

We have read the study by Takanari et al.1 published in Cardiovascular Research with interest, all the more so given that the reported results are in stark contrast with our own recent study.2
The report by Takanari et al.1 explored the effects of acute and chronic inhibition of the Ca2+/calmodulin dependent protein kinase II (CaMKII) pathway on ventricular conduction. The results obtained in the chronic setting do not have a counterpart in our own study, and will not be commented here. In the acute setting, the authors used calmodulin blocker W7 (10 μM) in isolated rabbit hearts subjected to cryoablation of the left ventricular (LV) endo- and midmyocardium, leaving only an ∼1 mm of viable subepicardial layer. They used optical mapping to estimate the longitudinal (CVL) and transverse (CVT) conduction velocity (CV) from isochronal activation maps obtained during pacing at a site in the centre of the LV free wall. They found that W7 significantly increased CVL and CVT by 6.0 and 12.6%, respectively (at the pacing cycle length of 800 ms) and decreased inducibility of ventricular tachycardia/fibrillation (VT/VF) by programmed stimulation. They linked these improvements in conduction to augmented localization of Cx43 in the intercalated disc caused by W7.
Our recent study using optical mapping in isolated (but otherwise intact) rabbit hearts has demonstrated that CaMKII blockade with 2.75 μM KN93 (KN93 directly targets CaMKII to inhibit its activity) resulted in a marked non-uniform slowing of conduction, with the largest effect occurring in or near the right ventricular (RV) outflow tract (RVOT).2 The vulnerable region in the RVOT subsequently became a preferred location for 2:1 conduction block, turbulent conduction, and initiation of VF upon subjecting the hearts to global ischemia.2 Rabbit ventricular myocytes isolated from RVOT exhibited intrinsically lower action potential upstroke velocity (dV/dtmax) than those isolated from the LV, and KN93 further depressed dV/dtmax in both locations. In the presence of KN93, RVOT myocytes were much more prone to 2:1 activation failure than LV myocytes after abrupt shortening of the pacing cycle length. We concluded that CaMKII blockade depresses ventricular excitability and is potentially pro-arrhythmic due to exacerbating intrinsically lower excitability in the RVOT, making it vulnerable to reentrant arrhythmias under conditions causing reduced depolarization reserve.
Assuming that the effects of W7 and KN93 are both due to inhibition of calmodulin/CaMKII pathway, the study by Takanari et al.1 and ours lead to opposite conclusions with regard to the role of this pathway in regulation of cardiac conduction. However, the assumption that W7 and KN93 should have the same effect could be incorrect. Note that whilst KN93 inhibits CaMKII by interfering with its binding to calmodulin,3 blockade of calmodulin with W-7 will inhibit other downstream calmodulin targets besides CaMKII.4 In addition, both W7 and KN93 have off-target effects.5–8 On the other hand, concordance between the effects of W7 and KN93 would strongly suggest that both these effects are due to blocking calmodulin/CaMKII pathway. With this logic in mind, we performed experiments using W7 instead of KN93 in otherwise the same experimental model as in our previous publication.2 Briefly, continuously perfused (normoxemic) rabbit hearts were exposed to W7 (10 or 20 μM) during sinus rhythm, and periodically subject to ventricular pacing at incremental rates from electrodes placed in the LV and RV. In the absence of mechanical uncouplers, fluorescence emitted by the voltage sensitive dye Di-4-ANEPPS staining the myocardium was collected with an EMCCD camera at a spatial resolution of 64 × 64 pixels (0.56 mm/pix) and frame interval of 2.06 ms, and the recorded signals used for determining the local conduction delays of the propagating wave fronts. We found that 10 μM W7 induced dramatic slowing of conduction, more pronounced in the RV than in the LV (Figure 1A). The conduction delays increased progressively with time of W7 perfusion, reaching statistical significance at 30 min (Figure 1B). Conduction depression was further enhanced by increasing W7 concentration (10 vs. 20 μM) and/or pacing frequency (3.3 vs. 5.0 vs. 5.9 Hz). Conduction slowing was accompanied by a progressive increase of the excitation threshold and culminated in failure to activate in a 1:1 manner, and/or turbulent conduction, and/or initiation of VT/VF. During the fastest pacing, these abnormal events were absent at baseline (0/5) but were present after 30 min 10 μM W7 perfusion in 4/5 hearts. Perfusion with 20 μM W7 led to sustained episodes of VT/VF maintained by reentrant waves. Time matched control experiments showed complete absence of arrhythmias and no changes in the conduction delays during relevant period of time (mean conduction delay at 0 min (post-equilibration) vs. 30 min: 1.24 ± 0.13 vs. 1.25 ± 0.18 ms/mm, paired Student’s t-test P = 0.88, n = 4). Thus, we found that the effects of W7 are indeed concordant to the effects of KN93 we observed previously in Ref. 2.
Figure 1.
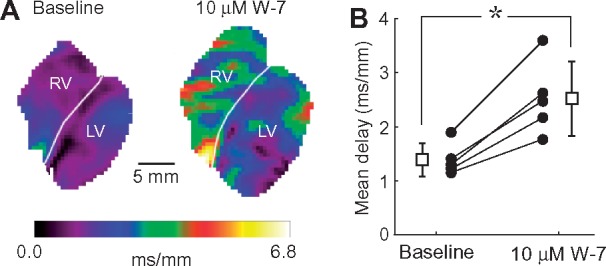
Calmodulin blocker W7 depresses cardiac conduction in intact rabbit hearts. (A) Distribution of local conduction delay values in the imaged anterior surface of both ventricles at baseline (left) and after 30-min perfusion with 10 μM W7 (right). Local delays (the inverse of the local conduction velocities) were computed as the magnitude of the gradient of the activation time. Hearts were paced simultaneously from two electrodes located at the lateral epicardial surface of each ventricle at a cycle length of 300 ms. White labels indicate the LV and RV location, and the white lines indicate the boundary between the chambers. (B). Mean conduction delays computed over the entire mapped surface at baseline and after 30 min perfusion with 10 μM W7: solid circles represent values for individual experiments; open square symbols and bars represent average and SD. *P = 0.00352 vs. baseline, paired Student‘s t-test (n = 5 hearts).
Despite using the same drug and the same species, there remain methodological differences between the Takanari‘s study1 and our own2 which could explain the discrepant outcomes. Our model might be more physiological not involving endomidmyocardial cryoablation and electromechanical uncoupler/non-specific phosphatase 2, 3-butandione monoxime (used by Takanari et al. at unusually high concentration of 100 mM). In addition, we analyzed local delays (inverse of CV) in all mapped RV and LV locations, not only in a selected region around the pacing electrode in the LV. This enabled us to find the epicenter of the depressant effect, located in the RV. Yet, even if we limited our analysis to the LV, we would still end up with results opposite to those by Takanari et al.1
Although various studies converge in considering CaMKII as a principle regulator of cardiac excitability, there is disagreement as to how this occurs. This controversy is particularly evident in single cell studies investigating how CaMKII regulates INa, with some suggesting that CaMKII activity favors an overall INa upregulation,9,10 whilst others argue that in fact CaMKII activity promotes an overall INa downregulation.11 Consideration of Takanari‘s study1 and our own2 clearly shows that this divergence has translated to the intact tissue level. In our view, the similarity of (adverse) effects exerted by W7 and KN93 on ventricular conduction strongly reinforces the notion that constitutive activity of the calmodulin/CaMKII pathway is necessary for normal ventricular excitation and conduction.2 Clearly, sorting this matter is important given that ineffective regulation of conduction can potentially lead to life-threatening arrhythmia. In the light of our observation that calmodulin/CaMKII blockade partially mimicked Brugada syndrome,2 patients having Brugada syndrome or other conduction diseases can be at enhanced risk of VF if treated with CaMKII blockers. Other conditions reducing depolarization reserve can also adversely interact with calmodulin/CaMKII blockade. It is important to ascertain the effect of calmodulin/CaMKII blockade on conduction in both normal and diseased heart before CaMKII blockers can be translated into clinical practice.
Conflict of interest: none declared.
Funding
This work was supported by a Nora Eccles Treadwell Foundation Research Grant (to A. V. Zaitsev) and National Heart, Lung, and Blood Institute Grants 1R0-1HL-103877 (to A. V. Zaitsev) and R0-1HL-128752 (to M. Warren).
References
- 1. Takanari H, Bourgonje VJ, Fontes MS, Raaijmakers AJ, Driessen H, Jansen JA, van der Nagel R, Kok B, van Stuijvenberg L, Boulaksil M, Takemoto Y, Yamazaki M, Tsuji Y, Honjo H, Kamiya K, Kodama I, Anderson ME, van der Heyden MA, van Rijen HV, van Veen TA, Vos MA.. Calmodulin/CaMKII inhibition improves intercellular communication and impulse propagation in the heart and is antiarrhythmic under conditions when fibrosis is absent. Cardiovasc Res 2016;111:410–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Warren M, Sciuto KJ, Taylor TG, Garg V, Torres NS, Shibayama J, Spitzer KW, Zaitsev AV.. Blockade of CaMKII depresses conduction preferentially in the right ventricular outflow tract and promotes ischemic ventricular fibrillation in the rabbit heart. Am J Physiol Heart Circ Physiol 2017;312:H752–H767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Pellicena P, Schulman H.. CaMKII inhibitors: from research tools to therapeutic agents. Front Pharmacol 2014;5:21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Saucerman JJ, Bers DM.. Calmodulin binding proteins provide domains of local Ca2+ signaling in cardiac myocytes. J Mol Cell Cardiol 2012; 52:312–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hegyi B, Chen-Izu Y, Jian Z, Shimkunas R, Izu LT, Banyasz T.. KN-93 inhibits IKr in mammalian cardiomyocytes. J Mol Cell Cardiol 2015;89:173–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Qu YJ, Bondarenko VE, Xie C, Wang S, Awayda MS, Strauss HC, Morales MJ.. W-7 modulates Kv4.3: pore block and Ca2+-calmodulin inhibition. Am J Physiol Heart Circ Physiol 2007;292:H2364–H2377. [DOI] [PubMed] [Google Scholar]
- 7. Rezazadeh S, Claydon TW, Fedida D.. KN-93 (2-[N-(2-hydroxyethyl)]-N-(4-methoxybenzenesulfonyl)]amino-N-(4-chlorocinnamyl)-N -methylbenzylamine), a calcium/calmodulin-dependent protein kinase II inhibitor, is a direct extracellular blocker of voltage-gated potassium channels. J Pharmacol Exp Ther 2006;317:292–299. [DOI] [PubMed] [Google Scholar]
- 8. Zhang XH, Jin MW, Sun HY, Zhang S, Li GR.. The calmodulin inhibitor N-(6-aminohexyl)-5-chloro-1-naphthalene sulphonamide directly blocks human ether a-go-go-related gene potassium channels stably expressed in human embryonic kidney 293 cells. Br J Pharmacol 2010;161:872–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Yoon JY, Ho WK, Kim ST, Cho H.. Constitutive CaMKII activity regulates Na+ channel in rat ventricular myocytes. J Mol Cell Cardiol 2009;47:475–484. [DOI] [PubMed] [Google Scholar]
- 10. Aiba T, Hesketh GG, Liu T, Carlisle R, Villa-Abrille MC, O'rourke B, Akar FG, Tomaselli GF.. Na+ channel regulation by Ca2+/calmodulin and Ca2+/calmodulin-dependent protein kinase II in guinea-pig ventricular myocytes. Cardiovasc Res 2010;85:454–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wagner S, Dybkova N, Rasenack EC, Jacobshagen C, Fabritz L, Kirchhof P, Maier SK, Zhang T, Hasenfuss G, Brown JH, Bers DM, Maier LS.. Ca2+/calmodulin-dependent protein kinase II regulates cardiac Na+ channels. J Clin Invest 2006; 116:3127–3138. [DOI] [PMC free article] [PubMed] [Google Scholar]