

Abstract
Human telomerase synthesizes telomeric DNA repeats (GGTTAG)n onto chromosome ends using a short template from its integral telomerase RNA (hTR). However, telomerase is markedly slow for processive DNA synthesis among DNA polymerases. We report here that the unique template‐embedded pause signal restricts the first nucleotide incorporation for each repeat synthesized, imparting a significantly greater K M. This slow nucleotide incorporation step drastically limits repeat addition processivity and rate under physiological conditions, which is alleviated with augmented concentrations of dGTP or dGDP, and not with dGMP nor other nucleotides. The activity stimulation by dGDP is due to nucleoside diphosphates functioning as substrates for telomerase. Converting the first nucleotide of the repeat synthesized from dG to dA through the telomerase template mutation, hTR‐51U, correspondingly shifts telomerase repeat addition activity stimulation to dATP‐dependent. In accordance, telomerase without the pause signal synthesizes DNA repeats with extremely high efficiency under low dGTP concentrations and lacks dGTP stimulation. Thus, the first nucleotide incorporation step of the telomerase catalytic cycle is a potential target for therapeutic enhancement of telomerase activity.
Keywords: deoxynucleoside diphosphate, DNA polymerase, processivity, reverse transcriptase, ribonucleoprotein
Subject Categories: DNA Replication, Repair & Recombination
Introduction
The ends of linear eukaryotic chromosomes are capped by telomeres, an array of short DNA repeats bound by specific telomeric proteins (Arnoult & Karlseder, 2015). Telomere length is crucial for chromosome integrity and maintained by the unique cellular reverse transcriptase, telomerase, that adds telomeric DNA repeats (GGTTAG)n processively onto chromosome ends (Wu et al, 2017b). Human telomerase utilizes a short 11‐nt template from the long non‐coding 451‐nt telomerase RNA (TR) for DNA repeat synthesis catalyzed by the telomerase reverse transcriptase (TERT) subunit (Podlevsky & Chen, 2012; Hockemeyer & Collins, 2015). The 5′ boundary of the TR template prevents non‐template usage to ensure telomeric DNA synthesis fidelity and is physically safeguarded in different species by divergent structural elements: a distal RNA helix and single‐stranded nucleotide span in vertebrates (Chen & Greider, 2003a), TERT‐binding TR elements in ciliates (Jansson et al, 2015; Jiang et al, 2015), or a template‐adjacent RNA helix in fungi (Tzfati et al, 2000; Seto et al, 2003; Qi et al, 2013). The human TR (hTR) template 5′ boundary is further protected by the template‐embedded pause signal, the first dT:rA base pair in the DNA product/RNA template hybrid, that restricts DNA synthesis beyond the 5′ boundary (Brown et al, 2014). Upon completing synthesis of a DNA repeat and reaching the 5′ template boundary, telomerase regenerates the RNA template for subsequent repeat synthesis (Wu et al, 2017a). There have been identified telomerase mutations which impair specifically repeat addition processivity and not catalytic activity, yet result in stem cell defects that manifest as a spectrum of short telomere syndromes (Robart & Collins, 2010; Alder et al, 2011; Gramatges et al, 2013; Zaug et al, 2013).
Telomerase repeat addition processivity relies on a highly complex and unique catalytic reaction cycle that comprises two distinct phases: (i) synthesis of a single telomeric repeat with six consecutive nucleotide incorporation steps and (ii) regeneration of the template for additional telomeric repeat synthesis (Podlevsky & Chen, 2012). Telomerase catalyzes the nucleotide incorporation reaction with an active site that comprises a triad of invariant aspartic acids universally conserved in DNA polymerases (Lingner et al, 1997). The template regeneration phase is unique to telomerase and is accomplished by a “template translocation” mechanism whereby the template dissociates from and then realigns with the newly extended DNA product (Autexier & Lue, 2006). Recent findings indicate that template translocation is a rapid process (Parks & Stone, 2014), although the precise mechanism remains enigmatic. Telomerase‐synthesized DNA products are predominantly released from the enzyme between two consecutive cycles of repeat synthesis, which generates the characteristic 6‐nt ladder banding pattern of the products (Greider, 1991). However, the specific steps that promote product release and limit processive repeat synthesis during telomerase catalytic cycle remain elusive.
Several intrinsic enzymatic determinants or reaction conditions that affect specifically repeat addition processivity have been identified. These reaction conditions, which include high dGTP concentration, stimulate telomerase repeat addition activity through an unknown mechanism (Hammond & Cech, 1997; Maine et al, 1999; Sun et al, 1999; Hardy et al, 2001; Wu et al, 2017a). Herein, we report that human telomerase exhibits lower kinetics specifically for the incorporation of the first nucleotide, a dG residue, of each telomeric repeat synthesized. This lower incorporation kinetics is mediated by the template‐embedded pause signal that arrests nucleotide synthesis at the end of the template and remains active following template translocation, therefore impairing processive repeat synthesis. Elevated dGTP concentrations increase the incorporation efficiency of the first nucleotide for each repeat synthesis and consequently stimulate both processivity and rate of telomerase repeat addition. These results reveal a critical step in the telomerase catalytic cycle that underlies the dGTP stimulation of human telomerase repeat addition.
Results
Human telomerase template‐embedded pause signal mediates high K M for nucleotide incorporation
Telomerase synthesizes DNA at an exceedingly lower rate than most DNA polymerases (Hwang et al, 2014), which is presumably due to the unique telomerase catalytic cycle for processive short DNA repeat synthesis. During each of the reiterated cycles, telomerase catalyzes the incorporation of six consecutive deoxynucleotides, dG1, dG2, dT3, dT4, dA5, and dG6, onto the 3′ hydroxyl of the DNA primer (Fig 1A), followed by template translocation to regenerate the template for the next cycle of repeat synthesis. With template translocation having been reported to be a rapid process (Parks & Stone, 2014), we investigated whether any of the six‐nucleotide incorporations limit overall telomerase repeat addition activity, especially as specific TR template residue mutations have been shown to affect telomerase enzymatic function (Gilley et al, 1995; Gilley & Blackburn, 1996; Drosopoulos et al, 2005; Brown et al, 2014). The kinetics of the individual nucleotide incorporation steps have not been previously assessed due to technical complications associated with analyzing processive telomerase enzymes. To overcome this technical difficulty, we employed template‐free (TF) human telomerase (Qi et al, 2012) that catalyzes a non‐processive DNA synthesis reaction using RNA templates supplied in trans, which permits a simplified and defined primer extension assay for determining the nucleotide incorporation efficiency at each individual position across the hTR template. Human TF telomerase was reconstituted by assembling in vitro‐expressed human TERT protein in rabbit reticulocyte lysate with the two essential hTR fragments, CR4/5 and a template‐free pseudoknot fragment lacking the template sequence (Figs 1B and EV1). This TF telomerase was assayed for nucleotide incorporation with a series of DNA/RNA duplex substrates comprising a DNA primer and an RNA oligo serving as the template (Fig EV2). The preassembled DNA/RNA hybrid substrates were specifically designed with permuted telomeric sequences and a short RNA template for measuring the K M of nucleotide incorporation specifically for dG1, dT3, dA5, and dG6, corresponding to positions 1, 3, 5, and 6 in the hTR template (Figs 1A and EV2). The K M measurements used extremely low concentrations of TF telomerase enzyme and excess nucleotide substrates ranging from 2 to 200 μM. With either 2 or 200 μM nucleotide substrate, the product formation over the incubation time remained linear indicating that the initial velocity of the reaction was measured (Appendix Fig S1). Remarkably, the K M for incorporating dG1 was approximately 120 μM, exceptionally higher than the K M for incorporating dT3, dA5, and dG6 that ranged from 3 to 31 μM (Fig 1C). To further investigate whether this was a result of nucleotide identity or the incorporation position within the template sequence, we altered the identity of the nucleotide incorporated at positions 1, 5, and 6 to dA1, dT1, dG5, and dT6 (Fig EV2). Altering the identity of the nucleotide incorporated at these positions did not substantially change the high K M for nucleotide incorporation at position 1, nor the low K M at other positions measured (Fig 1C). Interestingly, the dG‐to‐dT transversion at positions 1 and 6 noticeably increased K M from 120 to 400 μM and from 5 to 14 μM, respectively (Fig 1C). This is consistent with results reported for Tetrahymena telomerase, where the K M for incorporating dT was slightly higher than for dG (Lee & Blackburn, 1993; Collins & Greider, 1995). Overall, these results suggest that the high K M of the first nucleotide incorporation is position‐specific relative to the template sequence and independent of the identity of the nucleotide incorporated.
Figure 1. An elevated K M for the dG1 nucleotide incorporation by human telomerase.
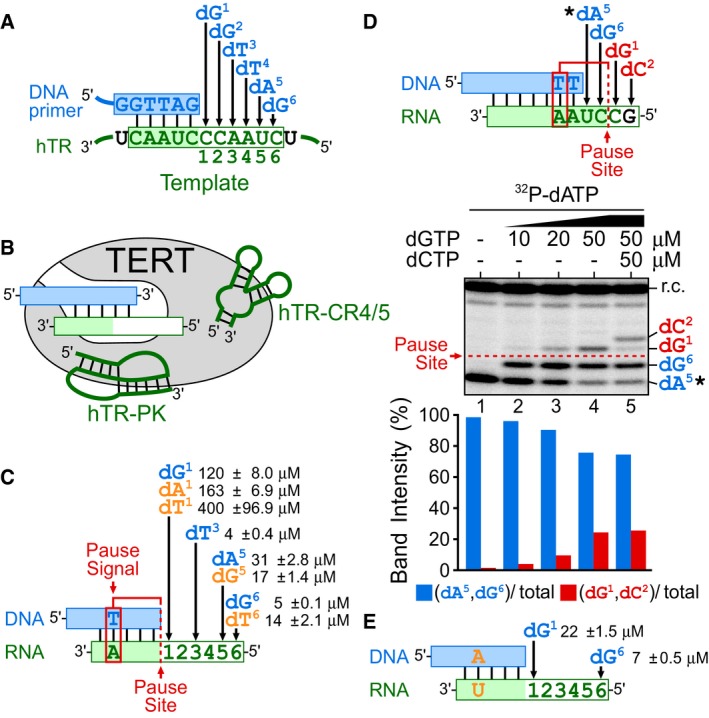
- Schematic of the six‐nucleotide incorporations catalyzed by human telomerase. Human telomerase catalyzes incorporations of six deoxynucleotides, dG1, dG2, dT3, dT4, dA5, and dG6, directed by the integral hTR template (green box). The hTR template contains two regions: an alignment sequence (shaded) for base pairing with the DNA primer and the actual templating sequence for specifying DNA polymerization. Numbers below the hTR template region denote the order and position of the six deoxynucleotide incorporations.
- Composition of template‐free (TF) telomerase. TF telomerase was reconstituted by assembling in vitro‐expressed human TERT protein with the two essential hTR fragments, CR4/5 and the pseudoknot (PK) that had the template region excised. Pre‐annealed DNA primer (blue box) and RNA template (green box) hybrids were used as substrates for the TF telomerase activity assay.
- K M for incorporating nucleotides dG1, dT3, dA5, and dG6 by TF telomerase. The dG1 incorporation is located adjacent to the pause site (red dashed line) specified by the pause signal (red box), a dT:rA base pair in the DNA/RNA hybrid. For measuring the K M of specific nucleotide incorporations, different DNA/RNA hybrid substrates with permuted sequences were used (Fig EV2). The K M for incorporating non‐telomeric nucleotides (orange) with corresponding template mutations are denoted.
- High dGTP concentrations overcome the pause signal‐mediated DNA synthesis arrest. Primer extension assays performed using the TF telomerase and a DNA/RNA hybrid substrate in the presence of 0.165 μM 32P‐dATP (asterisk) with a titration of dGTP (10, 20 and 50 μM). The DNA/RNA hybrid substrate allows for the incorporation of four nucleotides: dA5, dG6, dG1, dC2, as depicted. The pause signal (red box) and the pause site (red dashed line) in the DNA/RNA substrate arrests DNA synthesis after the incorporation of dA5 and dG6 (blue) reducing the incorporation efficiency for dG1 (red). A radiolabeled DNA recovery control (r.c.) was added before product purification and precipitation. Nucleotide incorporation beyond the pause site was quantitated by the intensity of dG1 and dC2 products (red) over the total intensity of products.
- Removal of pause signal decreases K M for dG1 incorporation. The DNA/RNA hybrid substrates with the pause signal dT:rA mutated to dA:rU (orange) were used to determine K M for nucleotide incorporation at positions dG1 and dG6 (Fig EV3).
Figure EV1. Comparison of wild‐type and template‐free (TF) telomerase reconstitution systems.
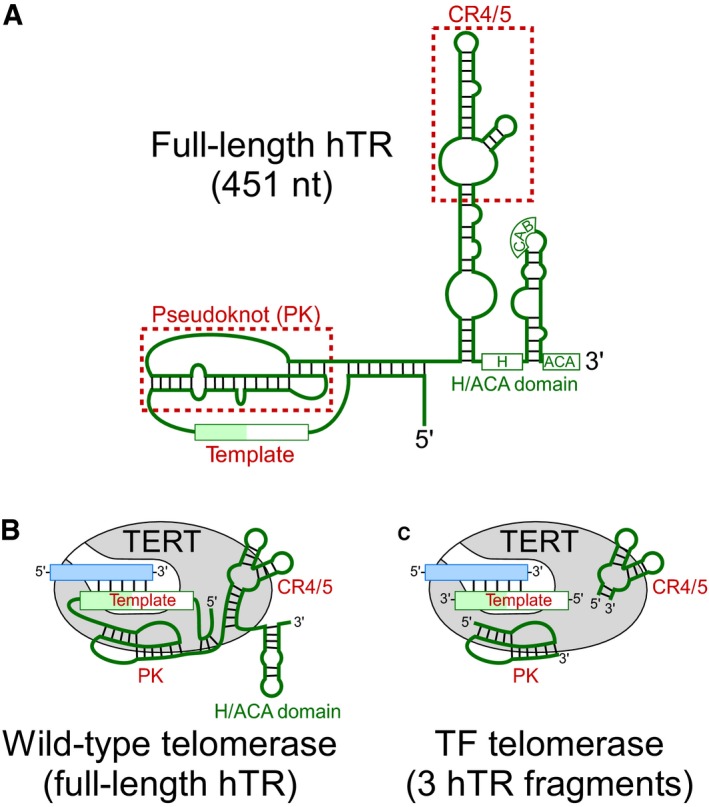
- Schematic of the 451‐nt full‐length hTR. The hTR secondary structure comprises three major structural domains: the pseudoknot (PK), conserved regions 4/5 (CR4/5), and box H/ACA domain. The template region and the two structural domains, PK, and CR4/5 (red), are minimally required for reconstituting telomerase activity in vitro.
- The wild‐type telomerase core enzyme comprises the full‐length hTR (green) and the catalytic TERT protein (gray). The substrate for wild‐type telomerase is a single‐stranded DNA primer (blue).
- TF telomerase comprises the minimally required PK and CR4/5 hTR fragments (green). The substrate for TF telomerase is duplex of a single‐stranded DNA primer (blue) pre‐annealed with an RNA template.
Figure EV2. Km measurement for nucleotide incorporations at specific template positions.
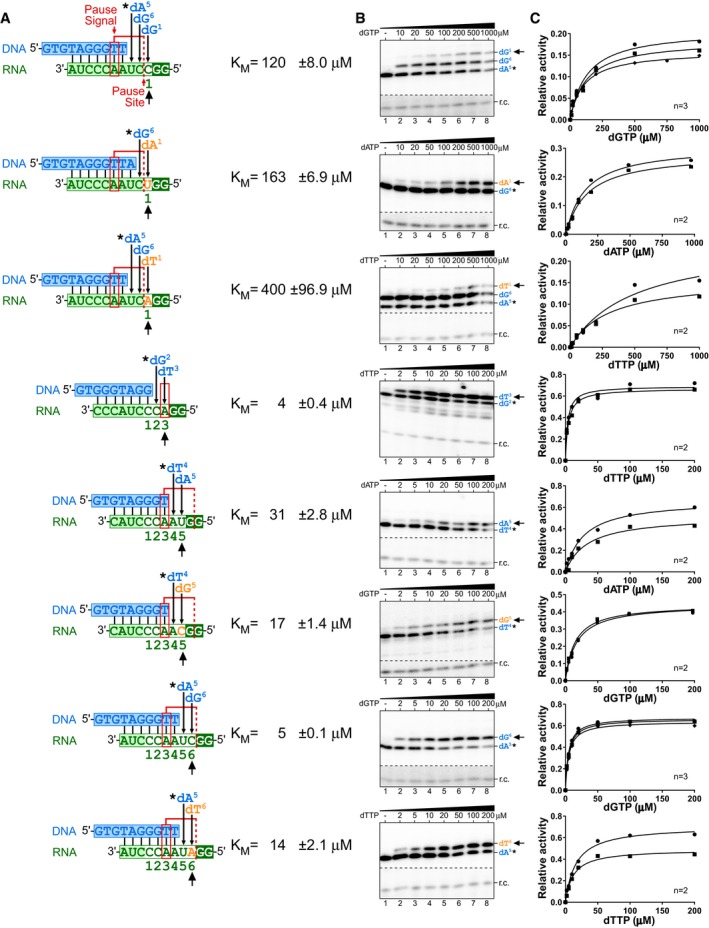
- Sequences of the DNA/RNA hybrid substrates, position of the pause signal, and pause site (red). In vitro‐reconstituted TF telomerase was analyzed with specific DNA/RNA hybrid substrates to determine the K M for nucleotide incorporation. Numbers below the DNA/RNA duplexes indicate positions corresponding to the telomerase template and the order of nucleotides incorporated. Non‐telomeric template residues (orange) are denoted. The nucleotide incorporation K M values measured are indicated (black arrow). The DNA products were labeled with a radioactive 32P‐dNTP (asterisk) followed by the nucleotide incorporation whose K M is specifically measured.
- Representative gels for K M measurements. A 32P end‐labeled DNA recovery control (r.c.) was added before product purification and precipitation. The nucleotide incorporation K M values measured are indicated (black arrow).
- Plots derived from the normalized intensity of the denoted (black arrow) nucleotide incorporation product over the total intensity of products for the specified nucleotide concentration. The Michaelis–Menten equation, Y = V max*X/(K M+X), was used to fit the nonlinear curve to determine the K M. Two or three replicates were performed for each measurement.
The incorporation of the first nucleotide, dG1, perfectly coincides with the DNA synthesis pause site governed by the pause signal embedded in the hTR template sequence (Fig 1C). The template‐embedded pause signal dT:rA base pair forms with the incorporation of dT3 and mediates DNA synthesis arrest after the incorporation of the three subsequent nucleotides, dT4, dA5, and dG6 (Brown et al, 2014). We hypothesized that the pause signal arresting DNA synthesis through an elevated K M for nucleotide incorporation at the pause site would be mitigated with increased dGTP concentrations. To test this hypothesis, we designed a DNA/RNA hybrid substrate with a dT:rA pause signal embedded within and a single‐stranded RNA template that allows for the incorporation of four nucleotides, corresponding to dA5, dG6, dG1, and a non‐telomeric dC2 (Fig 1D). The pause signal would arrest DNA synthesis at the pause site following the incorporation of 32P‐dA5 and dG6. In the presence of only 32P‐dATP, a single 32P‐dA5 residue was incorporated to the DNA primer, generating a single band (Fig 1D, lane 1). The inclusion of 5 μM dGTP in the reaction permitted the incorporation of dG6, while the subsequent dG1 incorporation was inhibited by the pause signal (Fig 1D, lane 2). Increasing the dGTP concentration to 20 or 50 μM increasingly overcame this DNA synthesis arrest and resulted in the dG1 incorporation, generating a pronounced third band (Fig 1D, lanes 3 and 4). These nucleotide incorporations were template‐dependent as the fourth band corresponding to a dC2 incorporation was generated only in the presence of dCTP (Fig 1D, lane 5). These results reveal that elevated dGTP concentrations effectively overcome the DNA synthesis arrest at the pause site, which is governed by the pause signal and mediated through the high K M of the dG1 incorporation.
To further explore the connection between the pause signal and the high K M for dG1 nucleotide incorporation, we eliminated the pause signal from the DNA/RNA hybrid substrate by mutating the base pair dT:rA to dA:rU and then measured K M for the dG1 nucleotide incorporation (Figs 1E and EV3). This transversion mutation has previously been shown to effectively inactivate the pause signal and permit DNA synthesis beyond the pause site (Brown et al, 2014). As expected, the removal of the pause signal significantly reduced the K M for dG1 incorporation from 120 to 22 μM (Figs 1E and EV3). In contrast, the loss of the pause signal in the DNA/RNA duplex did not affect the K M for incorporating dG6 (Fig 1C and E). We thus conclude that the template‐embedded pause signal has an inhibitory effect specifically on the dG1 nucleotide incorporation, mediated through a high K M exclusively for this incorporation.
Figure EV3. K M measurement for nucleotide incorporations in the presence or absence of the pause signal.
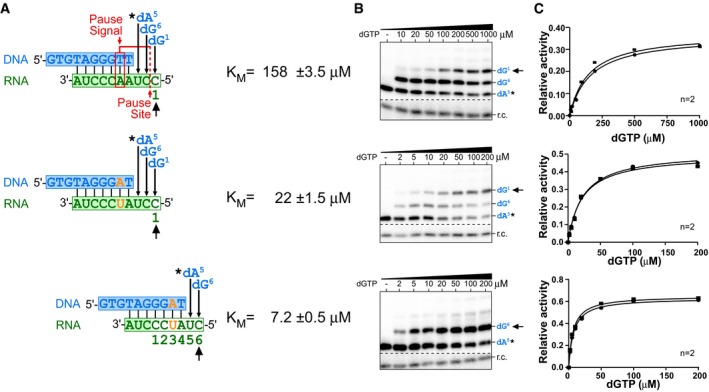
- Sequences of the DNA/RNA hybrid substrates, position of the pause signal, and pause site (red). In vitro‐reconstituted TF telomerase was analyzed with specific DNA/RNA hybrid substrates to determine the K M for each nucleotide incorporation. Numbers below the DNA/RNA duplexes indicate positions corresponding to the telomerase template and the order of nucleotides incorporated. Mutations to disrupt the pause signal denoted (orange). The DNA products were labeled by incorporating a 32P‐dATP (0.165 μM, asterisk) prior to the K M measurement.
- Representative gels for K M measurements. An 32P end‐labeled DNA recovery control (r.c.) was added before product purification and precipitation. The nucleotide incorporation K M values measured are indicated (black arrow).
- Plots derived from the normalized intensity of the denoted (black arrow) nucleotide incorporation product over the total intensity of products for the specified nucleotide concentration. The Michaelis–Menten equation, Y = V max*X/(K M+X), was used to fit the nonlinear curve to determine the K M. Two or three replicates were performed for each measurement.
dGTP stimulates telomerase repeat addition processivity and rate
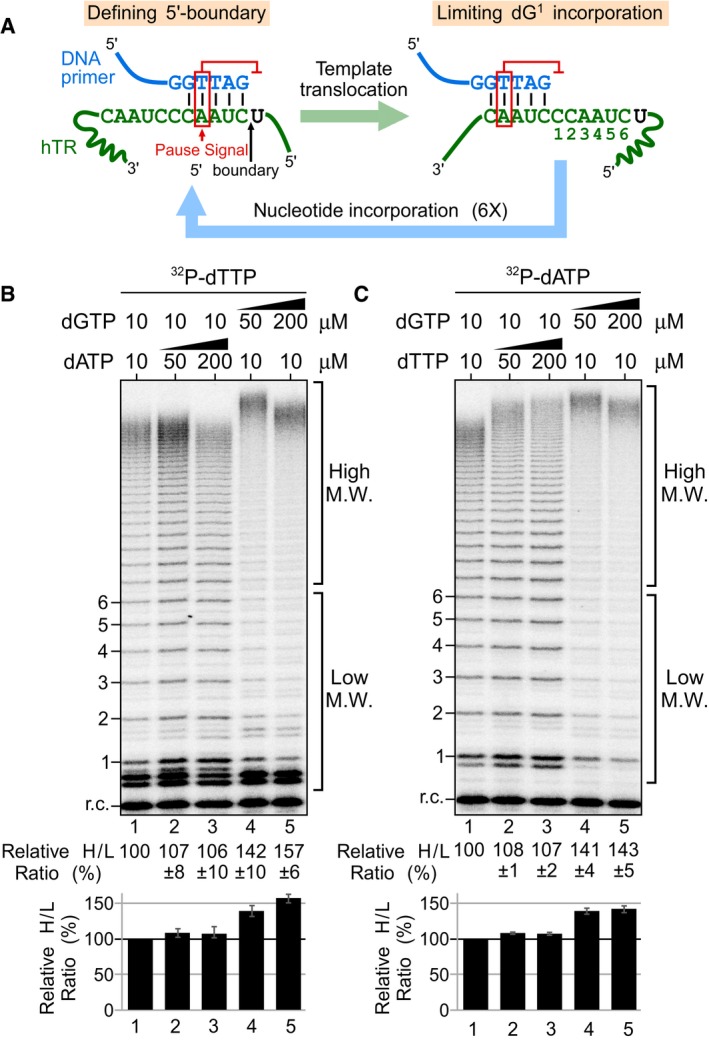
It has been previously proposed that the template‐embedded pause signal may have dual functions (Brown et al, 2014): arresting DNA synthesis at the end of the template and inhibiting the first nucleotide incorporation after template translocation (Fig 2A). Processive repeat addition by telomerase necessitates reiterated cycles of nucleotide addition. The inhibitory effect of the pause signal on the first nucleotide dG1 incorporation would impair initiating synthesis of each repeat, negatively affecting processive telomerase repeat addition. As high dGTP concentrations can effectively overcome the pause signal‐mediated inhibition of dG1 incorporation (Fig 1D), we hypothesized that high dGTP concentrations would facilitate progression into subsequent catalytic cycles and in turn promote telomerase repeat addition activity, generating more high molecular weight (M.W.) DNA products. In fact, dGTP‐dependent stimulation of repeat addition activity has been previously reported for human and ciliate telomerases (Hammond & Cech, 1997; Maine et al, 1999; Sun et al, 1999; Hardy et al, 2001). However, the underlying mechanism for dGTP stimulation of telomerase repeat addition remained unclear. To ascertain that this previously reported nucleotide stimulation of human telomerase is specific to dGTP, and not dATP or dTTP, we reconstituted wild‐type human telomerase with full‐length, template‐bearing hTR in human HEK293 cells and assayed the immuno‐purified telomerase enzyme for repeat addition activity with a telomeric DNA primer (TTAGGG)3 (Fig 2B and C). To determine the effects of dGTP and dATP concentrations on telomerase repeat addition, we performed the conventional telomerase primer extension assay in the presence of 32P‐dTTP with increasing concentrations (10, 50, and 200 μM) of dGTP or dATP individually (Fig 2B). Elevated dGTP concentrations at 50 or 200 μM significantly increased the relative ratio of high over low M.W. products by approximately 1.5‐fold (Fig 2B, lanes 1, 4, and 5), while varying the concentration of dATP had no significant effect on repeat addition (Fig 2B, lanes 1–3). To examine the effects of dTTP, a similar assay was performed in the presence of 32P‐dATP. Similarly, increased dGTP concentrations increased repeat addition efficiency by approximately 1.5‐fold (Fig 2C, lanes 1, 4, and 5), while elevated dTTP did not significantly increase repeat addition efficiency, but seemed to slightly increase the length of the highest M.W. products (Fig 2C, lanes 1–3). The slight increase of the highest M.W. products with elevated dTTP is consistent with a previous report (Drosopoulos & Prasad, 2010). Additionally, increasing nucleotide concentrations from 10 to 100 μM did not appear to alter single repeat synthesis, as nucleotide incorporation efficiency was saturated at 10 μM (Appendix Fig S2). These data clearly demonstrate that human telomerase repeat addition was stimulated specifically by increased concentrations of dGTP, and not dATP or dTTP. The increased ratio of higher over lower M.W. products could arise from an increase in the processivity and/or the rate of repeat addition, two independent attributes of the telomerase enzyme (Drosopoulos et al, 2005; Xie et al, 2010). To investigate the underlying mechanism for dGTP stimulation of telomerase repeat addition, we employed two specific telomerase activity assays to measure the processivity separately from the rate of repeat addition in the presence of elevated dGTP concentrations.
Figure 2. dGTP‐dependent repeat addition stimulation of human telomerase.
-
ASchematic of the dual function of the template‐embedded pause signal (red box). The pause signal defines the 5′ template boundary (black arrow) by inhibiting non‐telomeric DNA synthesis beyond the template boundary (Brown et al, 2014). A putative additional function of the pause signal is to limit dG1 incorporation post‐template translocation.
-
B, CDirect primer extension assays were performed with telomerase enzyme reconstituted in vivo and immuno‐purified. Telomerase was assayed in the presence of either (B) 0.165 μM 32P‐dTTP, 10 μM dTTP and a range of dGTP or dATP concentrations, or (C) 0.165 μM 32P‐dATP, 10 μM dATP and a range of dGTP or dTTP concentrations. A radiolabeled DNA recovery control (r.c.) was added before product purification and precipitation. Numbers to the left of the gel denote the number of repeats added to the telomeric primer. Ratio as a percent for the intensity of high over low M.W. DNA products generated relative to the reaction with the low nucleotide concentrations. A bar graph of the relative ratio of high/low M.W. DNA products are shown below the gel. The error bars represent standard error of the mean determined from three independent replicates.
To specifically measure the processivity of telomerase repeat addition, we designed a telomerase product release assay that quantitates repeat addition processivity based on the distribution of DNA products released from the enzyme (Fig 3A). Enzyme‐bound DNA products are still undergoing additional rounds of repeat addition, and the inclusion of these premature intermediates would influence the measurement of telomerase repeat addition processivity. For this assay, human telomerase was reconstituted in HEK 293FT cells and the immuno‐purified enzyme bound to beads was incubated with the telomeric DNA primer (TTAGGG)3 in the presence of different concentrations of dGTP or dATP. The DNA products released from the immobilized telomerase enzyme were isolated from the supernatant and analyzed to determine the probability of each successive DNA repeat addition. This probability was calculated from the slope of “products left behind” (Latrick & Cech, 2010) by plotting the intensity of major DNA products with the number of repeats (3–12) added (Fig 3A and B). The slope of the plot corresponds to the repeat addition processivity and was used to compare the relative repeat addition processivity across different assay conditions (Fig 3B). The results of this product release assay found that increasing dGTP concentration from 10 to 100 μM resulted in a twofold increase in processivity, while increasing dATP concentrations to 100 μM had no significant effect on processivity (Fig 3C). Thus, high dGTP concentration effectively stimulates the processivity of human telomerase repeat addition.
Figure 3. Effects of dGTP on repeat addition processivity and rate.
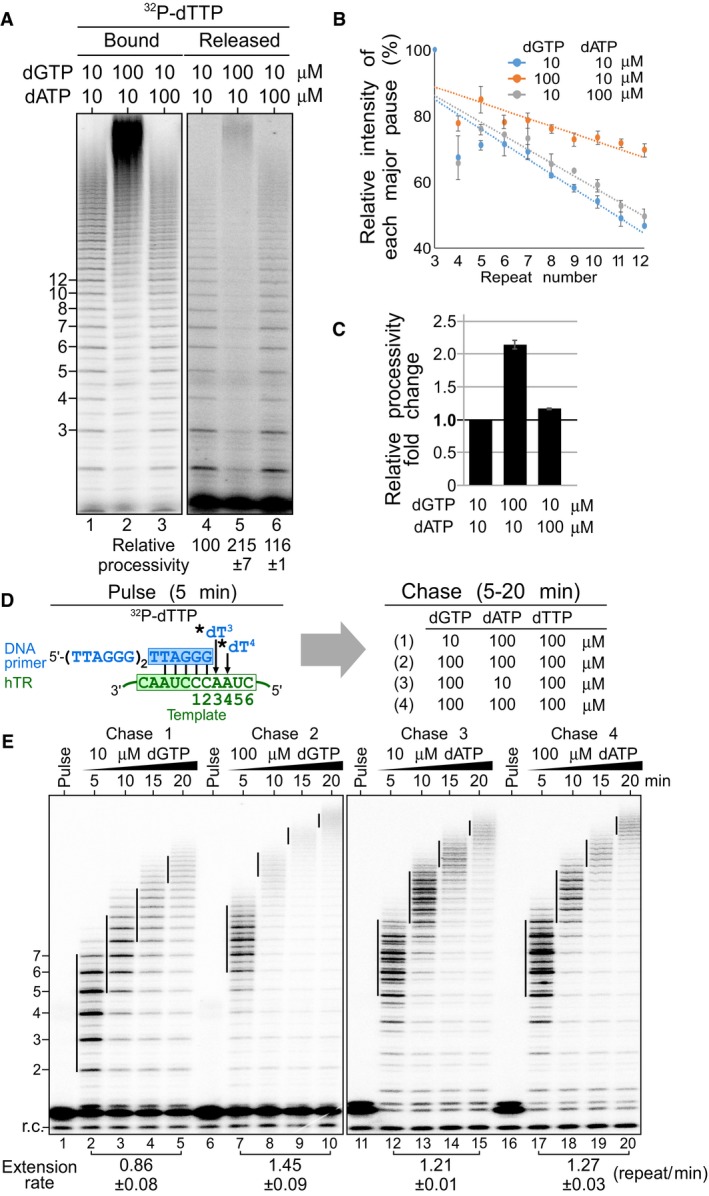
- Stimulation of repeat addition processivity with dGTP. In vivo‐reconstituted telomerase enzyme was assayed by the product release analysis in the presence of 0.165 μM 32P‐dTTP, 10 μM dTTP as well as either 10 or 100 μM of dGTP and dATP. The enzyme‐bound and released DNA products were separated and individually analyzed.
- Quantitation of repeat addition processivity. The intensities of the released DNA products with 3–12 repeats added were quantitated and normalized to the intensity of the product with 3 repeats added. The relative intensities of major bands were plotted against the number of repeats added to determine slopes that correspond to the relative processivity from each reaction (see Materials and Methods section). The error bars represent standard error of the mean determined from two independent replicates.
- Relative repeat addition processivity under different nucleotide concentrations. A bar graph indicates the fold change of the relative repeat addition processivity under either 10 or 100 μM of dGTP and dATP. The error bars represent standard error of the mean determined from two independent replicates.
- Schematic of pulse‐chase time course analysis. During the pulse reaction, the DNA primer (TTAGGG)3 was labeled with 0.165 μM 32P‐dTTP (asterisk) by the telomerase enzyme. During the chase reaction, the enzyme‐bound radiolabeled DNA primer was extended processively with telomeric repeats under 100 μM cold dTTP as well as either 10 or 100 μM of dGTP and dATP for 5, 10, 15, or 20 min.
- Repeat addition rate measured by the pulse‐chase time course analysis under differing nucleotide concentrations. The vertical lines on the gel indicate the major bands of telomere products synthesized during the chase reactions. Repeat addition rates are expressed as repeats per minute (see Materials and Methods) and indicated below the gel. The standard error of the mean was determined from two independent replicates.
We next investigated the effects of elevated dGTP concentrations on the rate of repeat addition. To specifically measure repeat addition rate, we employed a pulse‐chase time course assay to track the increasing size of the DNA products undergoing processive repeat addition over time (Fig 3D). For this pulse‐chase assay, immuno‐purified telomerase was initially incubated exclusively with 32P‐dTTP to radioactively label the DNA primer and this pulsed reaction was chased with cold nucleotides to track the processive enzyme–DNA complexes over time. The repeat addition rate was 0.86 repeat/min measured under the chase condition with 10 μM dGTP and 100 μM of dTTP and dATP. However, when chased with the dGTP concentration increased to 100 μM, the repeat addition rate nearly doubled to 1.45 repeat/min (Fig 3E, left panel). In contrast, when the reaction was chased with either 10 or 100 μM dATP, in the presence of 100 μM dGTP and dTTP, the repeat addition rate remained unchanged at 1.21 or 1.27 repeat/min, respectively (Fig 3E, right panel). Thus, dGTP concentration is a crucial determinant for the rate, in addition to the processivity, of human telomerase repeat addition.
First nucleotide incorporation following template translocation mediates nucleotide‐specific stimulation of telomerase repeat addition
The synthesis of a telomeric DNA repeat comprises three dG incorporations: dG1, dG2, and dG6 (Fig 4A). We hypothesized that the high dGTP concentration stimulated telomerase repeat addition by overcoming the high K M for dG1 incorporation. To determine whether the dG1 incorporation is specifically responsible for the dGTP stimulation of telomerase repeat addition, we generated three hTR template mutants hTR‐51U, hTR‐50U, and hTR‐46/52U that individually altered the three dG nucleotide incorporations to dA1, dA2, and dA6, respectively. We reconstituted these telomerase template mutants in human HEK293 cells and assayed the immuno‐purified enzyme by conventional telomerase primer extension assay in the presence of 32P‐dTTP with either 10 or 100 μM of dGTP or dATP. Remarkably, of these three mutants, only the hTR‐51U mutant had a pronounced increase of the higher M.W. products with increased dATP (Fig 4B, lanes 4 and 5) and was unaffected by increased dGTP (Fig 4B, lanes 4 and 6). Similar to wild type, the hTR‐50U and hTR‐46/52U mutant telomerases had increased repeat addition efficiencies with increased dGTP and were unaffected by increased dATP (Fig 4B, lanes 6–12). It is interesting to note that the hTR‐46/52U mutant telomerase had significantly lower repeat addition efficiency, producing less high M.W. products (Fig 4B, lanes 1 and 10). This was likely due to the less stable dA:rU base pairing located at the end of the primer/template duplex, which would negatively impact primer realignment efficiency during template translocation. Noticeably, the hTR‐51U and hTR‐50U mutant telomerases generated DNA products with altered banding profiles (Fig 4B). This is seemingly due to the substitution of a dA instead of a dG residue at the specified template positions affecting nucleotide incorporation efficiency, as it has been reported that telomerase is sensitive to template sequence alterations (Gilley et al, 1995; Drosopoulos et al, 2005). The template mutant hTR‐50U appeared to have a lower incorporation efficiency for dG6 residue, resulting in the accumulation of the DNA product after dA5 incorporation visible as an additional band (Fig 4B, lanes 7 and 8), which was effectively alleviated by high dGTP (Fig 4B, lane 9). Nonetheless, by changing the nucleotide incorporation from dG1 to dA1 with the hTR‐51U template mutation, we effectively changed the stimulation of human telomerase repeat addition from dGTP to dATP‐dependent. This further supports the hypothesis that the dG1 nucleotide incorporation is a critical step for human telomerase repeat addition.
Figure 4. The dG1 incorporation and telomerase repeat addition activity.

- Sequences of hTR mutant templates and DNA primers used in this assay. The wild‐type hTR template specifies for the incorporation of three dG residues, dG1, dG2, and dG6. The mutant hTR templates, 51U, 50U, and 46/52U, harbor rC‐to‐rU template mutations (orange) that specify incorporations of non‐telomeric dA1, dA2, and dA6 (orange), respectively. The telomerase template mutants were assayed with corresponding DNA primers as depicted.
- Direct primer extension assays of telomerase template mutants. Wild‐type and mutant telomerases were reconstituted in vivo and the immuno‐purified enzymes were assayed in the presence of 0.165 μM 32P‐dTTP, 10 μM dTTP as well as either 10 or 100 μM of dGTP and dATP. Ratio as a percent for the intensity of high over low M.W. DNA products generated relative to the wild‐type telomerase reaction with the low nucleotide concentrations. A bar graph of the relative ratio of high/low M.W. DNA products are shown below the gel. Standard error of the mean was determined from three independent replicates.
We next sought to determine whether the dATP‐dependent stimulation of hTR‐U51 mutant telomerase affects specifically the processivity and/or the rate of repeat addition. We examined the repeat addition processivity of the hTR‐51U mutant with our product release assay (Fig 5A). As expected, the hTR‐51U mutant had a 35% increase of telomerase repeat addition processivity in the presence of 100 μM dATP and no effect with 100 μM dGTP (Fig 5A–C). This result indicates that the first nucleotide incorporation in the telomerase catalytic cycle is an important determinant for repeat addition processivity. Following this, we then examined the contribution of the first nucleotide incorporation efficiency on the repeat addition rate. We performed a pulse‐chase time course assay with the hTR‐51U mutant to measure the repeat addition rate in the presence of either 10 or 100 μM of dATP or dGTP (Fig 5D). When the reactions were chased with 100 μM dATP, the repeat addition rate was dramatically increased to 3.15 repeat/min from 1.27 repeat/min with 10 μM dATP—a near twofold increase (Fig 5E). In contrast, increasing dGTP from 10 to 100 μM did not increase the repeat addition rate of hTR‐51U mutant, which remained similar from 2.99 to 2.95 repeat/min (Fig 5E). Our data indicate that the concentration of the nucleotide incorporated as the first residue of a telomeric DNA repeat is crucial for the processivity as well as the rate of human telomerase repeat addition.
Figure 5. Effects of dATP on repeat addition processivity and rate for the telomerase mutant hTR‐51U.
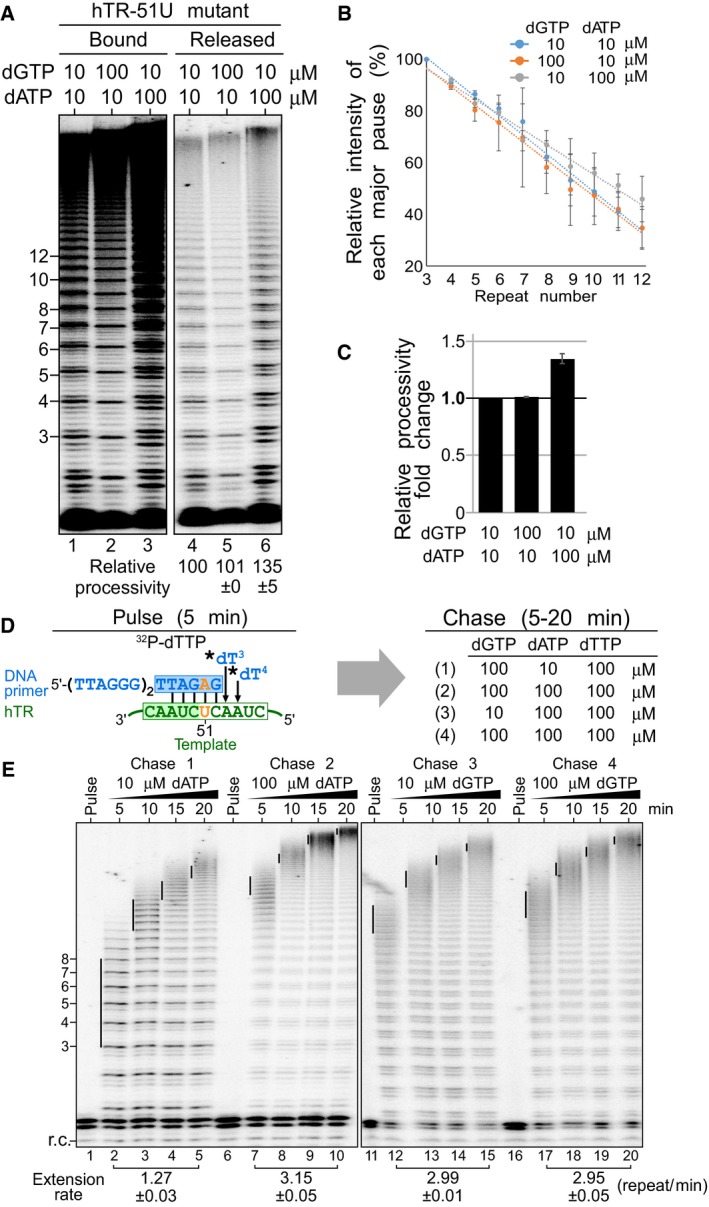
- dATP‐dependent stimulation of repeat addition processivity with the hTR‐51U mutant. In vivo‐reconstituted telomerase enzyme was assayed by the product release analysis in the presence of 0.165 μM 32P‐dTTP, 10 μM dTTP as well as either 10 or 100 μM of dGTP and dATP. The enzyme‐bound and released DNA products were separated and individually analyzed.
- Quantitation of repeat addition processivity. The intensities of the released DNA products with 3–12 repeats added were quantitated and normalized to the intensity of the product with 3 repeats added. The relative intensities of major bands were plotted against the number of repeats added to determine the slopes that correspond to the relative processivity from each reaction. The error bars represent standard error of the mean determined from two independent replicates.
- Relative repeat addition processivity under different nucleotide concentrations. A bar graph indicates the fold change of the relative repeat addition processivity under either 10 or 100 μM of dGTP and dATP. The error bars represent standard error of the mean determined from two independent replicates.
- Schematic of the pulse‐chase time course analysis for the telomerase mutant hTR‐51U. During the pulse reaction, the DNA primer (TTAGGG)2TTAGAG was labeled with 0.165 μM 32P‐dTTP (asterisk) by the telomerase hTR‐51U enzyme. During the chase reaction, the enzyme‐bound radiolabeled DNA primer was extended processively with telomeric repeats under 100 μM cold dTTP as well as either 10 or 100 μM dGTP and dATP for 5, 10, 15, or 20 min.
- Repeat addition rate measured by the pulse‐chase time course analysis under differing nucleotide concentrations. A radiolabeled DNA recovery control (r.c.) was added before product purification and precipitation. The vertical lines on the gel indicate the major bands of telomere products synthesized during the chase reactions. Repeat addition rates are expressed as repeats per minute and indicated below the gel. The standard error of the mean was determined from two independent replicates.
Telomerase can incorporate deoxynucleoside diphosphates as substrate
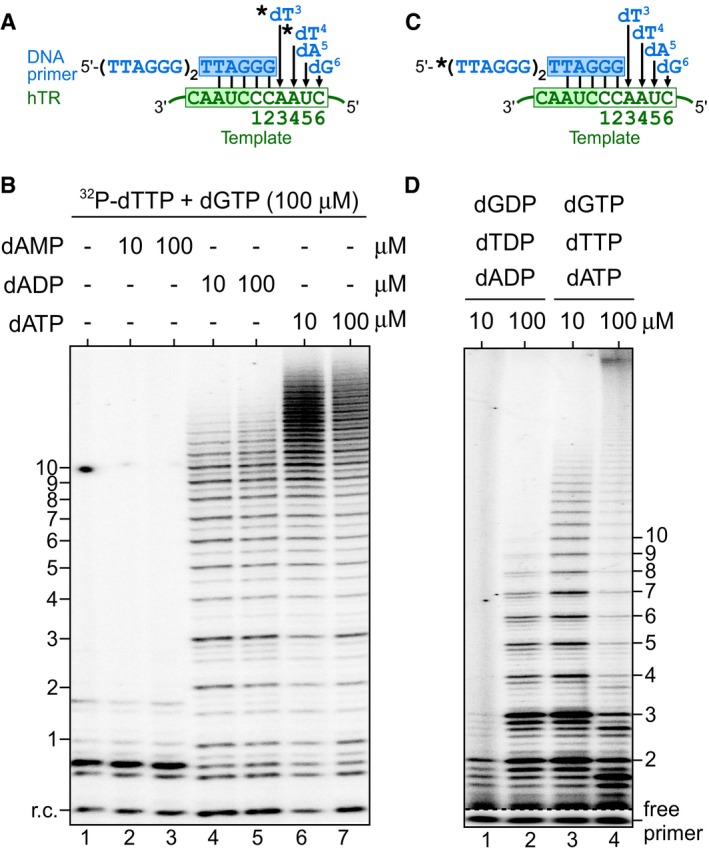
It was previously reported that Tetrahymena telomerase accumulates high M.W. DNA products in the presence of elevated dGTP, dGDP, or even dGMP, leading to the hypothesis that a secondary guanosine binding site was responsible for the dGTP‐dependent repeat addition stimulation (Hardy et al, 2001). However, our hTR51U telomerase mutant exhibited dATP‐dependent stimulation of repeat addition processivity, opposing a secondary guanosine binding site in human telomerase. To investigate the possibility of a more general purine nucleoside binding site for repeat addition stimulation, we examined whether dGMP or dGDP stimulates human telomerase repeat addition. Our result showed that dGMP, at either 10 or 100 μM, failed to stimulate telomerase repeat addition in a reaction containing 10 μM dGTP, 10 μM dTTP, and 100 μM dATP (Fig 6A, lanes 1 and 2). Interestingly, increasing dGDP concentrations from 10 to 100 μM generated a noticeable increase in telomerase repeat addition efficiency (Fig 6A, lanes 3 and 4). To investigate this dGDP‐dependent stimulation of repeat addition, we examined whether dGDP functions as a substrate for nucleotide incorporation by telomerase. By replacing dGTP with either dGDP or dGMP in the telomerase reaction, we showed that human telomerase can effectively incorporate dGDP as substrate for telomeric DNA synthesis (Fig 6A, lanes 8–9), yet not dGMP (Fig 6A, lanes 6–7). The purity of the dGMP, dGDP, and dGTP nucleotides was assessed by MALDI‐MS analysis to ensure no dGTP contamination (Appendix Fig S3). We further examined telomerase for utilizing dADP as substrate. Similarly, human telomerase can effectively incorporate dADP, and not dAMP (Fig EV4A and B). To eliminate the possibility of γ‐phosphate transfer from dATP in the reaction to dGDP, we performed a telomerase reaction lacking any nucleoside triphosphates using a 32P end‐labeled DNA primer with exclusively deoxynucleoside diphosphates: dGDP, dADP, and dTDP as substrates (Fig EV4C). This deoxynucleoside diphosphates‐only telomerase reaction generated a significant level of repeat addition activity (Fig EV4D, lanes 1–2), which, however, was consistently lower than the activity from the deoxynucleoside triphosphates reaction (Fig EV4D, lanes 3–4). These results suggest that deoxynucleoside diphosphates are sufficient substrates, yet less effective than deoxynucleoside triphosphates for human telomerase DNA synthesis.
Figure 6. Telomerase utilizes deoxynucleoside diphosphates as substrate.
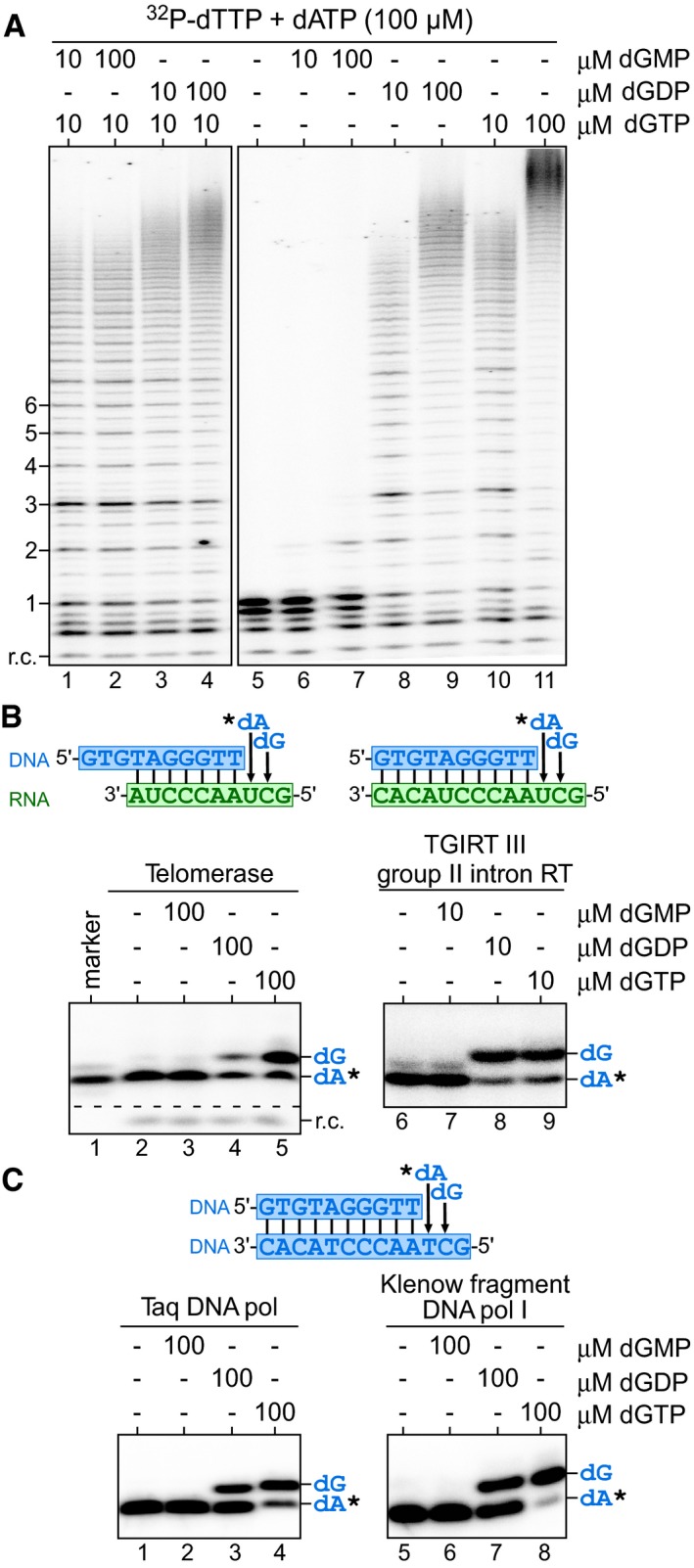
- Direct primer extension assays were performed with telomerase enzyme reconstituted in vivo and immuno‐purified. Telomerase was assayed in the presence of 0.165 μM 32P‐dTTP, 10 μM dTTP, 100 μM dATP as well as combinations of either 10 or 100 μM of dGMP, dGDP, or dGTP.
- Utilization of dGDP by RTs. Specific DNA/RNA hybrid substrates depicted harbor a template sequence for the incorporation of a single dA and a dG residue.
- Utilization of dGDP by DNA‐dependent DNA polymerases. The DNA/DNA hybrid substrate depicted harbors a template sequence to allow for incorporation of a single dA and a dG residue. Taq DNA polymerase (pol) and the Klenow fragment of DNA polymerases I were assayed in the presence of 0.165 μM 32P‐dATP (asterisk) and either dGMP, dGDP, or dGTP. The identity of nucleotides added are denoted to the right of the gel.
Figure EV4. The utilization of deoxynucleoside diphosphates as substrate for telomerase nucleotide addition.
- Schematic of nucleotide addition with wild‐type telomerase. The order of the nucleotides, dT3, dT4, dA5, and dG6, added prior to first template translocation is depicted. The DNA products were labeled with 0.165 μM 32P‐dTTP (asterisk) and 10 μM dTTP.
- Telomerase was assayed in the presence of 0.165 μM 32P‐dTTP and 100 μM dGTP with either 10 or 100 μM of dAMP, dADP, and dATP. An 32P end‐labeled DNA recovery control (r.c.) was added before product purification and precipitation.
- Schematic of nucleotide addition with a 32P end‐labeled DNA primer and wild‐type telomerase. The order of the nucleotides, dT3, dT4, dA5, and dG6, added prior to first template translocation, was depicted. The DNA primer was 32P end‐labeled (asterisk) to eliminate the need of including 32P‐dTTP in the dNDP‐only reaction.
- Telomerase was assayed in the presence of either 10 or 100 μM deoxynucleoside diphosphates (dGDP, dTDP, dADP) or deoxynucleoside triphosphates (dGTP, dTTP, dATP). Direct primer extension assays were performed with telomerase enzyme reconstituted in vivo and immuno‐purified.
We sought to assess the pervasiveness of DNA polymerases for utilizing deoxynucleoside diphosphates for DNA synthesis. In addition to telomerase examined in this study, it has been previously reported that the HIV RT and a bacteriophage DNA polymerase are capable of using deoxynucleoside diphosphates as substrate (Yang et al, 2002; Garforth et al, 2008). We examined three RTs: TF telomerase, AMV, and TGIRT III group II intron RTs, and three DNA‐dependent DNA polymerases: Taq, T4, and the Klenow fragment, for incorporating dGDP into DNA products using corresponding DNA/RNA or DNA/DNA duplex substrates and labeling 32P‐dATP (Figs 6B and C, and EV5). Except for the T4 DNA polymerase (Fig EV5, lane 7), all other examined enzymes showed significant incorporation of dGDP and not dGMP as substrate (Fig 6B, lane 4 and 8; Fig 6C, lanes 3; and 7; Fig EV5, lane 3). All enzymes analyzed showed template‐directed nucleotide incorporations lacking non‐specific terminal transferase activities. Therefore, the utilization of deoxynucleoside diphosphate as substrate for DNA synthesis is ubiquitous among DNA polymerases. Moreover, the dGDP‐dependent stimulation of human telomerase repeat addition is likely from active incorporation of dGDP as substrate rather than secondary‐site binding.
Figure EV5. Utilization of dGDP as substrate by the AMV RT and T4 DNA polymerase.
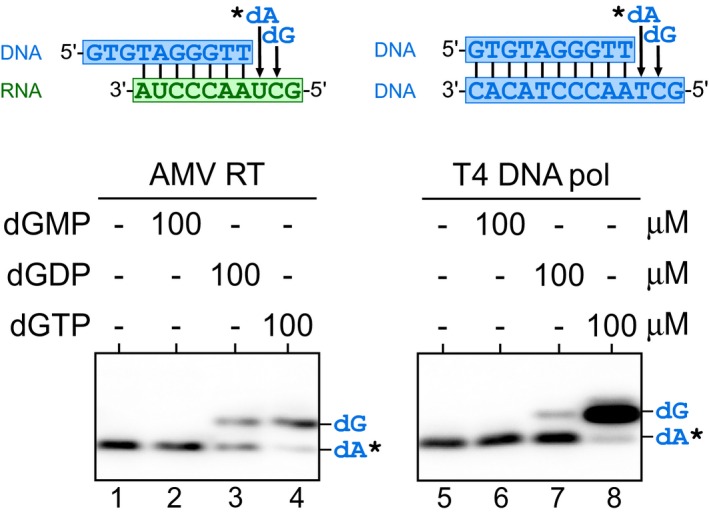
Sequences of the DNA/RNA or DNA/DNA hybrid substrates are shown above the gels. The templates of the substrates specify incorporation for a dA and a dG residue. The enzymes were assayed with 0.165 μM 32P‐dATP (asterisk) and either dGMP, dGDP, or dGTP at 100 μM.
Removal of the pause signal eliminates dGTP‐specific stimulation of human telomerase repeat addition
With our TF telomerase system, we showed that the template‐embedded pause signal is responsible for the high K M of the dG1 incorporation (Fig 1E), a crucial determinant for repeat addition processivity and rate (Fig 3). To assess whether the pause signal limits repeat addition in a processive telomerase reaction, we generated a human telomerase template mutant, termed ∆pause, that lacks the pause signal by mutating the four rA residues (nt positions 48, 49, 54, and 55) in the template to rU residues (Fig 7A). Our previous study indicated that each of the rA residues in the TR template must be altered to completely eliminate the pause signal (Brown et al, 2014). We reconstituted wild‐type and the Δpause mutant human telomerase in HEK293 cells and assayed the immuno‐purified enzyme with corresponding DNA primers and a titration of dGTP from 10 to 50 μM (Fig 7B). In sharp contrast to the wild‐type telomerase that had the expected dGTP‐dependent stimulation of repeat addition activity (Fig 7B, lanes 1–3), the Δpause mutant had strikingly high repeat addition activity with only 10 μM dGTP, which remained unchanged with 100 μM dGTP (Fig 7B, lanes 4–6). Interestingly, the Δpause template mutant had an altered major banding pattern compared with the wild‐type enzyme, which was presumably due to a shift of the limiting nucleotide incorporation from dG1 to the three dA residues for the mutant enzyme (Fig 7B, lane 4). We then analyzed the Δpause mutant for repeat addition processivity and rate separately (Fig 8). The Δpause mutant had an approximately onefold greater processivity at either 10 or 100 μM dGTP compared to the wild‐type enzyme at 10 μM dGTP (Fig 8A–C). The repeat addition rate of the Δpause mutant was found to be 1.70 repeat/min at 10 μM dGTP and 1.65 repeat/min with 100 μM dGTP (Fig 8D and E), which is significantly higher than the 0.7 and 1.3 repeat/min of the wild‐type enzyme at 10 and 100 μM dGTP, respectively (Fig 3D and E). These results strongly support the pause signal is responsible for the intrinsically low processivity and rate, as well as the dGTP‐specific stimulation of human telomerase repeat addition.
Figure 7. Effects of pause signal removal on telomerase repeat addition.
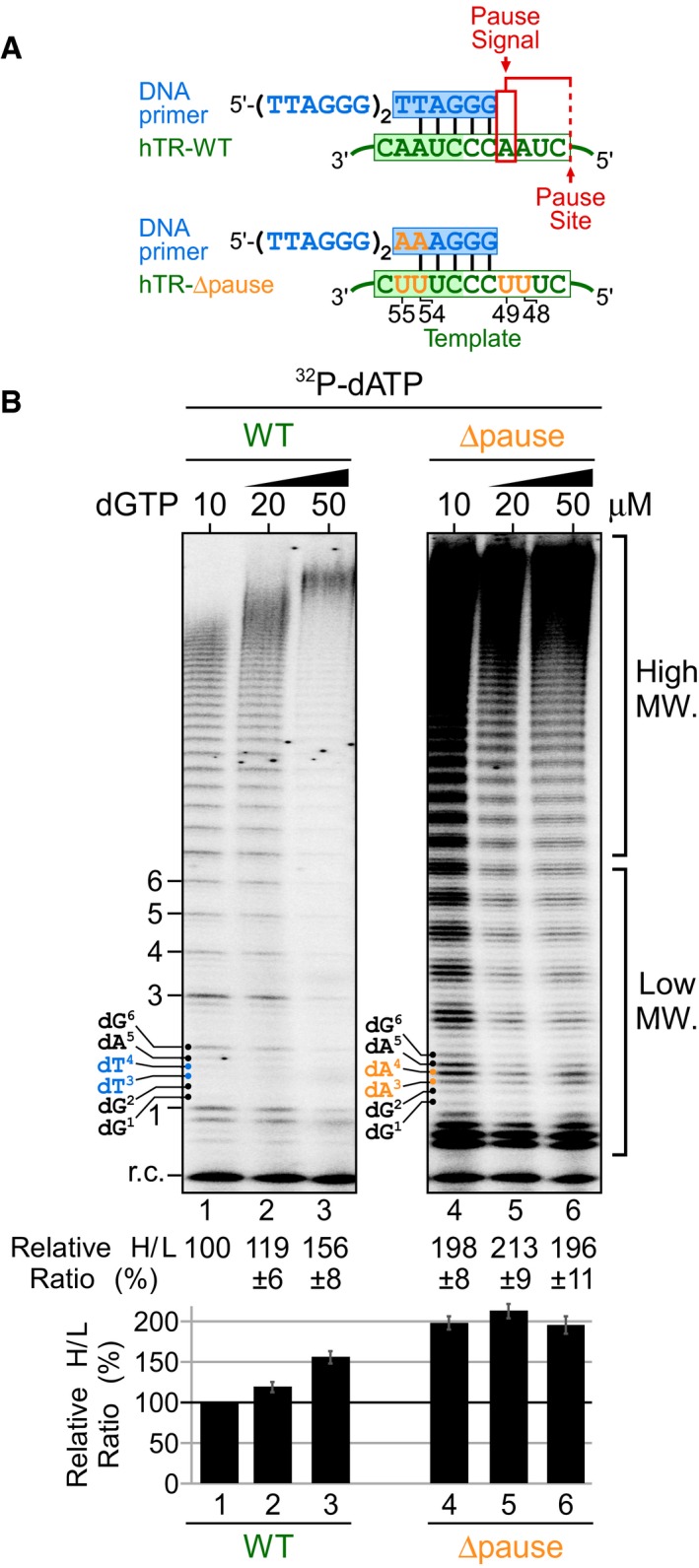
- Schematic of wild‐type and hTR Δpause template mutant telomerase with corresponding DNA primers. The hTR Δpause template mutant harbors rA‐to‐rU mutations (orange) at four residues: 48, 49, 54, and 55 to eliminate the template‐embedded pause signal (red box).
- Direct primer extension assays for the telomerase Δpause template mutant. Wild‐type and mutant telomerases were reconstituted in vivo and the immuno‐purified enzymes were assayed in the presence of 0.165 μM 32P‐dATP, 10 μM dATP as well as either 10, 20, or 50 μM of dGTP. Relative repeat addition activity was presented as the ratio of the intensity of high over low M.W. DNA products generated in each reaction normalized to the wild‐type enzyme reaction with 10 μM dGTP. A bar graph of the relative ratio of high/low M.W. DNA products are shown below the gel. Standard error of the mean was determined from three independent replicates.
Figure 8. Effects of dGTP on repeat addition processivity and rate of the hTR‐Δpause mutant telomerase.
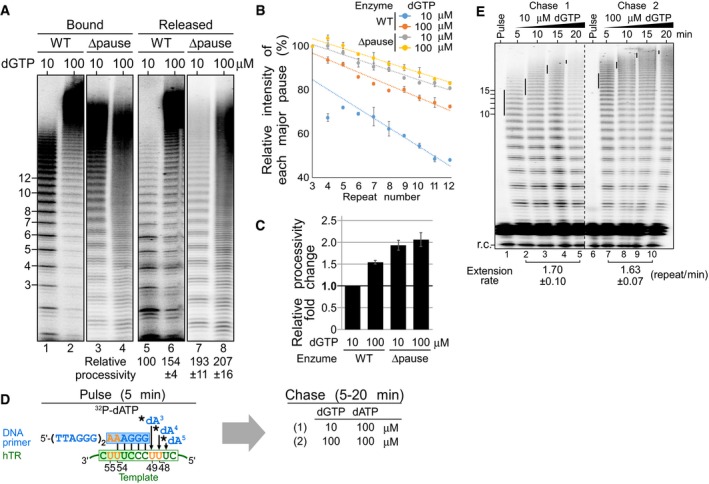
- Lack of stimulation of repeat addition processivity with dGTP for the telomerase mutant hTR‐Δpause. In vivo‐reconstituted telomerase enzyme was assayed by the product release analysis in the presence of 0.165 μM 32P‐dATP, 10 μM dATP as well as either 10 or 100 μM of dGTP. The enzyme‐bound and released DNA products were separated and individually analyzed.
- Quantitation of repeat addition processivity. The intensities of the released DNA products with 3‐12 repeats added were quantitated and normalized to the intensity of the product with 3 repeats added. The relative intensities of major bands were plotted against the number of repeats added to determine the slopes that correspond to the relative processivity from each reaction. The error bars represent standard error of the mean determined from two independent replicates.
- Relative repeat addition processivity under different nucleotide concentrations. A bar graph indicates the fold change of the relative repeat addition processivity under either 10 or 100 μM of dGTP for wild‐type and the hTR‐Δpause mutant. The error bars represent standard error of the mean determined from two independent replicates.
- Schematic of the pulse‐chase time course analysis for the telomerase mutant hTR‐51U. In the pulse reaction, the DNA primer (TTAGGG)2AAAGGG was labeled with 0.165 μM 32P‐dATP (asterisk) by the telomerase hTR‐Δpause enzyme. During the chase reaction, the enzyme‐bound radiolabeled DNA primer was extended processively with telomeric repeats under 100 μM cold dATP as well as either 10 or 100 μM dGTP for 5, 10, 15, or 20 min.
- Repeat addition rate measured by the pulse‐chase time course analysis under differing nucleotide concentrations. A radiolabeled DNA recovery control (r.c.) was added before product purification and precipitation. The vertical lines on the gel indicate the major bands of telomere products synthesized during the chase reactions. Repeat addition rates are expressed as repeats per minute and indicated below the gel. The standard error of the mean was determined from two independent replicates.
Discussion
Telomerase is an RNA‐dependent DNA polymerase specialized in adding telomeric DNA repeats onto chromosome ends (Podlevsky & Chen, 2016). Apart from other DNA polymerases, telomerase employs a highly orchestrated, yet poorly understood, catalytic cycle to regenerate the RNA template for processive synthesis of multiple telomeric DNA repeats (Wu et al, 2017b). The total number of DNA repeats added by a given telomerase enzyme in a single turnover is determined by the two tangibly separate attributes of the telomerase enzyme: the processivity and the rate of repeat addition (Podlevsky & Chen, 2012; Schmidt & Cech, 2015). The processivity of telomerase repeat addition corresponds to the probability of continuous DNA repeat synthesis over complete product release following each catalytic cycle of six‐nucleotide incorporations. Distinct from processivity, the rate of repeat addition corresponds to the number of telomeric repeat synthesized per unit time. The repeat addition rate is contributed by: (i) the rate of individual nucleotide additions to the primer and (ii) the rate of template regeneration by template translocation. In this work, we discovered a previously unknown role of the telomerase template‐embedded pause signal for inhibiting the dG1 incorporation step, the first nucleotide of each telomeric DNA repeat. This slow dG1 incorporation is a decisive step that affects the rate of the telomerase reaction. Failure to incorporate the dG1 residue prevents processive additional cycles of repeat synthesis, which prompts complete DNA product release and terminates the reaction (Fig 9).
Figure 9. A working model of the telomerase catalytic cycle.
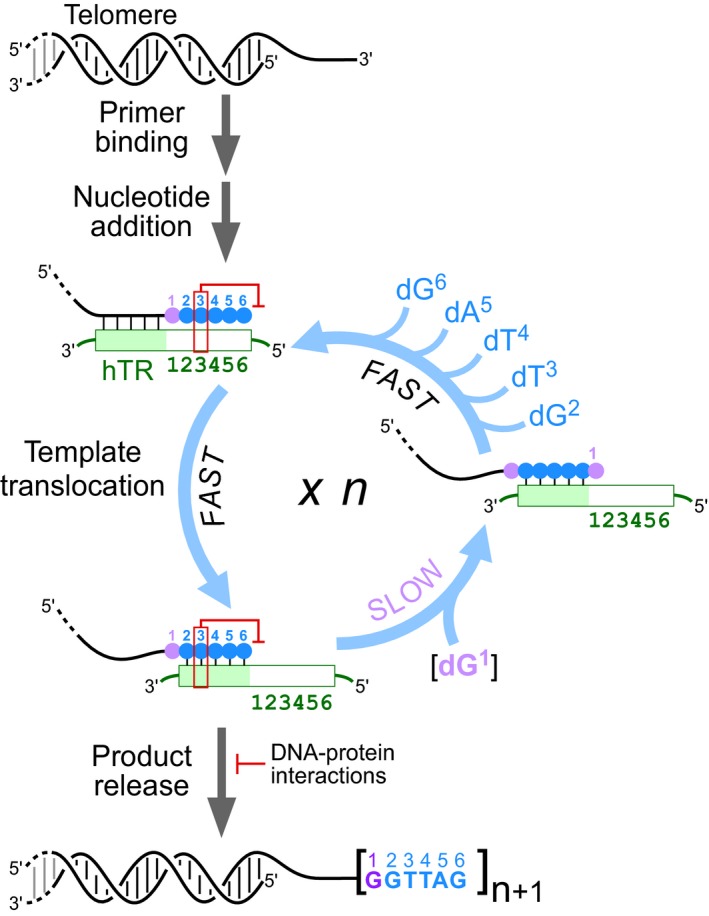
Following telomerase binding the 3′‐end of the telomere, nucleotide addition of six residues (violet and blue circles) proceeds to the end of the template and is arrested by the pause signal (red box) at the pause site. A fast template translocation process regenerates the template by realigning the template relative to the 3′‐end of the DNA primer. Progression to the next catalytic cycle is impeded by the still active pause signal from the DNA/RNA hybrid, causing a slow dG1 residue incorporation (violet) at the pause site. Failure to incorporate the dG1 residue promotes product release, which is counteracted by DNA–protein interactions through the TERT anchor sites and telomerase accessory POT1‐TPP1 protein complex. Successful dG1 residue incorporation is followed by the rapid incorporation of five additional residues, dG2, dT3, dT4, dA5, and dG6 (blue), completing a telomerase catalytic cycle. The number of repeats added corresponds to the number of catalytic cycles completed before complete disassociation of the telomeric DNA from the telomerase enzyme. Increasing the dGTP concentration increases the rate of the slow dG1 residue incorporation, which increases telomerase repeat addition processivity and rate.
As depicted in our working model of the telomerase catalytic cycle (Fig 9), each repeat synthesis comprises the addition of six nucleotides to the 3′ hydroxyl of the DNA primer and then arrested at the 5′ end of the hTR template by the physical boundary element (Chen & Greider, 2003a) and the unique template‐embedded pause signal (Brown et al, 2014). Following each repeat synthesis, telomerase regenerates the template through a template translocation step, which has recently been shown to be a rapid process (Parks & Stone, 2014) with a rate about 100‐fold faster than the overall repeat addition rate measured (Latrick & Cech, 2010). Thus, the template translocation step is unlikely a major determinant for the rate of repeat addition (Fig 9). Moreover, the high repeat addition activity and the lack of product release after the dG6 incorporation with the Δpause mutant suggest that template translocation is efficient (Fig 7B, lane 4). Recently, two models have been proposed for the mechanism of template translocation: single‐stranded DNA retention by DNA–protein interactions (Wu et al, 2017a) and DNA hairpin formation (Yang & Lee, 2015). Each of these models is possible for template translocation, yet additional work is necessary to establish the definitive mechanism. The data presented herein center on DNA synthesis post‐template translocation and neither support nor negate these proposed models. However, mutations across the template that alter the telomeric DNA sequence do not affect the functionality of the pause signal (Brown et al, 2014), which would imply that the putative DNA hairpin is largely unaffected by the DNA sequence or is not critical for pause signal‐mediated arrest of DNA synthesis. A myriad of intrinsic and extrinsic factors contribute synergistically to template translocation efficiency and repeat addition processivity. These intrinsic factors include the template length that affects template/primer realignment (Chen & Greider, 2003b), TERT anchor sites that bind DNA to prevent complete DNA dissociation (Jacobs et al, 2006; Wyatt et al, 2007; Finger & Bryan, 2008; Zaug et al, 2008; Akiyama et al, 2015), or TERT motifs that enable stable retention of the realigned DNA/RNA hybrid in the active site (Huard et al, 2003; Lue et al, 2003; Xie et al, 2010; Qi et al, 2012; Wu et al, 2017b). Moreover, telomerase accessory proteins, POT1 (Protection Of Telomeres 1) and TPP1 (TIN2 and POT1‐interacting protein 1), have been found to increase processivity by delaying product release through DNA–protein interactions (Lingner et al, 1997; Latrick & Cech, 2010). These DNA–protein interactions promote progression into the next cycle of repeat synthesis and augments telomerase repeat addition processivity (Fig 9). In addition to the intrinsic factors, extrinsic factor such as ionic strength, temperature, nucleotide, and primer concentrations (Sun et al, 1999). The sheer number of distinct factors that affect telomerase repeat addition processivity underlies the excessive complexity of template translocation.
After a successful template translocation event, the template is regenerated and ready for further nucleotide addition (Fig 9). Nevertheless, the pause signal remains effective following template translocation and inhibits the incorporation of the dG1 residue through an excessively high K M for this incorporation (Fig 1C). Our K M measurements for nucleotide incorporation at specific positions across template were only feasible by using the non‐processive TF telomerase (Qi et al, 2012) and specifically designed DNA/RNA hybrid substrates with permuted sequences (Fig EV2); these measurements were otherwise challenging and complex with a processive telomerase enzyme. By measuring individual nucleotide incorporation efficiencies with the TF telomerase, we found that removing the pause signal in the DNA/RNA hybrid effectively lowers the high K M of the dG1 incorporation to a value comparable to other positions across the template, suggesting that the pause signal is responsible for this inhibitory effect (Figs 1E and EV3). For the processive wild‐type telomerase enzyme, the removal of the pause signal from the hTR template dramatically increases repeat addition activity even in the presence of low dGTP concentration at 10 μM (Fig 7B). This suggests that the dG1 incorporation is no longer the limiting step for processive repeat addition. The template mutations introduced in the Δpause mutants modified the sequence of the DNA products, which may affect DNA–protein interactions with TERT anchor sites or accessory proteins. While altering DNA‐enzyme binding affinity has the potential to augment repeat addition processivity, it is unlikely that this would simultaneously enhance the rate of repeat addition (Fig 8). Therefore, it is more feasible that the increased rate of repeat addition with the Δpause mutant was caused by the lower K M of the dG1 incorporation. Under saturating dGTP concentrations, the Δpause mutant and wild‐type telomerases had equally high repeat addition activity (Drosopoulos et al, 2005), supporting our hypothesis that the dramatically high repeat addition activity of the Δpause mutant at low dGTP was mainly due to the alleviated dG1 incorporation. Moreover, the loss of the pause signal noticeably altered the banding pattern of DNA products, which was presumably from new limiting nucleotide incorporations at other template positions and premature product releases.
Upon recognition of the pause signal in the DNA/RNA hybrid, telomerase stalls DNA synthesis precisely at the pause site by the elevated K M for nucleotide incorporation (Fig 1C). While the underlying mechanism for pause signal recognition requires further investigation, we speculate that pause signal recognition induces a subtle conformational change in the DNA/RNA hybrid binding site and/or the catalytic site within the TERT protein that manifests as the elevated K M for nucleotide incorporation. Supporting this hypothesis, previous structural and biochemical studies suggest that telomerase undergoes conformational changes at distinct steps of the catalytic cycle (Mitchell et al, 2010; Tomlinson et al, 2015; Parks et al, 2017). However, determining the exact mechanism for pause signal‐mediated arrest of DNA synthesis would require high‐resolution structures of the telomerase enzyme complexed with various DNA/RNA hybrids and even the incoming nucleotide bound within the active site.
Beyond compensating for the high K M of the dG1 incorporation, high dGTP concentrations have no exotic stimulatory effects on human telomerase. Our results demonstrate that altering the dG1 incorporation to dA1 effectively shifted the stimulation of repeat addition processivity and rate from dGTP‐ to dATP‐dependent for human telomerase (Figs 4 and 5). However, the ciliate Tetrahymena thermophila telomerase has been reported to have repeat addition processivity stimulated by high concentrations of dGTP, dGDP or even dGMP (Hardy et al, 2001). In contrast, our assays with human telomerase failed to show dGMP‐dependent stimulation of repeat addition activity (Fig 6A). Thus, the effects of elevated dGTP concentrations on ciliate telomerases appear esoteric with an underlying mechanism distinct from human telomerase. It was interesting to observe that, in addition to dGTP, elevated dGDP also stimulates telomerase repeat addition (Fig 6A, lane 3). Rather than an allosteric effect from a secondary guanosine binding site, dGDP was directly utilized as substrate and effectively increased overall nucleotide concentrations (Fig 6A, lanes 8–9). Furthermore, telomerase assayed with solely deoxynucleoside diphosphates generated a significant amount of DNA products (Fig EV4D, lane 2), eliminating the possibility of phosphate transfer from deoxynucleoside triphosphates to diphosphates. Nonetheless, deoxynucleoside diphosphates are a poorer substrate to telomerase with fivefold to 10‐fold lower concentrations in cells than deoxynucleoside triphosphates (Bradshaw & Samuels, 2005). Thus, we do not expect telomerase to utilize deoxynucleoside diphosphates as substrate under cellular conditions. In addition to telomerase, all other RTs and most DNA‐dependent DNA polymerases examined in this study are capable to incorporate dGDP into DNA products (Figs 6B and C, and EV5). This, however, is not entirely unexpected as deoxynucleoside triphosphates and diphosphates both permit the same DNA polymerase catalysis with the 3′ hydroxyl of the DNA primer attacking the α‐phosphate of the incoming deoxynucleotide and the release of inorganic pyrophosphate or monophosphate, respectively (Kornberg, 1969). Interestingly, the Klenow fragment of Escherichia coli DNA polymerase that showed dGDP utilization activity in our assay (Fig 6C) has been previously reported inert with deoxynucleoside diphosphates (Kornberg, 1957; Garforth et al, 2008). This discrepancy is likely due to the lower nucleotide concentration used in previous studies.
Under estimated intracellular nucleotide concentrations (Bradshaw & Samuels, 2005), telomerase would be efficient for the synthesis of a single repeat, yet limited for the processive synthesis of multiple repeats through multiple catalytic cycles. Conventional telomerase assays have been performed under exceptionally high concentrations of dATP and dTTP in the range of 100–1,000 μM, with the radiolabeled dGTP in the very low μM range (Greider, 1991; Huard et al, 2003; Latrick & Cech, 2010; Wu et al, 2017a). These skewed assay conditions lead to significant DNA synthesis arrest at the end of the catalytic cycle and generated the characteristic six‐nucleotide ladder banding pattern, a hallmark of telomerase activity. Reducing individual nucleotide concentrations below 5 μM results in intermediate DNA repeat product accumulation due to the stalling of nucleotide addition before reaching the end of the template (Appendix Fig S2). The incorporation of dG1 is presumably slow under the low nucleotide concentrations in vivo, and telomerase would predominately release DNA products terminating with dG6 prior to the dG1 incorporation. The analysis of chromosome terminal sequences found a sharp increase of the specific dG6 terminal sequence from about 25% in telomerase‐null cells to about 40% in telomerase‐positive cells (Sfeir et al, 2005), which is consistent with our hypothesis that telomerase generates DNA products with this terminal sequence.
In contrast to the slow dG1 addition, the remaining five nucleotide incorporations appear to be efficient without accumulation of any significant intermediate products (Fig 3A, lane 4). However, in our telomerase product release assay, an intermediate DNA product with only three residues, dG1, dG2, and dT3, added was observed in the enzyme‐bound fraction (Fig 3A, lane 1), and not in the released product fraction (Fig 3A, lane 4). This intermediate DNA product likely resulted from a slightly slower incorporation of dT4. In support of the slower rate of the dT4 incorporation, increasing dTTP concentration to above 10 μM indeed increases repeat addition rate and generates products with a greater number of repeats added (Fig 2C, lanes 2 and 3), which is consistent with a previous report (Drosopoulos & Prasad, 2010). Together, these results support that the incorporation efficiencies of the remaining five residues, dG2, dT3, dT4, dA5, and dG6, minimally contribute to the overall repeat addition rate. The dG1 incorporation efficiency is the key determinant for both the processivity and the rate of telomerase repeat addition.
Elucidating the mechanism by which telomerase undergoes processive synthesis of hundreds of telomeric DNA repeats has remained challenging. The unique telomerase catalytic cycle relies on a highly orchestrated arrangement of TERT, TR, and accessory proteins to facilitate DNA product retention and effective template regeneration for processive repeat addition. Our findings herein redefine the telomerase catalytic cycle as three critical and distinct steps: (i) rapid template translocation followed by (ii) slow dG1 incorporation and (iii) efficient addition of the five remaining residues dG2, dT3, dT4, dA5, and dG6 (Fig 9). DNA synthesis arrest at the slow dG1 incorporation step promotes product release and limits processive repeat synthesis. The intrinsic low processivity of human telomerase is beneficial as it affords repeat addition regulation through DNA–protein interactions with TERT anchor sites and telomerase accessory proteins to control product release (Fig 9). Moreover, our findings suggest that telomerase products are released mainly from unsuccessful dG1 incorporation, instead of failed template translocation. Thus, the slow dG1 incorporation step and the inhibitory effect of the pause signal limit the processivity and rate of telomerase repeat addition, representing prime targets for therapeutic regulation of telomerase function.
Materials and Methods
In vitro reconstitution of TF human telomerase
Human TERT protein was expressed in rabbit reticulocyte lysate (RRL) from the pNFLAG‐hTERT plasmid DNA using the TnT T7 Quick Coupled transcription/translation kit (Promega) following manufacturer's instructions (Xie et al, 2010). The hTR pseudoknot (residues 64–184) and CR4/5 (residues 239–328) fragments were in vitro‐transcribed, gel‐purified, and assembled together with the TERT protein in RRL for 30 min at 30°C at a final concentration of 1.0 μM (Qi et al, 2012; Brown et al, 2014).
In vivo reconstitution of wild‐type human telomerase
HEK 293FT cells were grown in DMEM medium (Corning) supplemented with 10% FBS (Atlanta Biological), 1× Penicillin–Streptomycin–Amphotericin B mix (Lonza) and 5% CO2 at 37°C to 80–90% confluency. Cells in a 6‐well plate were transfected with 0.4 μg of pcDNA‐NFLAG‐hTERT, 1.6 μg of pBS‐U1‐hTR wild type or template mutants, and 6 μl of FuGENE HD transfection reagent (Promega) following manufacturer's instruction. Cells were harvested 48 h post‐transfection, homogenized in HEPES lysis buffer (20 mM HEPES‐KOH, pH 7.9, 400 mM NaCl, 0.2 mM EGTA, 2 mM MgCl2, 10% glycerol, 5 mM β‐mercaptoethanol, and 1× complete protease inhibitor cocktail (Roche), 1 mM PMSF), incubated on ice for 30 min and the lysate clarified by centrifugation. Two hundred microliters of cell lysate was combined with 30 μl Anti‐FLAG® M2 Beads (Sigma, Cat#A2220) pre‐washed with 1× TBS buffer (50 mM Tris–HCl, pH 7.4 and 150 mM NaCl) and incubated at 4°C with gentle rotation for 1 h. The beads were washed three times with 100 μl of 1× TBS buffer and once with 50 μl 1× telomerase reaction buffer (50 mM Tris–HCl, pH 7.5, 3 mM MgCl2, 50 mM KCl, 2 mM DTT, and 1 mM spermidine), followed by activity assay.
K M measurement using TF telomerase
One microliter of RRL reconstituted TF telomerase enzyme was assayed in a 10 μl reaction containing 1× telomerase reaction buffer, 40 μM pre‐annealed DNA/RNA duplex, specified dNTPs, and 0.165 μM of the denoted α‐32P‐dNTP. For measuring the K M values, the activity assays were performed with nucleotide concentrations varying from 0 to 200 μM, or up to 1 mM for high K M measurement. Reactions were incubated at 30°C for 60 min and terminated by phenol/chloroform extraction, followed by ethanol precipitation. The DNA products were resolved on a 15% (wt/vol) polyacrylamide/8M urea denaturing gel, dried, exposed to a phosphorstorage screen, and imaged on a Bio‐Rad FX‐Pro phosphorimager. The intensities of specific products were normalized to the total product intensity and plotted against the nucleotide concentrations with the Michaelis–Menten equation, Y = V max*X/(K M+X), used to fit the nonlinear curve to determine the K M (Prism 5, GraphPad Software).
Activity assay for dNDP and dNMP incorporation
One microliter of RRL reconstituted telomerase enzyme, 1 unit of AMV RT (Promega), 0.5 units of Taq DNA pol III (NEB), 1 unit of T4 DNA pol (Fermentas), or 0.5 units of Klenow fragment of DNA pol I (Invitrogen) were assayed in 10 μl reactions containing 1× telomerase reaction buffer, 40 μM of denoted pre‐annealed DNA/RNA or DNA/DNA hybrid substrates, 100 μM dGTP, dGDP, or dGMP, and 0.165 μM α‐32P‐dATP. The assay with TGIRT III group II intron RT (InGex) contained 50 units of enzyme, 1× reaction buffer (20 mM Tris–HCl, pH 7.5, 5 mM MgCl2, 0.45 M NaCl, 5 mM DTT) and 10 μM dGTP, dGDP, or dGMP, and 0.165 μM α‐32P‐dATP. Reactions were incubated at 30°C for 60 min and terminated by phenol/chloroform extraction, followed by ethanol precipitation. The size marker was prepared in a 10 μl reaction containing 1× reaction buffer (100 mM sodium cacodylate, pH 6.8, 1 mM CoCl2, and 0.1 mM DTT), 1 μM oligonucleotide as indicated, 10 units of terminal deoxynucleotidyl transferase (TdT, Affymetrix), and 0.165 μM α‐32P‐dATP (3,000 Ci/mmol, 10 mCi/ml; PerkinElmer). The reaction was incubated at 30°C for 5 s and terminated by addition of 10 μl 2× formamide loading buffer [10 mM Tris–HCl, pH 8.0, 80% (vol/vol) formamide, 2 mM EDTA, 0.08% bromophenol blue, and 0.08% xylene cyanol]. The DNA products were resolved on a 15% (wt/vol) polyacrylamide/8 M urea denaturing gel, dried, exposed to a phosphorstorage screen and imaged on a Bio‐Rad FX‐Pro phosphorimager.
Telomerase direct primer extension assay
Twenty microliters of immuno‐purified in vivo‐reconstituted telomerase enzyme on beads was assayed in a 10 μl reaction containing 1× telomerase reaction buffer, 1 μM DNA primer, specified dNTPs, and 0.165 μM of the denoted α‐32P‐dNTP. Reactions were incubated at 30°C for 60 min and terminated by phenol/chloroform extraction, followed by ethanol precipitation. The DNA products were resolved on a 10% (wt/vol) polyacrylamide/8 M urea denaturing gel, dried, exposed to a phosphorstorage screen, and imaged on a Bio‐Rad FX‐Pro phosphorimager. Repeat addition efficiency was estimated as the ratio of the high M.W. DNA products (> 6 repeats added) over the low M.W. DNA products (1–6 repeats added). The cutoff at 6 repeats was arbitrarily chosen to divide the gel into approximately two even sections. The relative high/low ratios of reactions with varying nucleotide concentrations were determined through normalization to the ratio from the low nucleotide concentration reaction.
Telomerase product release assay for repeat addition processivity determination
Twenty microliters of immuno‐purified in vivo‐reconstituted telomerase enzyme on beads was assayed in a 10 μl reaction containing 1× telomerase reaction buffer, 1 μM DNA primer, a range of dNTPs, and 0.165 μM of the denoted α‐32P‐dNTP. Reactions were incubated at 30°C for 60 min, and the DNA products in the supernatant were separated from the DNA products bound to the telomerase enzyme immobilized on the beads. Following ethanol precipitation, the DNA products were resolved on a 10% (wt/vol) polyacrylamide/8 M urea denaturing gel, dried, exposed to a phosphorstorage screen, and imaged on a Bio‐Rad FX‐Pro phosphorimager. Repeat addition processivity was calculated using the equation: (Latrick & Cech, 2010). The slope, k, was determined by plotting the intensity of each major band, normalized to the intensity of the first band, over the repeat number.
Pulse‐chase time course assay
Twenty microliters of immuno‐purified in vivo‐reconstituted telomerase enzyme on beads was initially pulsed with 0.165 μM α‐32P‐dTTP for 5 min and then chased with 100 μM dTTP and denoted concentrations of dATP and dGTP. Aliquots of the reactions were terminated by phenol/chloroform extraction at denoted time points, followed by ethanol precipitation. The DNA products were resolved on a 10 (wt/vol) polyacrylamide/8 M urea denaturing gel, dried, exposed to a phosphorstorage screen, and imaged on a Bio‐Rad FX‐Pro phosphorimager. To determine the rate of repeat addition, the longest DNA products with the highest intensity above initial pulse product bands were used to deduce a “modal band” to calculate the extension rate as previously described (Drosopoulos et al, 2005). The modal bands were determined from the intensity traces generated by the ImageJ (NIH) program, with the number of repeats comprising the modal band plotted against the time point of the chase reaction, and the slope of the best‐fit trend line determined the rate of repeat addition for the reaction (Appendix Fig S4).
Author contributions
JJ‐LC conceived the general ideas for this work and designed the experiments; YC performed the human telomerase analysis; DL performed MS analysis; YC, JDP, and DL and JJ‐LC analyzed the data. JJ‐LC and JDP wrote the manuscript with input from YC and DL.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Review Process File
Acknowledgements
This work was supported by National Institutes of Health Grant R01GM094450 (J.J.‐L.C.).
The EMBO Journal (2018) 37: e97953
References
- Akiyama BM, Parks JW, Stone MD (2015) The telomerase essential N‐terminal domain promotes DNA synthesis by stabilizing short RNA‐DNA hybrids. Nucleic Acids Res 43: 1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alder JK, Cogan JD, Brown AF, Anderson CJ, Lawson WE, Lansdorp PM, Phillips JA III, Loyd JE, Chen JJ‐L, Armanios M (2011) Ancestral mutation in telomerase causes defects in repeat addition processivity and manifests as familial pulmonary fibrosis. PLoS Genet 7: e1001352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnoult N, Karlseder J (2015) Complex interactions between the DNA‐damage response and mammalian telomeres. Nat Struct Mol Biol 22: 859–866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Autexier C, Lue NF (2006) The structure and function of telomerase reverse transcriptase. Annu Rev Biochem 75: 493–517 [DOI] [PubMed] [Google Scholar]
- Bradshaw PC, Samuels DC (2005) A computational model of mitochondrial deoxynucleotide metabolism and DNA replication. Am J Physiol Cell Physiol 288: C989–C1002 [DOI] [PubMed] [Google Scholar]
- Brown AF, Podlevsky JD, Qi X, Chen Y, Xie M, Chen JJ‐L (2014) A self‐regulating template in human telomerase. Proc Natl Acad Sci USA 111: 11311–11316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J‐L, Greider CW (2003a) Template boundary definition in mammalian telomerase. Genes Dev 17: 2747–2752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J‐L, Greider CW (2003b) Determinants in mammalian telomerase RNA that mediate enzyme processivity and cross‐species incompatibility. EMBO J 22: 304–314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins K, Greider CW (1995) Utilization of ribonucleotides and RNA primers by Tetrahymena telomerase. EMBO J 14: 5422–5432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drosopoulos WC, Direnzo R, Prasad VR (2005) Human telomerase RNA template sequence is a determinant of telomere repeat extension rate. J Biol Chem 280: 32801–32810 [DOI] [PubMed] [Google Scholar]
- Drosopoulos WC, Prasad VR (2010) The telomerase‐specific T motif is a restrictive determinant of repetitive reverse transcription by human telomerase. Mol Cell Biol 30: 447–459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finger SN, Bryan TM (2008) Multiple DNA‐binding sites in Tetrahymena telomerase. Nucleic Acids Res 36: 1260–1272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garforth SJ, Parniak MA, Prasad VR (2008) Utilization of a deoxynucleoside diphosphate substrate by HIV reverse transcriptase. PLoS ONE 3: e2074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilley D, Lee MS, Blackburn EH (1995) Altering specific telomerase RNA template residues affects active site function. Genes Dev 9: 2214–2226 [DOI] [PubMed] [Google Scholar]
- Gilley D, Blackburn EH (1996) Specific RNA residue interactions required for enzymatic functions of Tetrahymena telomerase. Mol Cell Biol 16: 66–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gramatges MM, Qi X, Sasa GS, Chen JJ‐L, Bertuch AA (2013) A homozygous telomerase T‐motif variant resulting in markedly reduced repeat addition processivity in siblings with Hoyeraal Hreidarsson syndrome. Blood 121: 3586–3593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greider CW (1991) Telomerase is processive. Mol Cell Biol 11: 4572–4580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammond PW, Cech TR (1997) dGTP‐dependent processivity and possible template switching of euplotes telomerase. Nucleic Acids Res 25: 3698–3704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy CD, Schultz CS, Collins K (2001) Requirements for the dGTP‐dependent repeat addition processivity of recombinant Tetrahymena telomerase. J Biol Chem 276: 4863–4871 [DOI] [PubMed] [Google Scholar]
- Hockemeyer D, Collins K (2015) Control of telomerase action at human telomeres. Nat Struct Mol Biol 22: 848–852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huard S, Moriarty TJ, Autexier C (2003) The C terminus of the human telomerase reverse transcriptase is a determinant of enzyme processivity. Nucleic Acids Res 31: 4059–4070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang H, Opresko P, Myong S (2014) Single‐molecule real‐time detection of telomerase extension activity. Sci Rep 4: 6391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs SA, Podell ER, Cech TR (2006) Crystal structure of the essential N‐terminal domain of telomerase reverse transcriptase. Nat Struct Mol Biol 13: 218–225 [DOI] [PubMed] [Google Scholar]
- Jansson LI, Akiyama BM, Ooms A, Lu C, Rubin SM, Stone MD (2015) Structural basis of template‐boundary definition in Tetrahymena telomerase. Nat Struct Mol Biol 22: 883–888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang J, Chan H, Cash DD, Miracco EJ, Ogorzalek Loo RR, Upton HE, Cascio D, O'Brien Johnson R, Collins K, Loo JA, Zhou ZH, Feigon J (2015) Structure of Tetrahymena telomerase reveals previously unknown subunits, functions, and interactions. Science 350: aab4070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornberg A (1957) Pathways of enzymatic synthesis of nucleotides and polynucleotides In The chemical basis of heredity, McElroy WD, Glass B. (eds), pp 579–608. Baltimore: The Johns Hopkins Press; [Google Scholar]
- Kornberg A (1969) Active center of DNA polymerase. Science 163: 1410–1418 [DOI] [PubMed] [Google Scholar]
- Latrick CM, Cech TR (2010) POT1‐TPP1 enhances telomerase processivity by slowing primer dissociation and aiding translocation. EMBO J 29: 924–933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MS, Blackburn EH (1993) Sequence‐specific DNA primer effects on telomerase polymerization activity. Mol Cell Biol 13: 6586–6599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lingner J, Hughes TR, Shevchenko A, Mann M, Lundblad V, Cech TR (1997) Reverse transcriptase motifs in the catalytic subunit of telomerase. Science 276: 561–567 [DOI] [PubMed] [Google Scholar]
- Lue NF, Lin Y‐C, Mian IS (2003) A conserved telomerase motif within the catalytic domain of telomerase reverse transcriptase is specifically required for repeat addition processivity. Mol Cell Biol 23: 8440–8449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maine IP, Chen SF, Windle B (1999) Effect of dGTP concentration on human and CHO telomerase. Biochemistry 38: 15325–15332 [DOI] [PubMed] [Google Scholar]
- Mitchell M, Gillis A, Futahashi M, Fujiwara H, Skordalakes E (2010) Structural basis for telomerase catalytic subunit TERT binding to RNA template and telomeric DNA. Nat Struct Mol Biol 17: 513–518 [DOI] [PubMed] [Google Scholar]
- Parks JW, Stone MD (2014) Coordinated DNA dynamics during the human telomerase catalytic cycle. Nat Commun 5: 4146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parks JW, Kappel K, Das R, Stone MD (2017) Single‐molecule FRET‐Rosetta reveals RNA structural rearrangements during human telomerase catalysis. RNA 23: 175–188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Podlevsky JD, Chen JJ‐L (2012) It all comes together at the ends: telomerase structure, function, and biogenesis. Mutation Res 730: 3–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Podlevsky JD, Chen JJ‐L (2016) Evolutionary perspectives of telomerase RNA structure and function. RNA Biol 13: 720–732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi X, Xie M, Brown AF, Bley CJ, Podlevsky JD, Chen JJ‐L (2012) RNA/DNA hybrid binding affinity determines telomerase template‐translocation efficiency. EMBO J 31: 150–161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi X, Li Y, Honda S, Hoffmann S, Marz M, Mosig A, Podlevsky JD, Stadler PF, Selker EU, Chen JJ‐L (2013) The common ancestral core of vertebrate and fungal telomerase RNAs. Nucleic Acids Res 41: 450–462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robart AR, Collins KL (2010) Investigation of human telomerase holoenzyme assembly, activity, and processivity using disease‐linked subunit variants. J Biol Chem 285: 4375–4386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt JC, Cech TR (2015) Human telomerase: biogenesis, trafficking, recruitment, and activation. Genes Dev 29: 1095–1105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seto AG, Umansky K, Tzfati Y, Zaug AJ, Blackburn EH, Cech TR (2003) A template‐proximal RNA paired element contributes to Saccharomyces cerevisiae telomerase activity. RNA 9: 1323–1332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sfeir AJ, Chai W, Shay JW, Wright WE (2005) Telomere‐end processing: the terminal nucleotides of human chromosomes. Mol Cell 18: 131–138 [DOI] [PubMed] [Google Scholar]
- Sun D, Lopez‐Guajardo CC, Quada J, Hurley LH, Von Hoff DD (1999) Regulation of catalytic activity and processivity of human telomerase. Biochemistry 38: 4037–4044 [DOI] [PubMed] [Google Scholar]
- Tomlinson CG, Moye AL, Holien JK, Parker MW, Cohen SB, Bryan TM (2015) Two‐step mechanism involving active‐site conformational changes regulates human telomerase DNA binding. Biochem J 465: 347–357 [DOI] [PubMed] [Google Scholar]
- Tzfati Y, Fulton TB, Roy J, Blackburn EH (2000) Template boundary in a yeast telomerase specified by RNA structure. Science 288: 863–867 [DOI] [PubMed] [Google Scholar]
- Wu RA, Tam J, Collins K (2017a) DNA‐binding determinants and cellular thresholds for human telomerase repeat addition processivity. EMBO J 36: 1908–1927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu RA, Upton HE, Vogan JM, Collins KL (2017b) Telomerase mechanism of telomere synthesis. Annu Rev Biochem 86: 439–460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyatt HDM, Lobb DA, Beattie TL (2007) Characterization of physical and functional anchor site interactions in human telomerase. Mol Cell Biol 27: 3226–3240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie M, Podlevsky JD, Qi X, Bley CJ, Chen JJ‐L (2010) A novel motif in telomerase reverse transcriptase regulates telomere repeat addition rate and processivity. Nucleic Acids Res 38: 1982–1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang G, Franklin M, Li J, Lin TC, Konigsberg W (2002) Correlation of the kinetics of finger domain mutants in RB69 DNA polymerase with its structure. Biochemistry 41: 2526–2534 [DOI] [PubMed] [Google Scholar]
- Yang W, Lee YS (2015) A DNA‐hairpin model for repeat‐addition processivity in telomere synthesis. Nat Struct Mol Biol 22: 844–847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaug AJ, Podell ER, Cech TR (2008) Mutation in TERT separates processivity from anchor‐site function. Nat Struct Mol Biol 15: 870–872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaug AJ, Crary SM, Jesse Fioravanti M, Campbell K, Cech TR (2013) Many disease‐associated variants of hTERT retain high telomerase enzymatic activity. Nucleic Acids Res 41: 8969–8978 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Review Process File