that T cells expressing CD28.ζ and 41BB.ζ CARs with short spacers had similar effector function, resulting in potent antitumor activity. In addition, incorporating the 41BB signaling domain into CD28.ζ CARs did not improve CAR T cell function. While we could not determine functional differences between CD28.ζ, 41BB.ζ, and CD28.41BB.ζ CAR T cells, we selected CD28.ζ CAR T cells for further clinical development based on safety consideration.
Keywords: GBM, CAR T cells, EphA2, brain tumor
Introduction
Glioblastoma is the most common and highly aggressive primary brain tumor.1 Due to a lack of effective and curative therapies, the median life expectancy is approximately 15 months.2, 3 Therefore, novel therapeutic approaches are needed to improve the outcomes of patients with glioblastoma. Tumor antigen-targeted T cell immunotherapy has the potential to meet this need and reduce treatment-related complications because it does not rely on the cytotoxic mechanisms of conventional therapies. For example, chemotherapeutic agents induce tumor cell killing by interfering with DNA synthesis, inhibiting mitosis, or directly damaging DNA.4 In contrast, the anti-tumor activity of T cells is predominately dependent on lytic proteins such as perforin and granzyme.5 Indeed, chimeric antigen receptor (CAR)-engineered T cells targeting CD19 have shown remarkable antitumor activity for chemorefractory B cell-derived hematological malignancies resulting in their recent US Food and Drug Administration (FDA) approval.6, 7
For glioblastoma, clinical studies have been conducted with CAR T cells targeting epidermal growth factor receptor variant III (EGFRvIII), interleukin-13 receptor alpha-2 (IL-13Rα2), or human epidermal growth factor receptor 2 (HER2).8, 9, 10 While the infusion of cells was safe, only limited anti-glioma activity was observed, except in one patient, who received intraventricular injections of IL-13Rα2-CAR T cells.9 Limited anti-glioma activity most likely reflects the immunosuppressive glioma environment,10, 11 but also highlights the need to develop CARs targeting additional glioma-associated antigens.10, 12
We have previously demonstrated that CAR T cells targeting the glioma-associated antigen ephrin type A receptor 2 (EphA2) have anti-glioma activity in vitro and in a preclinical glioma xenograft model.13 The CAR consisted of an EphA2-specifc single-chain variable fragment (scFv) derived from the monoclonal antibody (mAb) 4H5, which recognizes a conformational epitope that is exposed only on malignant cells,14 a CH2CH3 spacer, a CD28 transmembrane domain, and a CD28.ζ signaling domain. However, CH2CH3 spacers may limit the antitumor activity of CAR T cells in vivo by rendering T cells sensitive to Fc receptor-expressing immune cells.15, 16 In addition, T cells expressing CARs with 41BB.ζ or CD28.41BB.ζ endodomains might have superior antitumor activity than CD28.ζ CAR T cells.11
We therefore generated and compared a panel of EphA2-specific CARs that contain an IgG1-derived short spacer region, which is devoid of Fc receptor binding sites, and different signaling domains (CD28.ζ, 41BB.ζ, or CD28.41BB.ζ). We show that replacing the CH2CH3 spacer with an IgG1-derived short spacer increased the anti-glioma activity of CD28.ζ CAR T cells 20-fold. 41BB.ζ CAR T cells had similar anti-glioma activity, and including 41BB in CD28.ζ CARs did not further improve CAR T cell function. Based on our results we selected the CD28.ζ CAR for future phase 1 testing in humans.
Results
Generation of EphA2-CAR T Cells
To generate EphA2-specific CAR T cells, we designed retroviral vectors encoding two second-generation (CD28.ζ and 41BB.ζ) and one third-generation (CD28.41BB.ζ) CAR based on the humanized EphA2-specific mAb 4H5.14, 17 All CARs contained an N-terminal leader sequence, a codon-optimized 4H5 scFv, a short spacer consisting of the 16-amino acid IgG1 hinge, a CD28 transmembrane domain, and signaling domains derived from CD28.ζ, 41BB.ζ, or CD28.41BB.ζ (Figure 1A). In addition, all CAR-encoding retroviral vectors contained a truncated cluster of differentiation 19 (tCD19) gene at the C terminus of the CAR gene, separated by a 2A sequence, to allow detection of genetically modified T cells by fluorescence-activated cell sorting (FACS) analysis. As a control, we generated a CAR with a truncated endodomain (CAR.Δ) and/or used non-transduced (NT) T cells. CD3/CD28-activated T cells from healthy donors were transduced with RD114-pseudotyped retroviral particles, and genetically modified T cells were detected by FACS analysis 4–5 days later. T cells stably expressed tCD19 on their cell surface, with a mean transduction efficiency rate of 65.32% (SD ±12.43%) for CD28.ζ, 59.21% (SD ±10.7%) for 41BB.ζ, and 66.24% (SD ±5.76%) for CD28.41BB.ζ, and no significant differences in transduction efficiency among the constructs (Figures 1B and 1C). Expression of CARs was confirmed by western blot, using a CD3.ζ antibody for detection (Figure 1D). Phenotypic analysis revealed a mixture of CD4+ and CD8+ T cells with a CD4:CD8 ratio of 1:3, with no significant difference among the constructs (Figure S1).
Figure 1.

Developing Different Generation EphA2-Specific CAR T Cells
(A) Scheme of EphA2 CARs. All CARs contained an N-terminal leader sequence, a codon-optimized synthetic gene encoding for human 4H5, a spacer region (16-amino acid IgG1 hinge), a CD28 or CD8α transmembrane domain, signaling domains derived from CD28, 41BB, and CD3.ζ, and tCD19, separated by 2A sequence. (B and C) Genetic modification of T cells was confirmed by FACS analysis using a CD19 antibody. Representative plots (B) and summary data (C) are shown (mean 74.1%–93.3%, n = 5–6 per CAR construct). Error bars represent mean with SD. (D) Expression of full-length EphA2-CARs by western blot analysis using a CD3-ζ antibody under denaturing and non-denaturing conditions.
CD28.ζ, 41BB.ζ, and CD28.41BB.ζ T Cells Have Similar Effector Function as Judged by Cytokine Production and Cytolytic Activity
Having successfully generated EphA2-CAR T cells, we tested their specificity and effector function in vitro. First, EphA2-CAR T cells were incubated with EphA2+ (BV173-EphA2, U373, A549; Figure S2) and EphA2− (BV173) tumor cells at a 2:1 ratio for 24 hr. Supernatant was collected, and the concentrations of interferon-γ (IFNγ) and IL-2 were determined by ELISAs. T cells transduced with EphA2-specific CARs produced a significant (p < 0.01) amount of IFNγ in the presence of EphA2+ cells (BV173-EphA2, U373, A549), but not in the presence of EphA2− cells (BV173) (Figure 2A). CD28.ζ, 41BB.ζ, and CD28.41BB.ζ T cells also produced significant amounts of IL-2 when co-cultured with BV173-EphA2, U373, or A549 (Figure 2B), with no significant difference among constructs containing different signaling domains. NT T cells produced no IFNγ or IL-2 when co-cultured with any target cells.
Figure 2.
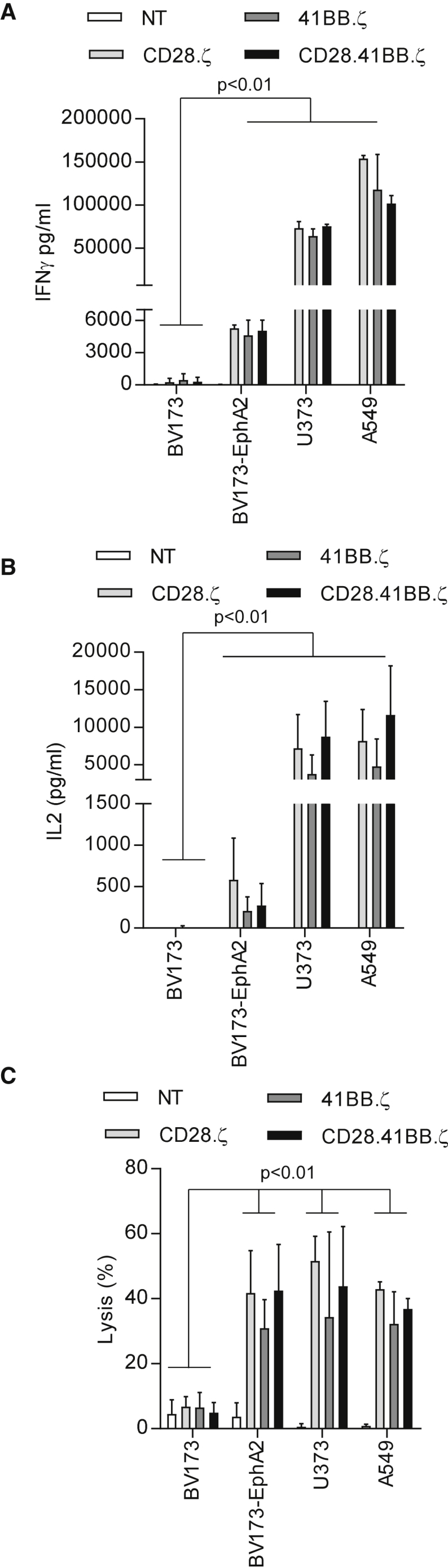
Effector Function Evaluation of EphA2-Specific CAR T Cells
EphA2-CAR or non-transduced (NT) T cells were co-cultured with BV173, BV173-EphA2, U373, and A549 cells at a 2:1 E:T ratio. NT T cells served as controls. After 24 hr, IFNγ (A) and IL-2 (B) were measured by ELISA (n = 4). (C) 4-hr cytotoxicity assay at an E:T ratio of 10:1 (n = 4). Error bars represent mean with SD.
To determine whether CAR T cells also produce other Th1 cytokines (granulocyte-macrophage colony-stimulating factor [GM-CSF], tumor necrosis factor alpha [TNF-α]) and/or Th2 cytokines (IL-4, IL-10) after target cell stimulation, we co-cultured EphA2-specific CAR T cells with BV173.EphA2 tumor cells. CAR T cells secreted high levels of GM-CSF (>2,000 pg/mL), intermediate levels of TNF-α and IL-10 (200 to <2,000 pg/mL), and low levels of IL-4 (<200 pg/mL). There was no significant difference in cytokine secretion levels among CARs except for TNF-α, which was secreted at significantly lower levels by 41BB.ζ CAR T cells in comparison with CD28.ζ (p < 0.01) or CD28.41BB.ζ (p < 0.05) T cells (Figure S3).
Lastly, we confirmed the lytic activity of EphA2-CAR T cells in a chromium (51Cr) release cytotoxicity assay at a 10:1 effector-to-target (E:T) ratio by using the EphA2+ and EphA2− tumor cells used in the co-culture experiments described above. EphA2-specific CAR T cells recognized and killed EphA2+ tumor cells, while control T cells showed no cytolytic activity against any tumor cells, indicating that killing of EphA2+ target cells depends on the expression of an EphA2-CAR (Figure 2A). There was no significant difference in cytolytic activity between different EphA2-CAR T cells. Thus, we could not discern a significant difference in the effector function of CD28.ζ, 41BB.ζ, and CD28.41BB.ζ CAR T cells as judged by cytokine production and cytolytic activity.
CD28.ζ, 41BB.ζ, and CD28.41BB.ζ CAR T Cells Proliferate Postexposure to EphA2+ Target Cells
To determine the effect of different co-stimulatory endodomains on EphA2-CAR T cell expansion in vitro, we performed a co-culture assay in which CAR T cells were weekly stimulated with EphA2+ target cells (U373, A549). All CAR T cells expanded significantly better when co-cultured with A549 in comparison with U373 cells. There was no difference in T cell expansion after A549 stimulation between CD28.ζ, 41BB.ζ, and CD28.41BB.ζ CAR T cells. However, after U373 stimulation, 41BB.ζ CAR T cells expanded significantly more than CD28.ζ and CD28.41BB.ζ CAR T cells (Figure 3).
Figure 3.

Addition of Extra Signaling Domain Does Not Improve the Proliferative Capacity of EphA2-CAR T Cells
EphA2-specific CAR T cells were co-cultured with A549 and U373 at a 2:1 E:T ratio. T cells were stimulated weekly with fresh A549 and U373 cells, and T cells were counted before addition of fresh target cells. Figure shows cumulative data of CAR T cell expansion (n = 4; post third and fourth stimulation with U373: 41BB CAR versus other CARs, p < 0.05, t test; error bars represent SEM).
CARs with Short Spacers Have Improved Anti-glioma Activity In Vivo
Because we observed no significant difference between CD28.ζ, 41BB.ζ, and CD28.41BB.ζ CAR T cells, we tested all three T cell populations in our orthotopic U373 glioma xenograft mouse model, in which T cells are directly injected into tumors.11, 13 The model allows for serial bioluminescence imaging because U373 are genetically modified to express an eGFP.ffLuc fusion protein (U373.eGFP.ffLuc). On day 0, U373.eGFP.ffLuc cells were injected stereotactically into the brains of severe combined immunodeficiency (SCID) mice, followed by CD28.ζ, 41BB.ζ, or CD28.41BB.ζ CAR T cells on day 7. As a negative control, we used CAR.Δ T cells, and as a positive control, T cells that expressed our original CAR (CH2CH3.CD28.ζ). Mice treated with CAR.Δ T cells showed continuous tumor growth within 4 days after T cell injection (Figure 4A). All T cell-expressing functional CARs had significant antitumor activity with a significant decrease (p < 0.05) in bioluminescence in comparison with the CAR.Δ T cell group within 3 weeks of T cell injection. Mice were followed for up to 100 days after tumor cell injection, and although CD28.ζ or 41BB.ζ CAR T cell-treated mice had a significant survival advantage (p < 0.05) in comparison with CH2CH3.CD28.ζ CAR T cell-treated mice, CD28.41BB.ζ CAR T cell-treated mice did not (Figure 4B).
Figure 4.

Treatment of Glioma Xenograft with T Cells Expressing EphA2-CARs Results in Tumor Regression and Improved Overall Survival
U373 glioma-bearing mice were treated on day 7 with CD28.ζ (n = 5), 41BB.ζ (n = 5), or CD28.41BB.ζ (n = 5) T cells. CH2CH3.CD28.ζ (n = 5) served as a positive control, and Δ CAR T cells (n = 5) served as a negative control. (A) Quantitative bioluminescence (radiance = photons/sec/cm2/sr) imaging data for all mice are shown. (B) Kaplan-Meier survival analysis (*p < 0.05).
Defining the Minimal Effective Dose of CD28.ζ, 41BB.ζ, and CD28.41BB.ζ CAR T Cells
Having established that 2 × 106 T cells expressing two out of three CAR constructs with short spacers have improved anti-tumor activity in comparison with CH2CH3.CD28.ζ CAR T cells, we next set out to define the lowest dose of T cells that can eradicate tumor cells. Glioma-bearing mice were injected with 4-fold (5 × 105), 20-fold (1 × 105), or 200-fold (1 × 104) lower T cell doses. CAR T cells sustained their potent anti-glioma activity at cell doses as low as 1 × 105 T cells with three out of five (CD28.ζ CAR) and two out of five (41BB.ζ and CD28.41BB.ζ CARs) mice surviving long term (Figure 5). At a cell dose of 1 × 104 T cells, only transient antitumor activity was observed (Figure 5).
Figure 5.

Defining Minimal Effective Dose
1 × 105 U373.eGFP.ffLuc cells were injected intracranially into mice. On day 7 mice received 5 × 105, 1 × 105, and 1 × 104 CD28.ζ (n = 5), 41BB.ζ (n = 5), or CD28.41BB.ζ (n = 5) CAR T cells. Quantitative bioluminescence (radiance = photons/sec/cm2/sr) imaging data for all mice are shown.
To investigate the mechanism of tumor recurrence, we measured the expression of EphA2 on recurring tumor as well as T cell persistence. Because the addition of 41BB (CD137) to CD28.ζ CARs did not improve the effector function of CAR T cells in any of the conducted experiments thus far, we restricted the following analyses to CD28.ζ and 41BB.ζ CAR T cells. Recurring tumors continued to express EphA2 at similar levels in comparison with untreated tumors as judged by FACS analysis (Figure S4). To determine T cell persistence, we injected 1 × 105 CD28.ζ or 41BB.ζ CAR T cells, which were also genetically modified to express eGFP.ffLuc, into U373 glioma-bearing mice. eGFP.ffLuc-expressing T cells served as control. No significant expansion of injected CAR T cells was observed, and T cells persisted for up to 6 days, with no significant difference between CD28.ζ and 41BB.ζ CAR T cells (Figure 6).
Figure 6.

Limited Persistence of EphA2-CAR T Cells In Vivo
CD28.ζ and 41BB.ζ CAR T cells were transduced to express eGFP.ffLuc. 1 × 105 unmodified U373 cells were injected intracranially into mice. On day 7, mice received 1 × 105 CD28.ζ.eGFP.ffLuc or 41BB.ζ.eGFP.ffLuc CAR T cells intracranially using the same tumor coordinates. (A) Bioluminescence imaging was used to monitor T cell persistence. (B) Area under the curve (AUC) of bioluminescence data.
Discussion
Here, we describe the generation of three CARs specific for the glioma-associated antigen EphA2. All three CARs contained a short spacer and either a CD28.ζ, 41BB.ζ, or CD28.41BB.ζ signaling domain. There was no significant difference between the phenotype and effector function of CAR T cells in vitro. In vivo, CAR T cell populations had potent anti-glioma activity that was significantly improved compared with our previously published EphA2-CAR with a long CH2CH3 spacer and a CD28.ζ signaling domain.13
Many groups, including ours, have shown that there is a complex interaction between the scFv, spacer region, transmembrane domain, and endodomain that determines optimal CAR T cell efficacy.11, 18, 19 For example, scFvs that recognize epitopes that are located proximal to the plasma membrane require long spacers, whereas distal epitopes require short spacers.18 In addition, long spacers that are based on CH2CH3 domains have been shown to render CAR T cells sensitive to innate immune cells,15 or can induce baseline (tonic) CAR signaling.16, 20 Because the limitations of CH2CH3-based spacers are independent of the targeted antigen, we did not repeat mechanistic studies with CH2CH3-based EphA2-CARs, but focused instead on EphA2-CARs with a short spacer. We generated three EphA2-CARs with a short spacer and different endodomains, all of which recognized EphA2+ target cells as judged by cytokine production and cytolytic activity. The epitope that is recognized by the EphA2-specific scFv 4H5 is not defined. However, our current and previous results indicate that for the 4H5 scFv epitope, location is not a critical factor that determines CAR function because T cells expressing a 4H5-based CAR with either a 16- or 293-amino acid spacer recognize EphA2+ target cells.
In vitro, we observed no significant difference in the effector function of T cells expressing CD28.ζ or 41BB.ζ CARs. This is consistent with our previous study in which we targeted the glioma-associated antigen IL-13Rα2.16 Others have demonstrated that GPC3-CARs with a 41BB.ζ endodomain preferentially induce the production of TH2 cytokines.21 However, we observed only low levels of IL-4 and IL-10 production with no significant differences between CD28.ζ and 41BB.ζ CAR T cells. Another group reported that CD28.ζ have greater functionality as judged by rapid tumor eradication, while 41BB.ζ mediates slower tumor killing but attains complete tumor eradication because of better persistence.22 In our study, we observed no significant differences in CAR T cell persistence in vivo between CD28.ζ and 41BB.ζ signaling domains. These opposing findings might be partially explained by the different tumor models that were used to compare CAR T cells (hepatocellular carcinoma, acute lymphoblastic leukemia, glioblastoma multiforme [GBM]). At present, it remains controversial whether CARs that encode two costimulatory endodomains endow T cells with superior effector function than CARs with a single costimulatory endodomain.23, 24, 25 Our finding adds to this controversy with showing no benefit of adding 41BB to CD28.ζ CAR T cells targeting EphA2. A recent study has demonstrated that expression of 41BBL on the cell surface of CD28.ζ CAR T cells results in more potent effector T cells than incorporating a 41BB signaling domain into the CAR itself.22, 26 We are planning to evaluate this approach in the future.
In vivo, T cells expressing our newly generated CARs with a short spacer had significantly greater antitumor activity in comparison with our original CH2CH3.CD28.ζ CAR T cells. Based on our experiment in which we evaluated lower T cell doses, short spacer CAR T cells were at least 20-fold more potent than T cells expressing our original CAR. Glioma recurrences in animals treated with low T cell doses continued to express EphA2, indicating that limited effector function of CAR T cells is responsible for this therapeutic failure.
Our results indicate that there is a limit to optimizing CAR T cell function by providing costimulation through CD28 and/or 41BB even in a xenograft model, which does not mimic the ‘complete’ immunosuppressive tumor microenvironment (TME) of gliomas because it is devoid of inhibitory T cells. Proper T cell activation and expansion requires not only signal 1 (activation) and signal 2 (costimulation), but also signal 3 (γ cytokines).26 Although costimulation induces γ-cytokine production by T cells, the observed lack of T cell expansion in vivo indicates that γ-cytokine production might be limited. Indeed, several investigators including ourselves have demonstrated that transgenic expression of IL-7, IL-12, or IL-15 in CAR T cells enhances their effector function in vivo.12, 27, 28 Transgenic expression of cytokines should also counteract the immunosuppressive TME. Besides transgenic expression of cytokines, expression of a constitutive IL-7 receptor in CAR T cells has also shown to enhance their effector function, including T cells expressing the 41BB.ζ CAR designed in this study.29 Besides transgenic expression of ‘immune stimulatory genes’, silencing negative regulators in CAR T cells is another promising strategy to render them resistant to the immunosuppressive (TME) glioma microenvironment.30, 31 While these strategies enhance CAR T cell function, they also increase the risk of unwanted side effects. To mitigate this risk, several groups of investigators have developed and tested inducible suicide genes in preclinical models and early-phase clinical studies.32, 33, 34
The goal of our studies was to optimize an EphA2-CAR to select for phase 1 testing in humans. Because adding 41BB to CD28.ζ did not enhance the effector function of T cells, the developed CD28.ζ or 41BB.ζ CAR are prime candidates for further testing. Ideally, both CARs could be compared in patients by infusing two T cell products that are modified with the CD28.ζ or 41BB.ζ CAR. Although such a trial design has been done in the past to compare CD19-CAR.ζ and CD19-CAR.CD28.ζ35 or CD19-CAR.CD28.ζ and CD19-CAR.CD28.41BB.ζ (NCT01853631), such studies are currently cost prohibitive because they require the manufacturing of two clinical-grade vectors and production of two genetically modified T cell products for each patient.
Additional practical consideration of selecting a CAR includes whether CAR expression by itself affects CAR T cell expansion during cell product manufacturing. We therefore compared the expansion of CD28.ζ or 41BB.ζ CAR and observed no significant difference (Figure S5). Lastly, there are safety considerations when an overexpressed self-antigen like EphA2 is targeted. We are mitigating the risk of “on target/off cancer toxicity” by using a scFv that recognizes a conformational epitope exposed only on malignant cells.17 Nevertheless, for EphA2-CAR T cells, limiting T cell persistence for first in human testing seems an advisable strategy. Based on the results of the conducted clinical studies with CD19-CAR T cells, which indicate that 41BB.ζ CAR T cells persist longer in humans than in CD28.ζ CAR T cells,36 we have selected EphA2-specific CD28.ζ CAR T cells for further clinical development. No dose-limiting toxicity has been observed with IL-13Rα2- and HER2-CAR T cells in early-phase clinical studies for GBM patients.8, 9, 10 However, our EphA2-CAR T cells target a conformational epitope. We therefore decided to replace tCD19 with CD20 in the retroviral vector encoding our EphA2-CAR (Figure S6A) so that CAR T cells can be eliminated with rituximab if unwanted side effects occur.37, 38 T cells expressing CD28.ζ CAR and CD20 had similar effector function as CD28.ζ CAR T cells and were readily eliminated in the presence of Rituxan and complement (Figures S6B–S6D).
In conclusion, we have optimized EphA2-CARs for the adoptive T cell therapy of EphA2+ glioblastoma. While we could not discern a functional difference between CD28.ζ, 41BB.ζ, and CD28.41BB.ζ CAR T cells, we selected CD28.ζ CAR T cells for further clinical development based on safety consideration.
Materials and Methods
Cell Lines
Blood samples from healthy donors were obtained in accordance to protocols approved by the Institutional Review Board of Baylor College of Medicine. U373 (GBM), A549 (lung cancer), and 293T (human embryonic kidney) were purchased from the American Type Culture Collection (ATCC, Manassas, VA, USA). BV173 (Ph+ chronic B lymphoblastic leukemia) was purchased from the German Collection of Microorganisms and Cell Cultures (DSMZ). U373 cells expressing EGFP and firefly luciferase (U373.eGFP.FFLuc) and BV173-expressing EphA2 were generated by lentiviral transduction. T cells and cell lines were grown in RPMI or DMEM (GE Healthcare Life Sciences HyClone Laboratories, Logan, UT, USA) with 10% fetal bovine serum (FBS; GE Healthcare Life Sciences HyClone) and 2 mM GlutaMAX-I (Invitrogen, Carlsbad, CA, USA). The “Characterized Cell Line Core Facility” at MD Anderson Cancer Center (Houston, TX, USA) performed cell line validation.
Generation of Retroviral Vectors Encoding EphA2-Specific CARs
Generation of the EphA2-specific scFv was previously described.13 In brief, a codon-optimized gene, encoding the humanized version of EphA2 mAb EA2,17 was synthesized by GeneArt (Invitrogen, Carlsbad, CA, USA) containing the immunoglobulin heavy-chain leader peptide and the 4H5 heavy chain flanked by 5′ NcoI and 3′ BamHI sites. This mini gene was subcloned into SFG retroviral vector containing human IgG1 hinge and CD28.ζ, 41BB.ζ, or CD28.41BB.ζ endodomains.11, 39 All CARs contained a CD28 transmembrane domain except for EphA2-CAR.SSR.41BB.ζ, which had a CD8α transmembrane domain. All plasmids also contained a tCD19 at a C terminus, which was used for CAR detection. EphA2-CAR.SSR.Δ without an endodomain was generated by In-Fusion cloning (Takara Bio USA, Mountain View, CA, USA). All cloning of the CARs was verified by sequencing (Seqwright, Houston, TX, USA). RD114-pseudotyped retroviral particles were generated by transient transfection of 293T cells as previously described.13
Transduction of T Cells
Human peripheral blood mononuclear cells (PBMCs) from healthy donors were obtained under a Baylor College of Medicine institutional review board (IRB)-approved protocol, after informed consent was obtained in accordance to the Declaration of Helsinki. To generate EphA2-CAR T cells, we isolated PBMCs by Lymphoprep (Abbott Laboratories, Chicago, IL, USA) gradient centrifugation and then stimulated on treated non-tissue culture 24-well plates, which were precoated with OKT3 (CRL-8001; ATCC) and CD28 (BD Biosciences, San Jose, CA, USA) antibodies. Recombinant human IL-7 and IL-15 (IL-7, 10 ng/mL; IL-15, 5 ng/mL; Proleukin; Chiron, Emeryville, CA, USA) were added to cultures on day 2.40 On day 3, OKT3/CD28-stimulated T cells (2.5 × 105 cells/well) were transduced on RetroNectin (Clontech, Mountain View, CA)-coated plates in the presence of IL-7 and IL-15. On day 5 or 6, transduced T cells were transferred into new tissue culture 24-well plates and subsequently expanded with IL-7 and IL-15. NT T cells were activated with OKT3/CD28 and expanded in parallel with IL-7 and IL-15. EphA2-specific CAR T cell expression was determined 4–5 days post-transduction.
Flow Cytometry
A FACSCanto (BD Biosciences, San Jose, CA, USA) instrument was used to acquire immunofluorescence data, which were analyzed with CellQuest (BD Biosciences). FlowJo v.7 (FlowJo, Ashland, OR, USA) was used for final data analysis and graphic representation. Isotype controls were immunoglobulin G1-fluorescein isothiocyanate (IgG1-FITC; BD), IgG1-phycoerythrin (IgG1-PE; BD), IgG1-peridinin chlorophyll protein (IgG1-PerCP; BD), and isotype Cy5 (Jackson ImmunoResearch Laboratories, West Grove, PA, USA). Genetically modified T cells were detected using a CD19-PE antibody (BD) along with CD8-PerCP, CD4-Pacific Blue, CCR7-FITC, and CD45RA-AF750. Forward and side scatter gating were used to discriminate live cells from dead cells.
Cells were collected and washed once with PBS (Sigma, St. Louis, MO, USA) containing 1% FBS (GE Healthcare Life Sciences HyClone Laboratories; FACS buffer) prior to the addition of antibodies. Cells were incubated with antibodies for 30 min on ice in the dark, washed once, and fixed in FACS buffer containing 0.5% paraformaldehyde (BD Biosciences) prior to analysis.
Western Blot
Cells were dissociated with PBS + 3 mM EDTA and lysed in a buffer containing 50 mM Tris, 150 mM NaCl, 5 mM EDTA, 1% Triton X-100 (all from Sigma, St. Louis, MO, USA), and protease inhibitors (Thermo Scientific, Waltham, MA, USA). Protein concentrations were determined using a Bio-Rad protein assay (Bio-Rad, Hercules, CA, USA) with BSA as the standard. Samples were denatured in Laemmli buffer (Bio-Rad) with βME (2-mercaptoethanol; reducing conditions; Bio-Rad) or without βME (non-reducing condition) at 95°C for 5 min. Cell lysates (5 μg per lane) were run on a 10% SDS polyacrylamide gel and transferred to nitrocellulose membranes (Bio-Rad). Membranes were blocked with 5% milk powder in Tris-buffered saline (TBS) + 0.1% Tween 20 (all from Sigma) and then probed with anti-CD3.ζ (sc-1239; Santa Cruz Biotechnology, Santa Cruz, CA, USA) or GAPDH (sc-47724; Santa Cruz Biotechnology) mouse monoclonal antibodies followed by a horseradish peroxidase (HRP)-conjugated goat mouse IgG antibody (sc-2005; Santa Cruz Biotechnology). Blots were developed using SuperSignal West Dura Extended Duration Substrate (Thermo Scientific) and exposed to GeneMate Blue Basic Autoradiography Film (BioExpress, Kaysville, UT, USA).
Co-culture Assays
For co-culture assays, each CAR was expressed in T cells from the same donor. Biological repeats were done using different donors, and data presented in the figures are the average of three to five donors.
Cell Co-culture Assay
CAR T cells were co-cultured with target cells at a 2:1 E:T ratio in a 24-well plate. NT T cells served as controls. After 24 hr, culture supernatants were harvested, and the presence of cytokines was determined by ELISA or Multiplex assay (HSTCMAG28SPMX13; EMD Millipore, Billerica, MA, USA).
Repeated Stimulation Assay
EphA2-CAR T cells were co-cultured with EphA2+ tumor cells (U373 and A549) at a 2:1 E:T ratio in the absence of cytokines. 7 days later, CAR T cells were counted and re-challenged with fresh tumor cells at the same ratio. NT T cells served as a control, and CAR T cell incubation without tumor cells (media only) allowed us to evaluate for any unspecific CAR T cell expansion. CAR T cell expansion was assessed by calculating fold changes in absolute CAR T cell numbers at the indicated time points.
Cytotoxicity Assay
Standard 51Cr Release Assay
CAR T cells were tested for specific cytotoxicity against EphA2+ and EphA2− tumor cell lines (target cells). A total of 1 × 106 target cells were labeled with 0.1 mCi (3.7MBq) 51Cr and mixed with decreasing numbers of effector cells (CAR T cells) to give E:T ratios of 40:1, 20:1, 10:1, and 5:1. Target cells incubated in complete medium alone or in 1% Triton X-100 were used to determine spontaneous and maximum 51Cr release, respectively. After 4 hr, supernatants were collected and radioactivity was measured in a gamma counter (Cobra Quantum; PerkinElmer, Wellesley, MA, USA). The mean percentage of specific lysis of triplicate wells was calculated according to the following formula: [test release − spontaneous release]/[maximal release − spontaneous release] × 100.
Complement-Dependent Cytotoxicity Assay
T cells transduced with CD28.ζ.CD20 or Δ were labeled with 0.1 mCi 51Cr and then treated with 10 μg/mL rituximab (Roche, San Francisco, CA, USA), 10% baby rabbit complement (Cedarlane Labs, Burlington, NC, USA), or 10 μg/mL rituximab and 10% baby rabbit complement. Cells were incubated for 4 hr. Targets in media alone or 1% Triton X-100 were used for spontaneous and maximum 51Cr release, respectively. Supernatants were collected and radioactivity measured on a gamma counter. Mean percentage of specific lysis of triplicate samples was calculated as in the standard chromium release assay.
Orthotopic Xenograft SCID Mouse Model
All animal experiments followed a protocol approved by the Baylor College of Medicine Institutional Animal Care and Use Committee. Experiments were performed as described previously12, 13 with a few modifications. ICR-SCID mice were purchased from Taconic (IcrTac:ICR-Prkdcscid; Fox Chase C.B-17 SCID ICR; Taconic, Hudson, NY, USA). Male 8- to 9-week-old mice were anesthetized, the head was shaved, and the mice were immobilized in a Cunningham Mouse/Neonatal Rat Adaptor (Stoelting, Wood Dale, IL, USA) stereotactic apparatus fitted into an E15600 Lab Standard Stereotaxic Instrument (Stoelting) and then scrubbed with 1% povidone-iodine. A 10-mm skin incision was made along the midline. The tip of a 30G half-inch needle mounted on a Hamilton syringe (Hamilton, Reno, NV, USA) served as the reference point. A 1-mm burr hole was drilled into the skull 1 mm anterior and 2 mm to the right of the bregma. 1 × 105 U373.eGFP.ffLuc cells in 2.0 μL were injected 3 mm deep over 5 min. The needle was left in place for 3 min, to avoid tumor cell extrusion, and then withdrawn over 5 min. Seven days after tumor cell injection, animals were treated with 2 × 106 effector cells in 2 μL to the same tumor coordinates. The incision was closed with two to three interrupted 7.0 Ethilon sutures (Ethicon, Somerville, NJ, USA). A subcutaneous injection of 0.03–0.1 mg/kg buprenorphine (Buprenex; RBH, Hull, UK) was given for pain control.
Bioluminescence Imaging
Isoflurane-anesthetized animals were imaged using the IVIS system (IVIS; Xenogen, Alameda, CA, USA) 10–15 min after 150 mg/kg D-luciferin (Xenogen) was injected per mouse intraperitoneally. The photons emitted from the luciferase-expressing tumor cells were quantified using Living Image software (Caliper Life Sciences, Hopkinton, MA, USA). A pseudo-color image representing light intensity (blue least intense and red most intense) was generated and superimposed over the grayscale reference image. Mice were euthanized when the tumor radiance was greater than 1 × 109 on two occasions or when they met euthanasia criteria (neurological deficits, weight loss, signs of distress) in accordance with the Center for Comparative Medicine at Baylor College of Medicine.
Statistical Analysis
All in vitro experiments were performed at least in triplicate; GraphPad Prism 5 software (GraphPad Software, La Jolla, CA, USA) was used for statistical analysis. Measurement data were presented as mean ± SD. The differences between means were tested by appropriate tests. The significance level used was p < 0.05. For the mouse experiments, changes in tumor radiance from baseline at each time point were calculated. Survival determined from the time of tumor cell injection was analyzed by the Kaplan-Meier method.
Author Contributions
Z.Y., B.L.P., F.C., and G.K. conducted the experiments. G.K. and S.G. designed and supervised the study. All authors contributed to data analysis and manuscript preparation.
Conflicts of Interest
The Center for Cell and Gene Therapy has a research collaboration with Celgene and Bluebird Bio. Z.Y., F.C., and S.G. have patent applications in the field of T cell and gene therapy for cancer and/or EphA2-targeted therapies.
Acknowledgments
This work was supported by American Brain Tumor Association Basic Research Fellowship in honor of Joel A. Gingras, Jr. (BRF160004), the Alex Lemonade Stand Foundation, NIH grants CA203270 and T32 GM008231, and the James McDonald Foundation and CPRIT grant RP110553. The Characterized Cell Line Core Facility at MD Anderson Cancer Center is funded by NIH grant CA16672.
Footnotes
Supplemental Information includes six figures and can be found with this article online at https://doi.org/10.1016/j.omtm.2018.01.009.
Supplemental Information
References
- 1.Omuro A., DeAngelis L.M. Glioblastoma and other malignant gliomas: a clinical review. JAMA. 2013;310:1842–1850. doi: 10.1001/jama.2013.280319. [DOI] [PubMed] [Google Scholar]
- 2.Konar S.K., Bir S.C., Maiti T.K., Nanda A. A systematic review of overall survival in pediatric primary glioblastoma multiforme of the spinal cord. J. Neurosurg. Pediatr. 2017;19:239–248. doi: 10.3171/2016.8.PEDS1631. [DOI] [PubMed] [Google Scholar]
- 3.Gittleman H., Lim D., Kattan M.W., Chakravarti A., Gilbert M.R., Lassman A.B., Lo S.S., Machtay M., Sloan A.E., Sulman E.P. An independently validated nomogram for individualized estimation of survival among patients with newly diagnosed glioblastoma: NRG Oncology RTOG 0525 and 0825. Neuro Oncol. 2017;19:669–677. doi: 10.1093/neuonc/now208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kaufmann S.H., Earnshaw W.C. Induction of apoptosis by cancer chemotherapy. Exp. Cell Res. 2000;256:42–49. doi: 10.1006/excr.2000.4838. [DOI] [PubMed] [Google Scholar]
- 5.Martínez-Lostao L., Anel A., Pardo J. How do cytotoxic lymphocytes kill cancer cells? Clin. Cancer Res. 2015;21:5047–5056. doi: 10.1158/1078-0432.CCR-15-0685. [DOI] [PubMed] [Google Scholar]
- 6.Sheridan C. First approval in sight for Novartis’ CAR-T therapy after panel vote. Nat. Biotechnol. 2017;35:691–693. doi: 10.1038/nbt0817-691. [DOI] [PubMed] [Google Scholar]
- 7.Brudno J.N., Kochenderfer J.N. Chimeric antigen receptor T-cell therapies for lymphoma. Nat. Rev. Clin. Oncol. 2018;15:31–46. doi: 10.1038/nrclinonc.2017.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ahmed N., Brawley V., Hegde M., Bielamowicz K., Kalra M., Landi D., Robertson C., Gray T.L., Diouf O., Wakefield A. HER2-specific chimeric antigen receptor-modified virus-specific T cells for progressive glioblastoma: a phase 1 dose-escalation trial. JAMA Oncol. 2017;3:1094–1101. doi: 10.1001/jamaoncol.2017.0184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brown C.E., Alizadeh D., Starr R., Weng L., Wagner J.R., Naranjo A., Ostberg J.R., Blanchard M.S., Kilpatrick J., Simpson J. Regression of glioblastoma after chimeric antigen receptor T-cell therapy. N. Engl. J. Med. 2016;375:2561–2569. doi: 10.1056/NEJMoa1610497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.O’Rourke D.M., Nasrallah M.P., Desai A., Melenhorst J.J., Mansfield K., Morrissette J.J.D., Martinez-Lage M., Brem S., Maloney E., Shen A. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci. Transl. Med. 2017;9:eaaa0984. doi: 10.1126/scitranslmed.aaa0984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Krenciute G., Krebs S., Torres D., Wu M.F., Liu H., Dotti G., Li X.N., Lesniak M.S., Balyasnikova I.V., Gottschalk S. Characterization and functional analysis of scFv-based chimeric antigen receptors to redirect T cells to IL13Rα2-positive glioma. Mol Ther. 2016;24:354–363. doi: 10.1038/mt.2015.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Krenciute G., Prinzing B.L., Yi Z., Wu M.F., Liu H., Dotti G., Balyasnikova I.V., Gottschalk S. Transgenic expression of IL15 improves antiglioma activity of IL13Rα2-CAR T cells but results in antigen loss variants. Cancer Immunol. Res. 2017;5:571–581. doi: 10.1158/2326-6066.CIR-16-0376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chow K.K., Naik S., Kakarla S., Brawley V.S., Shaffer D.R., Yi Z., Rainusso N., Wu M.F., Liu H., Kew Y. T cells redirected to EphA2 for the immunotherapy of glioblastoma. Mol. Ther. 2013;21:629–637. doi: 10.1038/mt.2012.210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Damschroder M.M., Widjaja L., Gill P.S., Krasnoperov V., Jiang W., Dall’Acqua W.F., Wu H. Framework shuffling of antibodies to reduce immunogenicity and manipulate functional and biophysical properties. Mol. Immunol. 2007;44:3049–3060. doi: 10.1016/j.molimm.2006.12.019. [DOI] [PubMed] [Google Scholar]
- 15.Hudecek M., Sommermeyer D., Kosasih P.L., Silva-Benedict A., Liu L., Rader C., Jensen M.C., Riddell S.R. The nonsignaling extracellular spacer domain of chimeric antigen receptors is decisive for in vivo antitumor activity. Cancer Immunol. Res. 2015;3:125–135. doi: 10.1158/2326-6066.CIR-14-0127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jonnalagadda M., Mardiros A., Urak R., Wang X., Hoffman L.J., Bernanke A., Chang W.C., Bretzlaff W., Starr R., Priceman S. Chimeric antigen receptors with mutated IgG4 Fc spacer avoid fc receptor binding and improve T cell persistence and antitumor efficacy. Mol Ther. 2015;23:757–768. doi: 10.1038/mt.2014.208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Coffman K.T., Hu M., Carles-Kinch K., Tice D., Donacki N., Munyon K., Kifle G., Woods R., Langermann S., Kiener P.A., Kinch M.S. Differential EphA2 epitope display on normal versus malignant cells. Cancer Res. 2003;63:7907–7912. [PubMed] [Google Scholar]
- 18.Hudecek M., Lupo-Stanghellini M.T., Kosasih P.L., Sommermeyer D., Jensen M.C., Rader C., Riddell S.R. Receptor affinity and extracellular domain modifications affect tumor recognition by ROR1-specific chimeric antigen receptor T cells. Clin. Cancer Res. 2013;19:3153–3164. doi: 10.1158/1078-0432.CCR-13-0330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Haso W., Lee D.W., Shah N.N., Stetler-Stevenson M., Yuan C.M., Pastan I.H., Dimitrov D.S., Morgan R.A., FitzGerald D.J., Barrett D.M. Anti-CD22-chimeric antigen receptors targeting B-cell precursor acute lymphoblastic leukemia. Blood. 2013;121:1165–1174. doi: 10.1182/blood-2012-06-438002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Watanabe N., Bajgain P., Sukumaran S., Ansari S., Heslop H.E., Rooney C.M., Brenner M.K., Leen A.M., Vera J.F. Fine-tuning the CAR spacer improves T-cell potency. OncoImmunology. 2016;5:e1253656. doi: 10.1080/2162402X.2016.1253656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li W., Guo L., Rathi P., Marinova E., Gao X., Wu M.F., Liu H., Dotti G., Gottschalk S., Metelitsa L.S., Heczey A. Redirecting T cells to glypican-3 with 4-1BB zeta chimeric antigen receptors results in Th1 polarization and potent antitumor activity. Hum. Gene Ther. 2017;28:437–448. doi: 10.1089/hum.2016.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhao Z., Condomines M., van der Stegen S.J.C., Perna F., Kloss C.C., Gunset G., Plotkin J., Sadelain M. Structural design of engineered costimulation determines tumor rejection kinetics and persistence of CAR T cells. Cancer Cell. 2015;28:415–428. doi: 10.1016/j.ccell.2015.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Milone M.C., Fish J.D., Carpenito C., Carroll R.G., Binder G.K., Teachey D., Samanta M., Lakhal M., Gloss B., Danet-Desnoyers G. Chimeric receptors containing CD137 signal transduction domains mediate enhanced survival of T cells and increased antileukemic efficacy in vivo. Mol. Ther. 2009;17:1453–1464. doi: 10.1038/mt.2009.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhong X.S., Matsushita M., Plotkin J., Riviere I., Sadelain M. Chimeric antigen receptors combining 4-1BB and CD28 signaling domains augment PI3kinase/AKT/Bcl-XL activation and CD8+ T cell-mediated tumor eradication. Mol. Ther. 2010;18:413–420. doi: 10.1038/mt.2009.210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Carpenito C., Milone M.C., Hassan R., Simonet J.C., Lakhal M., Suhoski M.M., Varela-Rohena A., Haines K.M., Heitjan D.F., Albelda S.M. Control of large, established tumor xenografts with genetically retargeted human T cells containing CD28 and CD137 domains. Proc. Natl. Acad. Sci. USA. 2009;106:3360–3365. doi: 10.1073/pnas.0813101106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yeku O.O., Brentjens R.J. Armored CAR T-cells: utilizing cytokines and pro-inflammatory ligands to enhance CAR T-cell anti-tumour efficacy. Biochem. Soc. Trans. 2016;44:412–418. doi: 10.1042/BST20150291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yeku O.O., Purdon T.J., Koneru M., Spriggs D., Brentjens R.J. Armored CAR T cells enhance antitumor efficacy and overcome the tumor microenvironment. Sci. Rep. 2017;7:10541. doi: 10.1038/s41598-017-10940-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Markley J.C., Sadelain M. IL-7 and IL-21 are superior to IL-2 and IL-15 in promoting human T cell-mediated rejection of systemic lymphoma in immunodeficient mice. Blood. 2010;115:3508–3519. doi: 10.1182/blood-2009-09-241398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shum T., Omer B., Tashiro H., Kruse R.L., Wagner D.L., Parikh K., Yi Z., Sauer T., Liu D., Parihar R. Constitutive signaling from an engineered IL7 receptor promotes durable tumor elimination by tumor-redirected T cells. Cancer Discov. 2017;7:1238–1247. doi: 10.1158/2159-8290.CD-17-0538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vega E.A., Graner M.W., Sampson J.H. Combating immunosuppression in glioma. Future Oncol. 2008;4:433–442. doi: 10.2217/14796694.4.3.433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Razavi S.M., Lee K.E., Jin B.E., Aujla P.S., Gholamin S., Li G. Immune evasion strategies of glioblastoma. Front. Surg. 2016;3:11. doi: 10.3389/fsurg.2016.00011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hoyos V., Savoldo B., Quintarelli C., Mahendravada A., Zhang M., Vera J., Heslop H.E., Rooney C.M., Brenner M.K., Dotti G. Engineering CD19-specific T lymphocytes with interleukin-15 and a suicide gene to enhance their anti-lymphoma/leukemia effects and safety. Leukemia. 2010;24:1160–1170. doi: 10.1038/leu.2010.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kaneko S., Mastaglio S., Bondanza A., Ponzoni M., Sanvito F., Aldrighetti L., Radrizzani M., La Seta-Catamancio S., Provasi E., Mondino A. IL-7 and IL-15 allow the generation of suicide gene-modified alloreactive self-renewing central memory human T lymphocytes. Blood. 2009;113:1006–1015. doi: 10.1182/blood-2008-05-156059. [DOI] [PubMed] [Google Scholar]
- 34.Brown C.E., Badie B., Barish M.E., Weng L., Ostberg J.R., Chang W.C., Naranjo A., Starr R., Wagner J., Wright C. Bioactivity and safety of IL13Rα2-redirected chimeric antigen receptor CD8+ T cells in patients with recurrent glioblastoma. Clin Cancer Res. 2015;21:4062–4072. doi: 10.1158/1078-0432.CCR-15-0428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Savoldo B., Ramos C.A., Liu E., Mims M.P., Keating M.J., Carrum G., Kamble R.T., Bollard C.M., Gee A.P., Mei Z. CD28 costimulation improves expansion and persistence of chimeric antigen receptor-modified T cells in lymphoma patients. J. Clin. Invest. 2011;121:1822–1826. doi: 10.1172/JCI46110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Porter D.L., Levine B.L., Kalos M., Bagg A., June C.H. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N. Engl. J. Med. 2011;365:725–733. doi: 10.1056/NEJMoa1103849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bonifant C.L., Szoor A., Torres D., Joseph N., Velasquez M.P., Iwahori K., Gaikwad A., Nguyen P., Arber C., Song X.T. CD123-engager T cells as a novel immunotherapeutic for acute myeloid leukemia. Mol Ther. 2016;24:1615–1626. doi: 10.1038/mt.2016.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Introna M., Barbui A.M., Bambacioni F., Casati C., Gaipa G., Borleri G., Bernasconi S., Barbui T., Golay J., Biondi A., Rambaldi A. Genetic modification of human T cells with CD20: a strategy to purify and lyse transduced cells with anti-CD20 antibodies. Hum. Gene Ther. 2000;11:611–620. doi: 10.1089/10430340050015798. [DOI] [PubMed] [Google Scholar]
- 39.Vera J., Savoldo B., Vigouroux S., Biagi E., Pule M., Rossig C., Wu J., Heslop H.E., Rooney C.M., Brenner M.K., Dotti G. T lymphocytes redirected against the kappa light chain of human immunoglobulin efficiently kill mature B lymphocyte-derived malignant cells. Blood. 2006;108:3890–3897. doi: 10.1182/blood-2006-04-017061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Xu Y., Zhang M., Ramos C.A., Durett A., Liu E., Dakhova O., Liu H., Creighton C.J., Gee A.P., Heslop H.E. Closely related T-memory stem cells correlate with in vivo expansion of CAR.CD19-T cells and are preserved by IL-7 and IL-15. Blood. 2014;123:3750–3759. doi: 10.1182/blood-2014-01-552174. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.