

Abstract
Aims
Spontaneous Ca2+ waves in cardiomyocytes are potentially arrhythmogenic. A powerful controller of Ca2+ waves is the cytoplasmic H+ concentration ([H+]i), which fluctuates spatially and temporally in conditions such as myocardial ischaemia/reperfusion. H+-control of Ca2+ waves is poorly understood. We have therefore investigated how [H+]i co-ordinates their initiation and frequency.
Methods and results
Spontaneous Ca2+ waves were imaged (fluo-3) in rat isolated ventricular myocytes, subjected to modest Ca2+-overload. Whole-cell intracellular acidosis (induced by acetate-superfusion) stimulated wave frequency. Pharmacologically blocking sarcolemmal Na+/H+ exchange (NHE1) prevented this stimulation, unveiling inhibition by H+. Acidosis also increased Ca2+ wave velocity. Restricting acidosis to one end of a myocyte, using a microfluidic device, inhibited Ca2+ waves in the acidic zone (consistent with ryanodine receptor inhibition), but stimulated wave emergence elsewhere in the cell. This remote stimulation was absent when NHE1 was selectively inhibited in the acidic zone. Remote stimulation depended on a locally evoked, NHE1-driven rise of [Na+]i that spread rapidly downstream.
Conclusion
Acidosis influences Ca2+ waves via inhibitory and stimulatory signals (the latter facilitating intracellular Ca2+-loading through modulation of sarcolemmal Na+/Ca2+ exchange activity). During spatial [H+]i-heterogeneity, -inhibition dominates in acidic regions, while rapid diffusion stimulates waves in downstream, non-acidic regions. Local acidosis thus simultaneously inhibits and stimulates arrhythmogenic Ca2+-signalling in the same myocyte. If the principle of remote H+-stimulation of Ca2+ waves also applies in multicellular myocardium, it raises the possibility of electrical disturbances being driven remotely by adjacent ischaemic areas, which are known to be intensely acidic.
Keywords: Calcium cycling, Cell signalling, Membrane transport, Intracellular sodium, Intracellular pH
Introduction
An aberrant form of Ca2+ signalling is the Ca2+ wave, commonly observed in ventricular myocytes during periods of Ca2+-overload.1 Ca2+ waves can occur spontaneously, initiated by a localised SR Ca2+ release that propagates spatially, via a ‘fire-diffuse-fire’ form of Ca2+-induced Ca2+ release from the SR2. Ca2+ waves are believed to facilitate triggered arrhythmias, by driving delayed after-depolarizations (DADs) that may transition to ectopic action potentials.3,4 Spontaneous Ca2+ waves are common events during clinical conditions such as myocardial ischaemia. One factor that may trigger Ca2+ waves is a fall of pHi, although wave initiation has been variously reported to be stimulated or inhibited by acidosis.5,6 Ca2+ waves are also promoted by other factors, in particular by elevations of [Na+]i that occur when the Na+/K+ pump is inhibited.7 It is notable, therefore, that a significant [Na+]i rise occurs when the sarcolemmal Na+/H+ exchanger (NHE1) is stimulated by acidosis.8 In the present work, we have investigated the mechanisms coupling Ca2+ waves to changes of pHi.
Intracellular H+ ions are powerful modulators of cell function. In ventricular myocytes they are generated metabolically, but maintained at low cytoplasmic levels ([H+]i ∼60 nM, equivalent to pHi 7.2), most commonly via extrusion on NHE1.9 Despite regulation, reversible increases of [H+]i of about 30 nM occur physiologically (equivalent to a fall of ∼0.15 pH units), while much larger increases of about 400 nM (equivalent to a fall of ∼0.6 pH units) occur during myocardial ischaemia.10 These [H+]i elevations represent a form of intracellular H+ signalling. Prominent among the targets for such signalling are Ca2+ handling proteins, namely the ryanodine receptor (RyR),11–13 the sarco/endoplasmic reticulum Ca2+ ATPase (SERCA),14,15 Na+/Ca2+ exchanger (NCX),16 and the L-type calcium channel.17 Although direct H+ interaction with these proteins is predominantly inhibitory, an elevation of [H+]i can enhance the electrically evoked calcium transient (CaT),18 an effect that helps to protect contractility during acidosis. As mentioned above, this latter stimulation occurs indirectly through H+ activation of NHE1. The resulting rise of [Na+]i slows Ca2+ efflux on sarcolemmal NCX, which boosts sarcoplasmic reticulum (SR) Ca2+ loading via SERCA, thereby increasing CaT amplitude.18,19 Quite how the balance between inhibitory and excitatory effects of on Ca2+ signalling impacts on the generation of arrhythmogenic Ca2+ waves has yet to be examined.
The importance of spatial interactions among [H+]i, [Na+]i and [Ca2+]i in ventricular myocytes has been highlighted in recent work.18 Because of high intracellular buffering, cytoplasmic H+ mobility is low,20 so that localised [H+]i microdomains can form.21 A microdomain of elevated [H+]i, by locally stimulating NHE1, elevates , which increases the CaT amplitude. But because Na+ diffuses rapidly in cytoplasm,18,22 it can enhance the CaT globally within the cell, including in regions not experiencing acidosis. In the present work we investigate if spontaneous Ca2+ waves are similarly controlled through interacting spatial and signals.
In order to investigate the coupling of Ca2+ waves to pHi, we have manipulated intracellular pH, Na+, and Ca2+ in rat ventricular myocytes. Our data indicate that H+ ions exert direct inhibitory and indirect excitatory effects on Ca2+ waves. The excitatory effect is mediated via intracellular Na+. When excitation is induced by a localised acidic microdomain, it is typically expressed remotely in non-acidic regions of the cell, driven by fast diffusion. Our data provide evidence that acidosis, and its spatial heterogeneity, is a powerful substrate for the initiation and spatial organisation of pro-arrhythmic Ca2+ waves.
Methods
Detailed methods are available in the Supplementary material online.
Ventricular myocyte isolation
All procedures were performed in accordance with UK Home Office and local guidelines. Ventricular myocytes were isolated from 46 male Sprague-Dawley rats, as previously described,23 or from neonatal rats (1 day old).
Fluorescence measurements of intracellular pH and Ca2+
pHi: myocytes were loaded with carboxy-seminaphthorhodofluor-1 (cSNARF-1, 10 μM) at room temperature. Cells were imaged confocally on a Leica SP5 inverted microscope. Intracellular calibration of cSNARF-1 was performed in separate experiments using nigericin.23
Ca2+ waves and sparks: myocytes were AM-loaded with fluo-3 (18 μM). Cells were superfused and quiescent cells were imaged in linescan (xt) mode along the cell’s longitudinal axis at 400 lines per second. Ca2+ sparks were imaged in solution containing 1 mM free Ca2+. Switching the superfusate to one containing raised (5 mM) free Ca2+ induced a modest calcium overload that triggers Ca2+ waves.
Generating intracellular pH gradient
A pHi gradient was induced using a microfluidic device consisting of a square-bore double-barrelled micropipette, which released two parallel microstreams of solution perpendicular to the cell.24 One microstream contained Tyrode with 5 mM CaCl2 plus 10 mM sucrose to help visualize the interstream boundary; the other microstream contained 80 mM acetate with 5 mM free Ca2+. The position of the microstream boundary across the cell was recorded in xy before and after each linescan experiment. The smooth pHi gradient induced is illustrated in Figure 4B.
Figure 4.

An acidic microdomain affects Ca2+ wave initiation both locally and remotely. (A) A longitudinal pHi gradient was generated by regional superfusion of the myocyte with 80 mM acetate. Top: schematic of dual microperfusion apparatus; middle; representative pHi gradient along a myocyte, xy image with SNARF-1; bottom; representative Ca2+ wave recorded in linescan mode during an imposed pHi gradient. (B) Magnitude of pHi gradient (n = 12 cells/4 animals) was unaffected by inhibiting NHE1 in the acidic microdomain (n = 11 cells/2 animals). (Ci) Exemplar linescans recorded from 2 rat ventricular myocytes, under control conditions (left panel of each cell), and during imposition of a longitudinal pHi gradient by regional superfusion of the myocyte with 80 mM acetate (right panel of each cell). In order to compress the results, the timecourse for the line-scans has been broken into three parallel columns. Note that, during control conditions, Ca2+ waves initiate randomly along the length of the myocyte, whereas, during imposition of the pHi gradient, waves initiate almost exclusively in the non-acidic microdomain (represented by the blue bar above each column of line-scans), and their frequency increases. The acidic zone is represented by the red bar above each column of linescans. (Cii) Exemplar linescans recorded from 2 cells under equivalent conditions, but with 30 µM DMA included in the regional acetate superfusion (to inhibit NHE activity). During imposition of the pHi gradient, wave initiations remain restricted to the non-acidic microdomain, but now their overall frequency does not change compared with control. (D) Ca2+ wave initiation was inhibited in the acidic (acetate, A) microdomain and remotely stimulated in the non-acidic (control, C) microdomain (n = 6 cells/3 animals). (E) Ca2+ wave initiation was attenuated in the acidic microdomain, but absence of NHE1 activity prevented the remote stimulation of Ca2+ waves in the non-acidic microdomain (n = 5 cells/2 animals). Paired t-tests.
Data analysis
Data are expressed as mean ± SE. Statistical significance was tested using paired Student’s t-test or nested ANOVA, depending on the experimental protocol. Where relevant, the Holm correction for multiple testing was applied. Significance is denoted as * for P < 0.05, ** for P < 0.01, *** for P < 0.001.
Wave propagation velocity was calculated from linescan images using the angle of incidence of the wavefront, relative to the longitudinal axis of the cell.
Linescan recordings of Ca2+ sparks were normalized using a custom-built Matlab macro, followed by spark frequency analysis using an algorithm developed by Kong et al.25
Results
Acidosis affects Ca2+ waves via and signals
We explored the -sensitivity of Ca2+ waves, by first imposing whole-cell changes of pHi, at constant pHo. Individual ventricular myocytes, when subjected to modest Ca2+ overload (5 mM ), displayed spontaneous Ca2+ waves (Figure 1Ai), with a frequency of 18.7 ± 2.5 min−1. At times these waves converted into whole-cell CaTs (Supplementary material online, Figure S1), induced by the Ca2+-driven transient inward current (Iti) triggering an action potential.3,4 Cells displaying wave-triggered CaTs were not included in the analysis, because of their confounding effects on wave properties. Intracellular acidification (from pHi 7.3 to 6.6), induced by superfusion of 80 mM acetate, initially reduced wave frequency (Figure 1Aii). After 30 s, wave frequency then progressively increased above control levels (Figure 1Ai, ii, Supplementary material online, FigureS2A). The increase could be graded by varying the magnitude of intracellular acidosis (superfusion with 20, 40, or 80 mM acetate; Figure 1Aiv). The increase was attributable to an -dependent stimulation of NHE1 activity, as it was abolished in the presence of 5-(N,N-dimethyl) amiloride (DMA, 30 μM), a high-affinity NHE1 inhibitor. Under these conditions, reducing pHi from 7.3 to 6.6 now produced a decrease in Ca2+ waves (Figure 1Bi, ii, Supplementary material online, FigureS2B), which was also graded with the severity of acidosis (Figure 1Biv; note that, in these latter experiments, extreme alkalosis, produced by 20 mM trimethylamine superfusion, also reduced wave frequency). Abolition of -evoked Ca2+ waves was confirmed with a structurally different NHE1 inhibitor, cariporide, which targets NHE1 more specifically among NHE isoforms than DMA26; Supplementary material online, Figure S2C, D. Thus, for the pHi range from 7.3 to 6.6, raising [H+]i exerts opposing effects on Ca2+ wave frequency; an NHE1-independent inhibition, and a delayed, NHE1-dependent stimulation.
Figure 1.

Intracellular acidosis stimulates Ca2+ waves in an NHE1-dependent manner. (Ai) Ca2+ waves (fluo-3) were triggered at 5 mM extracellular [Ca2+]. Images recorded in linescan (xt) mode along the longitudinal axis of the cell at 400 lines per second. F/F0 = fluorescence divided by resting fluorescence (F0 averaged over first 1 s of recording). (Aii) Decreasing pHi (cSNARF-1, mean pHi trace, error bars are SEM, n = 13 cells/2 animals) by 80 mM acetate superfusion initially decreased, then increased wave frequency (n = 18 cells/5 animals). Waves were counted in 10 s bins and normalized to the mean frequency in the first 10 s bin. (Aiii) [Na+]i timecourse, on the same timescale as Aii and taken under comparable experimental conditions, replotted from Figure 2B of reference 18. SBFI ratio acquired every 4 s, n = 6 cells/2 animals, error bars are SEM. (Aiv) Ca2+ wave frequency showed a positive relationship with decreasing pHi. Error bars are SD (pHi), SEM (frequency). Wave frequency and pHi averaged over 1 min, from 30 s after onset of acetate superfusion. For 80 mM acetate n = 18 cells/5 animals (waves), 13 cells/2 animals (pHi); 40 mM acetate n = 5 cells/1 animal (waves), 8 cells/2 animals (pHi); 20 mM TMA (for extreme intracellular alkalosis) n = 10 cells/2 animals (waves), 8 cells/2 animals (pHi). (Bi) Ca2+ wave frequency during acidosis with NHE1 inhibitor (30 μM DMA). (Bii) NHE1 inhibition attenuates pHi recovery during acidosis (n = 13 cells/3 animals) and reveals an underlying inhibitory effect of intracellular acidosis on Ca2+ wave frequency (n = 12 cells/3 animals). (Biii) [Na+]i timecourse, on the same timescale as Bii and taken under comparable experimental conditions, replotted from Figure 2B of reference 18. SBFI ratio acquired every 4 s, n = 7 cells/2 animals, error bars are SEM. (Biv) Relationship between wave frequency and pHi in absence of NHE1 activity. Error bars are SD (pHi), SEM (frequency). For 80 mM acetate + DMA n = 12 cells/3 animals (waves), 17 cells/3 animals (pHi); 40 mM acetate + DMA n = 9 cells/2 animals (waves), 6 cells/2 animals (pHi); 20 mM acetate + DMA n = 9 cells/2 animals (waves), 8 cells/2 animals (pHi); 20 mM TMA n = 10 cells/2 animals (waves), 8 cells/2 animals (pHi). Paired t-tests.
H+-evoked stimulation of Ca2+ waves is accompanied by a DMA-sensitive rise of [Na+]i (cf. Figures 1Aiii, 1Biii), consistent with enhanced Na+ influx on NHE1. The [Na+]i rise leads, via a slowing of Ca2+ efflux on sarcolemmal NCX, to retention of intracellular Ca2+ 7 and, ultimately, to increased Ca2+ wave frequency. The [Na+]i measurements have been extracted from our previously published work, measured under comparable experimental conditions18 (as the published [Na+]i-rise was recorded in 1 mM [Ca2+]o, whereas current experiments were conducted in 5 mM [Ca2+]o, we confirmed that the rise was not influenced by the different [Ca2+]o levels; see Supplementary material online, Figure S6). For clarity, [Na+]i data have been replotted in Figures 1Aiii and 1Biii. The data emphasize that preventing the H+-evoked rise of [Na+]i with DMA, prevents wave stimulation. Stimulation is thus dependent on the rise of [Na+]i, while acid-induced wave inhibition, observed in the presence of an NHE1 inhibitor, is attributable directly to the rise of [H+]i. N.B. since the rise of [Na+]i is completely prevented by inhibiting NHE, we do not anticipate a role for H+-induced inhibition of the Na+/K+ pump under these conditions.
Ca2+ wave stimulation has been quantified in Figure 2Ai, which plots, on logarithmic axes, wave frequency vs. [Na+]i (data amalgamated from Figure 1Aii, Aiii). Wave frequency rises steeply with [Na+]i (to a first approximation, wave frequency ∝ [Na+]i4). In contrast, wave frequency in the absence of NHE1 activity decreased with a rise of [H+]i, displaying a more shallow H+-dependence (Figure 2Aii). The net result for whole-cell acidosis on wave frequency will be a combination of these stimulatory and inhibitory components. At steady-state, the combination is net stimulatory (Figure 1Aii, iv), reflecting the steep positive dependence of wave frequency on [Na+]i.
Figure 2.

Acidosis-induced stimulation of Ca2+ waves depends on sarcolemmal NCX. (Ai) Ca2+ wave initiation frequency is steeply dependent on [Na+]i. Wave frequency ∝ [Na+]i4. Note the logarithmic axes. (Aii) In the absence of an NHE-driven [Na+]i rise, Ca2+ waves show a linear, inverse relationship with [H+]i. Note the logarithmic axes. Fit using linear regression: 50.5 × [H+]−1. (B) Proposed interactions between intracellular H+, Na+, and Ca2+ modify SR Ca2+ load, and thus Ca2+ wave probability. Pharmacological inhibitors are in red. (C) Wave frequency averaged during 2nd min of acetate superfusion. Inhibiting mitochondrial NCX (‘CGP’: 20 μM CGP-37157) had no effect on acidosis-induced Ca2+ wave stimulation (n = 11 cells/2 animals). Inhibiting both mitochondrial NCX and the mitochondrial Ca2+-uniporter (‘Ru360’: 10 μM ruthenium-360) enhanced wave stimulation (n = 11 cells/5 animals). Inhibiting sarcolemmal NCX (10 μM SN-6) prevented acidosis-induced wave stimulation (n = 4 cells/2 animals). Nested ANOVA followed by pairwise comparison with Holm correction for multiple testing.
Mitochondrial NCX does not promote Ca2+ waves during acidosis
Although sarcolemmal NCX is likely to mediate the [Na+]i-dependence of Ca2+ wave frequency, this does not exclude an additional role for mitochondrial NCX (mNCX). Elevating [Na+]i has been proposed to promote mitochondrial Ca2+ efflux on mNCX,27 in addition to slowing the forward mode of sarcolemmal NCX. Promoting mitochondrial Ca2+ efflux may therefore supplement the domain of cytoplasmic Ca2+ that eventually enhances Ca2+ wave frequency. To test this hypothesis, myocytes were first subjected to intracellular acidosis in the presence of the sarcolemmal NCX inhibitor, SN-6. The drug fully inhibited the H+-evoked stimulation of Ca2+ wave frequency (Figure 2C), indicating a necessary role for the sarcolemmal exchanger. In contrast, selective inhibition of mNCX with CGP-37157 had no effect on H+-evoked waves (Figure 2C), indicating that mNCX is unlikely to be supplementing Ca2+ wave stimulation. Indeed, it is more likely that mitochondria attenuate Ca2+ waves. This is supported by the observation that when the mitochondrial uniporter (MCU), which mediates Ca2+ uptake, was blocked by pre-incubation with ruthenium-360 (Ru360), followed by inhibition of mNCX with CGP-37157, then H+-induced Ca2+ wave frequency was enhanced by 70% (Figure 2C). The result suggests that mitochondrial Ca2+ uptake normally plays a protective role during intracellular acidosis, by sequestering some of the intracellular Ca2+-overload.
Acidosis affects multiple properties of Ca2+ waves
In addition to Ca2+ wave frequency, we investigated whether raising [H+]i influenced other features, notably, Ca2+ wave propagation velocity (vprop), amplitude, and time-course. Raising [H+]i increased wave velocity, both in the presence and absence of NHE1 activity (Figure 3A, B). As shown in Figure 3C, velocity rose with a fall of pHi from 7.7 to 6.65 (equivalent to a 250 nM rise in [H+]i), increasing by ∼40% over the range. According to a recent model of Ca2+ wave propagation,28 a plausible explanation for the velocity increase is an H+-dependent reduction in cytoplasmic Ca2+ buffering, for example, by troponin C and other proteins, as well as by cytoplasmic histidyl-dipeptides.24 Decreased Ca2+ buffering may reflect a reduction in the Ca2+ binding constant (kon), or an increase in the unbinding constant (koff). Increasing [H+]i also increased resting [Ca2+]i (as reported previously24) and peak F/F0 fluorescence, and slowed Ca2+ wave relaxation (Suuplementary material online, Figure S3).
Figure 3.

Intracellular acidosis alters Ca2+ wave velocity. (A) Intracellular acidosis increases velocity of Ca2+ waves in the presence (n = 18 cells/5 animals) (i) and absence (i.e. with 30 µM DMA; n = 11 cells/2 animals) (ii) of NHE1 activity. (B) Normalized wave velocity measured during 2nd min of 80 mM acetate superfusion. (C) Wave velocity and pHi were averaged over 100 s, starting 20 s after onset of acetate superfusion with 30 μM DMA (80 mM acetate + DMA n = 11 cells/2 animals (waves), 5 cells/1 animals (pHi); 40 mM acetate + DMA n = 4 cells/1 animal (waves), 6 cells/2 animals (pHi); 20 mM acetate + DMA n = 8 cells/2 animals (waves), 8 cells/2 animals (pHi); 20 mM TMA n = 12 cells/2 animals (waves), 8 cells/2 animals (pHi)). Fit: y=-0.5223x + 4.7943 (R2 = 0.996). Paired t-tests.
Local acidosis stimulates downstream Ca2+ waves in remote regions of the myocyte
Acidosis was imposed locally at one end of an isolated myocyte using a dual micro-superfusion device positioned at right-angles to the cell (see Methods). One microstream contained 80 mM acetate while the other contained acetate-free Tyrode (Figure 4A). This generated a stable end-to-end pHi gradient of ∼0.6 units (Figure 4B).
Under control conditions (before imposing the pHi gradient), no significant difference was observed in Ca2+ wave frequency between ROIs positioned at opposite ends of the cell (P = 0.23; these regions were defined as segments along the line-scan at either end of the myocyte, of length equal to one third of total cell length). In contrast, after imposing the pHi gradient, Ca2+ waves were initiated, and became clustered in the non-acidic end of the cell, even though this region was many sarcomere-lengths away from the acidic zone. Thus, over the first 2 min of an imposed pHi gradient, Ca2+ waves were rarely initiated in the acidic zone, but their frequency increased up to five-fold in the non-acidic zone (Figure 4Ci, D). Selective pharmacological inhibition (DMA) of NHE1 activity in the non-acidic zone did not prevent the local increase in Ca2+ wave frequency (P = 0.01, n = 6), indicating that the relevant NHE1 activity driving wave initiation was in the acidic zone. This was tested further by selectively superfusing DMA over the acidic end of the myocyte. Although the pHi gradient itself was unaffected by this manoeuvre, the stimulation of Ca2+ waves in the non-acidic zone was now greatly attenuated (Figure 4Dcf. 4E). Thus, locally enhancing NHE1 activity remotely stimulates Ca2+ waves.
In summary, during uniform acidosis, all regions of a myocyte are equally likely to support a Ca2+ wave, driven by NHE1 activity. However, when NHE1 is stimulated locally (by inducing a localised acidosis within the cell) there is selective stimulation and clustering of Ca2+ waves in downstream non-acidic zones.
Local acidosis inhibits local Ca2+ sparks
Since H+ ions affect multiple proteins involved in Ca2+ signalling, the mechanism by which Ca2+ waves are inhibited in the acidic microdomain, but remotely stimulated in the non-acidic microdomain, is not immediately obvious. One possibility is that H+ ions locally decrease RyR open probability (Po), which could account for inhibition of wave initiation. A decreased Po may also enhance SR Ca2+ retention.29 This could contribute to remote wave stimulation if the locally elevated SR Ca2+ load then diffused rapidly into non-acidic regions of the SR lumen.
To explore whether the spatial effects of acidosis on wave frequency are all due to RyR inhibition, Ca2+ spark frequency was measured. Under resting conditions, spark frequency at either end of the myocyte was not different (P = 0.33, n = 9). However, when a pHi gradient was imposed, Ca2+ spark frequency in the acidic microdomain decreased by 72 ± 11% (P = 0.0002, n = 9), while that in the non-acidic microdomain was not significantly altered (Figure 5A, P = 0.24, n = 9). This phenomenon was insensitive to NHE1 inhibition (Figure 5Aii). The results argue for local H+ inhibition of RyRs, but also indicate that any resulting local rise in luminal SR [Ca2+] does not affect the Po of remote RyRs.
Figure 5.

Inhibition of RyR channels by H+ ions can account for regional Ca2+ wave inhibition during pHi heterogeneity. (Ai) 3D surface plots of Ca2+ sparks (recorded in linescan mode) illustrate a uniform distribution under resting conditions. Imposed pHi gradient inhibits spark events in acidic (acetate, A) microdomain. (Aii) Sparks were inhibited in the acidic microdomain in the presence (n = 9 cells/2 animals) and absence (n = 5 cells/1 animal) of NHE1 activity. (Bi) Top panel represents dual microperfusion apparatus. Linescan of a Ca2+ wave during regional superfusion with tetracaine. (Bii) Wave initiation and propagation were suppressed in the tetracaine-exposed microdomain (remote effect is not significant; n = 12 cells/3 animals, P = 0.1175). Paired t-tests.
To verify the effect on Ca2+ waves of regional RyR inhibition, tetracaine (an RyR antagonist) was applied to one half of a myocyte to simulate the inhibitory effect of H+ ions, while maintaining pHi uniformly at resting levels throughout the cell. Figure 5B shows that Ca2+ wave initiation frequency observed with locally applied tetracaine was very similar to that observed with local acidosis and NHE1 inhibition (cf.Figure 4E). In both cases, Ca2+ wave frequency was suppressed in the tetracaine/acetate-exposed region of the cell, and did not significantly change in the downstream (unexposed) region. These findings confirm that, while local RyR blockade by H+ ions or tetracaine can explain local (upstream) inhibition of Ca2+ waves, it cannot explain wave stimulation in downstream regions. This latter effect requires the local, upstream activity of NHE1.
Properties of the Ca2+ wave map onto pHi gradients
To investigate whether properties of Ca2+ waves, other than initiation frequency, also map onto pHi heterogeneity, Ca2+ wave velocity (vprop), amplitude and time-course were measured in the acidic and non-acidic microdomains. For the few Ca2+ waves that successfully propagated from the non-acidic to the acidic zones of the cell, their velocity increased in the acidic zone (Figure 6Aii, 40 ± 7% faster, P = 0.0002, n = 8). A similar trend was observed when DMA was superfused over the acidic end of the cell, indicating that the faster propagation was independent of NHE1 activity (Figure 6Aii, 40 ± 6% faster vprop than non-acidic microdomain, P = 0.0002, n = 5).
Figure 6.

Ca2+ wave velocity and propagation map on to local pHi non-uniformities. (Ai) During a stable longitudinal pHi gradient, Ca2+ wave is faster in the acidic (acetate) microdomain. (Aii) Wave propagation velocity in the acidic (acetate, A) and non-acidic (control, C) microdomains in the presence (n = 8 cells/4 animals) or absence (n = 5 cells/3 animals) of NHE1 activity. (B) Wave propagation was suppressed in the acidic microdomain, while remaining unaffected in the non-acidic microdomain (n = 8 cells/4 animals). (C) NHE1 inhibition had no effect on the success of wave propagation in the acidic microdomain (n = 9 cells/2 animals). Dashed lines represent control. Paired t-tests.
The peak F/F0 of the Ca2+ wave was also significantly higher in the acidic microdomain compared with the non-acidic microdomain (Supplementary material online, Figure S4), while wave relaxation was significantly slower in the acidic microdomain (Supplementary material online, Figure S4). It was also noted that Ca2+ waves frequently failed to propagate within the acidic microdomain (irrespective of the initiation site), unlike the negligible failure rate observed under control conditions (Figure 6B, C). Taken together, our results show that multiple properties of the Ca2+ wave (velocity, propagation-failure, amplitude, and relaxation rate, in addition to initiation frequency) are all subservient to the local pHi. Spatial non-uniformity of pHi thus induces dramatic heterogeneity of Ca2+ waves.
pHi gradients across confluent monolayers of myocytes induce [Na+]i gradients
To explore whether spatial pHi gradients can be established in multicellular cardiac structures, as well as in isolated cells, dual microperfusion was used to deliver a microstream containing 40 mM lactate in parallel to a lactate-free microstream over a confluent monolayer of neonatal rat ventricular myocytes. Under control conditions (i.e. uniform exposure to lactate-free solution), no pHi heterogeneity was observed when imaging the cSNARF-1 fluorescence ratio (Figure 7Ai). However, when the monolayer was regionally superfused with 40 mM lactate (to represent the metabolic acidosis seen during myocardial ischaemia), a stable pHi gradient of ∼0.3 units formed in the region of the solution boundary, of width ∼100 µm, equivalent to several neonatal cell-lengths (Figure 7Aii; cell-length is typically 20–40 µm). Cytoplasmic acidification in lactate-exposed cells activates NHE1 locally, which produces a rise in [Na+]i, detected using SBFI fluorescence.30 In the monolayers analysed in Figure 7, cytoplasmic diffusion and gap junctional permeation of Na+ ions then resulted in a smooth gradient of elevated [Na+]i, ∼2.5 mM in magnitude, co-located with the pHi gradient but extending up to 100 µm beyond the acidic zone. Note that, although SBFI fluorescence is modestly pH-sensitive,31 the ensuing SBFI response cannot be explained as an artefact generated by the underlying pHi gradient, because the shapes of the pHi and profiles are not superimposable. The observations in monolayers are thus consistent with those made in isolated ventricular myocytes, showing that intracellular Na+ signalling can be stimulated downstream of a localised acidic domain.
Figure 7.

pHi gradients are sustained in multicellular syncytial networks, and generate gradients. (A) Exemplar cSNARF-1 fluorescence ratio map, calibrated in units of pH, of a confluent monolayer of neonatal rat ventricular myocytes under (i) control conditions and (ii) during regional superfusion with 40 mM lactate. (B) Exemplar fluorescence map showing [Na+]i and SBFI ratio during regional superfusion with 40 mM lactate, normalized to starting levels. (C) SBFI ratio normalised to control conditions, absolute [Na+]i (uniform exposure to lactate-free solution), and the change in pHi (calculated from cSNARF-1 ratio) plotted across an axis perpendicular to the boundary between microstreams recorded at three different time-points during dual microperfusion. Measurements obtained from four monolayers. The rise in [Na+]i in cells under the lactate-containing microstream is due to Na+ entry via NHE1, stimulated by the ensuing intracellular acidosis. The pHi gradient across the inter-stream boundary is sharp due to slow H+ ion diffusion and rapid transmembrane H+-equivalent fluxes (lactate-H+ transport by MCT). The [Na+]i gradient, in contrast, is shallower due to fast Na+ permeation through gap junctions and diffusion within the cytoplasm.
Discussion
We have demonstrated that acidosis powerfully stimulates arrhythmogenic Ca2+ waves. While this is consistent with earlier phenomenological descriptions,5 we have now quantified the relationship among [H+]i, [Na+]i, and Ca2+ waves, and demonstrated antagonistic control by and . Furthermore, since H+ and Na+ diffuse at different rates in myocyte cytoplasm, the control of Ca2+ wave generation becomes a complex spatio-temporal process. A key finding is that a localised source of intracellular acid inhibits Ca2+ waves in the immediate vicinity, but remotely stimulates them in distal, non-acidic regions, an effect mediated via the diffusible messenger, .
Ca2+ wave frequency is controlled by inhibitory and excitatory signals
During uniform acidosis, spontaneous Ca2+ wave initiation depends on a balance between the stimulatory effects of H+-activated Na+ influx via pHi regulatory transporters such as NHE1, and the inhibitory effects of H+ ions on Ca2+ handling proteins, notably RyRs. Wave frequency is steeply stimulated by a rise of [Na+]i compared with a more shallow inhibition by [H+]i. Thus, when NHE1 flux is enhanced, [Na+]i rises, and waves can be profoundly stimulated. When, however, NHE1 flux is tempered, inhibitory effects of H+ can dominate, thus reducing Ca2+ wave frequency. This latter inhibitory effect has been suggested to be cardioprotective.32
A previous report concluded that intracellular acidosis caused only Ca2+ wave suppression,6 in contrast to our findings. This apparent discrepancy can be explained partly by the lower experimental temperature in the earlier study, which would decrease NHE1 activity,33 and by the briefer exposure to weak acid, which would limit any rise of [Na+]i. In our own work, we have clearly identified both -dependent stimulation and H+-dependent inhibition of Ca2+ waves.
We further demonstrate that increased [Na+]i couples to Ca2+ waves via the modulation of sarcolemmal NCX, since the increase in wave frequency was prevented by pharmacological inhibition of this transporter. In principle, mitochondrial NCX could also make a contribution, however inhibiting mNCX provided no evidence for this. Instead, we confirm a cardioprotective role for mitochondria in buffering excess cytoplasmic Ca2+.34
Rapidly diffusing Na+ ions couple local acidosis to remote Ca2+ wave stimulation
Microdomains of pHi are intuitively expected to affect local Ca2+ signals, under conditions such as vascular perfusion heterogeneity. The unexpected finding from our study is that an acidic microdomain can affect more than just local signalling; it drives downstream Ca2+ waves remotely, a phenomenon that depends on NHE1 activity in the acidic zone. For clarity, our results supporting this stimulatory mechanism have been amalgamated in Figure 8A.
Figure 8.
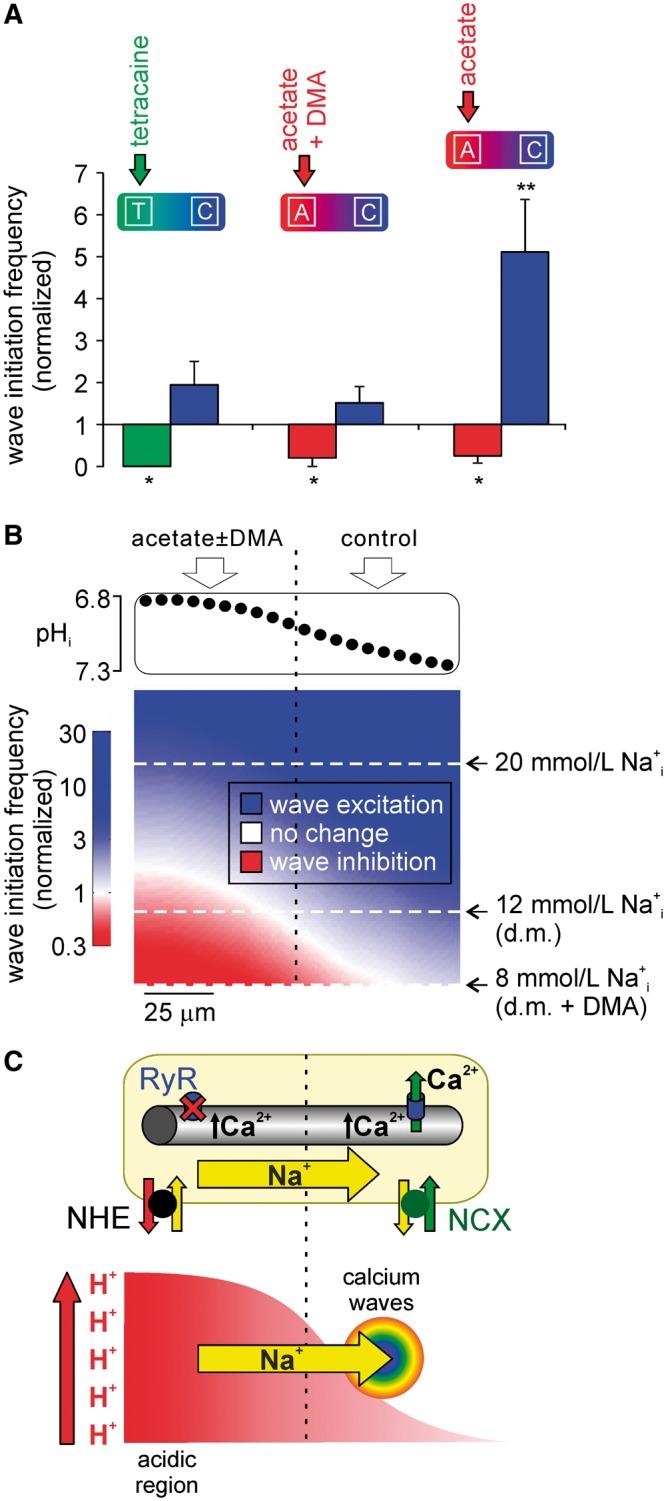
Local and remote pH-Ca2+ interactions. (A) Comparing spatial Ca2+ wave initiation data demonstrates that local inhibition of waves is NHE1-independent, and can be attributed to RyR inhibition, while remote stimulation only occurs in the presence of locally activated NHE1. For clarity, data from Figures 4 and 5 replotted here to permit comparison. (B) The frequency of wave initiation depends on the balance between inhibition by H+ and stimulation by Na+. Top: the experimentally measured pHi gradient illustrated here (cf. Figure 4B) was used in the model. Bottom: Results of a simulation based on the numerical relationships described in Supplementary material online, Figures 1 and 2. The model was used to predict wave initiation frequency (color-coded: red = wave inhibition, white = no change, blue = wave stimulation) during a pHi gradient, for a range of [Na+]i (y-axis, with representative values indicated). The simulation predicts (i) for resting [Na+]i (8 mM, bottom white dashed line, i.e. when NHE is inhibited with DMA), waves are inhibited in the acidic region and remain at control frequency in the non-acidic region; (ii) for a rise in [Na+]i comparable to the load during whole-cell acidosis18 (20 mM, top white dashed line), wave frequency is stimulated throughout the cell; (iii) for a modest rise in [Na+]i (12 mM, middle white dashed line), as seen during an imposed pHi gradient18, waves are inhibited in the acidic zone and stimulated downstream in non-acidic regions. d.m. = dual microperfusion. (C) Schematic of differential effects of pHi non-uniformity on Ca2+ wave frequency. Local intracellular acidosis (represented in bottom part of schematic; x-axis is distance along cell) activates NHE1, leading to a rapid and global rise in [Na+]i (fast diffusion—yellow arrow). This loads the SR throughout the cell, but waves preferentially emerge in the non-acidic microdomain due to the absence of H+ inhibition of RyRs.
One explanation for remote Ca2+ wave triggering is that the NHE1-driven increase in [Na+]i raises luminal SR Ca2+ within the acidic zone (via -slowing of sarcolemmal Ca2+ efflux on NCX, and subsequent Ca2+-sequestration into the SR by SERCA). Elevated luminal Ca2+ might then diffuse throughout the whole SR, enhancing Po for RyRs in non-acidic regions,35 thereby facilitating downstream Ca2+ wave generation. Measurements of SR Ca2+ diffusivity, however, are in the range of 9 to 60 µm2/s,36,37 values that predict a luminal transit time of about 6 and 1 min, respectively over a distance of 80 µm (mean distance between mid-point of microdomain ROIs within a single myocyte). Since the remote stimulatory effects on Ca2+ waves are already well established within 1 min of imposing a local acidosis, the contribution from diffusive re-distribution of luminal SR Ca2+ can, at best, only partially account for our observed results. Additionally, the re-balancing of flux between SERCA activity and passive leak would tend to return SR Ca2+ to physiological levels in regions away from the acidic source.
An alternative explanation is that rapid downstream diffusion of cytoplasmic Na+ ions, initially imported by NHE1 into the acidic microdomain, is the signal that raises SR [Ca2+] throughout the cell. This global stimulation by Na+ is then superimposed with local inhibitory effects of H+ in the acidic microdomain, leading to Ca2+ wave suppression in acidic zones, but wave initiation and clustering in downstream, non-acidic zones. Our recent measurements of high cytoplasmic Na+ diffusivity (∼680 μm2/s)18 are consistent with the time-frame of our Ca2+ wave responses.
To test the quantitative feasibility of the above hypothesis, a simple model was constructed (see Figure 8), based on the [Na+]i- and [H+]i-dependencies of Ca2+ wave frequency (Figure 2Ai, Aii). This was then used to predict the frequency of Ca2+ waves during imposition of a local acid load (Figure 8B). In the simulation, if [Na+]i is restrained at resting levels of ∼8 mM (analogous to NHE1 inhibition), the model predicts that wave frequency is unchanged in areas of normal pHi, but inhibited in the acidic microdomain. If [Na+]i is allowed to rise uniformly to around 20 mM, comparable to that observed under whole-cell acidosis (cf. Figure 1Aiii), the model predicts Ca2+ wave stimulation globally. Thus, regardless of the pHi gradient, a significant overload now promotes stimulation of waves in all cellular regions. However, the model prediction is radically different for an intermediate [Na+]i-rise to 12 mM. This rise is comparable to that measured experimentally during imposition of the pHi gradient18 (note that, because of rapid diffusion, this [Na+]i-rise is near-uniform throughout the cell18). Under these conditions, the model predicts an inhibition of Ca2+ waves in the acidic microdomain and stimulation in the more alkaline microdomain (Figure 8B). Quantitative analysis therefore supports the mechanistic interpretation of our data.
We conclude that rapid spread of cytoplasmic Na+ ions, resulting in a modest, global rise of [Na+]i, is responsible for remote triggering of Ca2+ waves in regions where the pHi is normal. Although [Na+]i will also be raised at the source of acid, the superimposed inhibitory effects of H+ ions will dominate in that region. Our data suggest that local Ca2+ wave suppression is, at least in part, a result of RyR inhibition by H+ ions, on the basis that, in separate experiments, regional acidosis also suppresses local spark frequency.
Ca2+ waves organized by spatial H+ and Na+ signals: cellular consequences
The above data and modelling indicate that spatial domains for H+ and Na+ will control both the site and frequency of spontaneous Ca2+ waves within a ventricular myocyte. Spatial pHi non-uniformity will also grade wave propagation velocity, which will grade the magnitude of any consequent DAD.38 Because of low mobility, myocytes readily develop non-uniformity of pHi during enhanced sarcolemmal H+ flux, compounded by the spatial distribution of H+-transporters, with NHE1 predominantly located at the lateral sarcolemma and intercalated discs.21 As a result, NHE1 activation results transiently in both longitudinal and radial pHi gradients.21,39 Under conditions of Ca2+ overload, this pHi pattern predicts preferential Ca2+ wave emergence in less acidic sub-sarcolemmal regions. This will influence the spatial distribution of excitation-contraction coupling within the cell, and may also modulate the gating of connexin channels at gap junctions.9 At present, in an isolated cell, it would be experimentally challenging to map Ca2+ waves to such dynamic pHi gradients, but our current observations with larger, stable pHi gradients show clear spatial wave initiation and clustering at the alkaline border of an acidic zone.
In our previous study, regional acidosis was found to influence globally the amplitude of the CaT within a ventricular myocyte, via rapidly diffusing , which co-ordinates the magnitude of electrically evoked SR Ca2+ release throughout the cell. This response suggested a mechanism for spatially unifying the contractile signal in the face of pHi heterogeneity.18 In the present work, however, we find that the inhibitory and stimulatory effects of a local acidosis on Ca2+ wave frequency remain spatially separated. While this is reminiscent of the effect of local acidosis on diastolic [Ca2+],24 the mechanisms are dissimilar, since the diastolic effects are independent of changes in Na+, whereas remote wave stimulation is entirely dependent on the H+-driven rise in . The apparent paradox in the difference in spatial behaviour between Ca2+ waves and CaTs can be explained by considering the difference in nature of the events. CaT amplitude is a graded, continuous variable, and so we see a graded influence of [Na+]i and [H+]i. In contrast, Ca2+ wave initiation is an all-or-none event. Although waves are, again, H+- and Na+-sensitive, we see a binary effect, dependent on the balance between the two probability distributions of H+-dependent inhibition and Na+-dependent stimulation. This difference between CaTs and Ca2+ waves means that, when pHi is spatially heterogeneous, CaT amplitude can be fine-tuned in different regions of the cell, but the pH-dependent thresholding of wave initiation can result in spatially separated effects with wave suppression in acidic zones and wave triggering in non-acidic zones.
Ca2+ waves organized by spatial H+ and Na+ signals: role in ischaemic myocardium?
Accumulation of metabolic weak acids such as CO2 and lactate is well documented within regionally ischaemic areas of myocardium,40–42 and this accumulation is expected to generate large and stable pHi gradients across borderzones. In experimental models, gradients of extracellular myocardial pH can be as steep as 0.8 units, expressed over a few myocyte lengths.42 Indeed, here we have shown that a multicellular neonatal preparation can sustain a steep pHi gradient across several cell-lengths, and that this induces an intercellular gradient (Figure 7). The question is whether myocardial pHi gradients will remotely trigger Ca2+ wave initiation in a way comparable to that observed in single myocytes. In the electrically coupled myocardium, because and diffusivities differ substantially (100 μm2/s20 vs. 680 μm2/s18), the resulting myocardial spread of will be more restricted than that for . Consequently, any H- dependent inhibition of aberrant Ca+-signalling within the ischaemic zone should also be spatially restricted, coupled with a wider spatial stimulation by Na+ ions, as illustrated schematically in Figure 8C. Propagation of Ca2+ waves through myocardial gap junctions has a high failure rate,43,44 while junctional H+ permeation is slow and requires chaperoning by carrier-molecules.20 In contrast, Na+ ions readily permeate gap junctions,45 which remain open even at relatively acidic pHi.46 Thus, in a regionally ischaemic myocardium, there is potential for a spatially defined borderzone of vulnerability, characterized by only modest acidosis, but an elevated [Na+]i that is fuelled by Na+ diffusion from more acidic (ischaemic) zones (Figure 8C). The elevated [Na+]i may contribute to the Ca2+ waves and arrhythmias that have been observed consistently at borderzones.47,48
Recent studies47,49–51 demonstrate that Ca2+ waves are most likely to be arrhythmogenic under conditions of local heterogeneity50 that favour the synchronous emergence of waves within a specific area of tissue. It is therefore tempting to suggest that pHi non-uniformity in the myocardium52 may provide an important substrate for such synchronous Ca2+ waves. The arrhythmogenic risk will also be increased by the enhanced wave propagation velocity induced by acidosis. While many factors, other than H+ ions, also contribute to arrhythmogenic activity in ischaemia,53 the principle of remote H+-stimulation of Ca2+ waves, established here in our single cell studies, now merits experimental evaluation in the regionally ischaemic myocardium.
Conclusions
We have shown that, in ventricular myocytes, H+ ions inhibit Ca2+ waves, via inhibition of Ca2+-handling proteins such as RyRs, but they also stimulate waves by activating Na+ influx on NHE1. The steep dependence of Ca2+ wave initiation on [Na+]i often results in overall stimulation. When spatial pHi heterogeneity is introduced into a myocyte, we find that acidic zones remotely trigger Ca2+ waves in non-acidic zones. This is explained by the different-sized spatial domains for elevated inhibitory [H+] and stimulatory [Na+], predicted by their different ionic diffusivities. As well as providing a stimulus for aberrant Ca2+ signalling in acidic myocytes, such spatial signalling may also have relevance in regional myocardial ischaemia, where the formation of pHi gradients across groups of myocytes is likely. Remote H+-triggering of Ca2+ waves emphasizes the importance of Na+ as a spatial messenger in the heart.
Supplementary material
Supplementary material is available at Cardiovascular Research online.
Conflict of interest: none declared.
Funding
This work was supported by the British Heart Foundation (Programme Grant to RDVJ; RG/08/016 and RG15/9/31534, CRE Travelling Fellowship to RDVJ and MB); the Wellcome Trust (PhD Studentship to RDVJ and EM); and the Royal Society (University Research Fellowship to PS).
Supplementary Material
References
- 1. Orchard CH, Eisner DA, Allen DG.. Oscillations of intracellular Ca2+ in mammalian cardiac muscle. Nature 1983;304:735–738. [DOI] [PubMed] [Google Scholar]
- 2. Keizer J, Smith GD.. Spark-to-wave transition: saltatory transmission of calcium waves in cardiac myocytes. Biophys Chem 1998;72:87–100. [DOI] [PubMed] [Google Scholar]
- 3. Kass RS, Lederer WJ, Tsien RW, Weingart R.. Role of calcium ions in transient inward currents and aftercontractions induced by strophanthidin in cardiac Purkinje fibres. J Physiol 1978;281:187–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Capogrossi MC, Houser SR, Bahinski A, Lakatta EG.. Synchronous occurrence of spontaneous localized calcium release from the sarcoplasmic reticulum generates action potentials in rat cardiac ventricular myocytes at normal resting membrane potential. Circ Res 1987;61:498–503. [DOI] [PubMed] [Google Scholar]
- 5. Orchard CH, Houser SR, Kort AA, Bahinski A, Capogrossi MC, Lakatta EG.. Acidosis facilitates spontaneous sarcoplasmic reticulum Ca2+ release in rat myocardium. J Gen Physiol 1987;90:145–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. O'Neill SC, Eisner DA.. pH-dependent and -independent effects inhibit Ca2+-induced Ca2+ release during metabolic blockade in rat ventricular myocytes. J Physiol 2003;550:413–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Antoons G, Willems R, Sipido KR.. Alternative strategies in arrhythmia therapy: evaluation of Na/Ca exchange as an anti-arrhythmic target. Pharmacol Therapeutics 2012;134:26–42. [DOI] [PubMed] [Google Scholar]
- 8. Bountra C, Vaughan-Jones RD.. Effect of intracellular and extracellular pH on contraction in isolated, mammalian cardiac muscle. J Physiol 1989;418:163–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Vaughan-Jones RD, Spitzer KW, Swietach P.. Intracellular pH regulation in heart. J Mol Cell Cardiol 2009;46:318–331. [DOI] [PubMed] [Google Scholar]
- 10. Elliott AC, Smith GL, Eisner DA, Allen DG.. Metabolic changes during ischaemia and their role in contractile failure in isolated ferret hearts. J Physiol 1992;454:467–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Rousseau E, Pinkos J.. pH modulates conducting and gating behaviour of single calcium release channels. Pflugers Archiv 1990;415:645–647. [DOI] [PubMed] [Google Scholar]
- 12. Xu L, Mann G, Meissner G.. Regulation of cardiac Ca2+ release channel (ryanodine receptor) by Ca2+, H+, Mg2+, and adenine nucleotides under normal and simulated ischemic conditions. Circ Res 1996;79:1100–1109. [DOI] [PubMed] [Google Scholar]
- 13. Balnave CD, Vaughan-Jones RD.. Effect of intracellular pH on spontaneous Ca2+ sparks in rat ventricular myocytes. J Physiol 2000;528:25–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Mandel F, Kranias EG, Grassi de Gende A, Sumida M, Schwartz A.. The effect of pH on the transient-state kinetics of Ca2+-Mg2+-ATPase of cardiac sarcoplasmic reticulum. A comparison with skeletal sarcoplasmic reticulum. Circ Res 1982;50:310–317. [DOI] [PubMed] [Google Scholar]
- 15. Hulme JT, Orchard CH.. Effect of acidosis on Ca2+ uptake and release by sarcoplasmic reticulum of intact rat ventricular myocytes. Am J Physiol 1998;275:H977–987. [DOI] [PubMed] [Google Scholar]
- 16. Boyman L, Hagen BM, Giladi M, Hiller R, Lederer WJ, Khananshvili D.. Proton-sensing Ca2+ binding domains regulate the cardiac Na+/Ca2+ exchanger. J Biol Chem 2011;286:28811–28820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Saegusa N, Moorhouse E, Vaughan-Jones RD, Spitzer KW.. Influence of pH on Ca2+ current and its control of electrical and Ca2+ signaling in ventricular myocytes. J Gen Physiol 2011;138:537–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Swietach P, Spitzer K, Vaughan-Jones RD.. Na+ ions as spatial intracellular messengers for co-ordinating Ca2+ signals during pH heterogeneity in cardiomyocytes. Cardiovasc Res 2015;105:171–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Garciarena CD, Youm JB, Swietach P, Vaughan-Jones RD. H. +- activated Na+ influx in the ventricular myocyte couples Ca2+-signalling to intracellular pH. J Mol Cell Cardiol 2013;61:51–59. [DOI] [PubMed] [Google Scholar]
- 20. Swietach P, Spitzer KW, Vaughan-Jones RD.. pH-dependence of extrinsic and intrinsic H+-ion mobility in the rat ventricular myocyte, investigated using flash photolysis of a caged-H+ compound. Biophys J 2007;92:641–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Garciarena CD, Ma Y-l, Swietach P, Huc L, Vaughan-Jones RD.. Sarcolemmal localisation of Na+/H+ exchange and Na+– co-transport influences the spatial regulation of intracellular pH in rat ventricular myocytes. J Physiol 2013;591:2287–2306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kushmerick MJ, Podolsky RJ.. Ionic mobility in muscle cells. Science 1969;166:1297–1298. [DOI] [PubMed] [Google Scholar]
- 23. Villafuerte FC, Swietach P, Youm J-B, Ford K, Cardenas R, Supuran CT, Cobden PM, Rohling M, Vaughan-Jones RD.. Facilitation by intracellular carbonic anhydrase of Na+- co-transport but not Na+/H+ exchange activity in the mammalian ventricular myocyte. J Physiol 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Swietach P, Youm J-B, Saegusa N, Leem C-H, Spitzer KW, Vaughan-Jones RD.. Coupled Ca2+/H+ transport by cytoplasmic buffers regulates local Ca2+ and H+ ion signaling. Proc Natl Acad Sci U S A 2013;110:E2064–E2073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kong CHT, Soeller C, Cannell MB.. Increasing sensitivity of Ca2+ spark detection in noisy images by application of a matched-filter object detection algorithm. Biophys J 2008;95:6016–6024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Williams IA, Xiao X-h, Ju Y-k, Allen DG.. The rise of [Na+]i during ischemia and reperfusion in the rat heart - underlying mechanisms. Pflügers Archiv 2007;454:903–912. [DOI] [PubMed] [Google Scholar]
- 27. Maack C, Cortassa S, Aon MA, Ganesan AN, Liu T, O’Rourke B.. Elevated cytosolic Na+ decreases mitochondrial Ca2+ uptake during excitation-contraction coupling and impairs energetic adaptation in cardiac myocytes. Circ Res 2006;99:172–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Swietach P, Spitzer KW, Vaughan-Jones RD.. Modeling calcium waves in cardiac myocytes: importance of calcium diffusion. Front Biosci 2010;15:661–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Overend CL, O'Neill SC, Eisner DA.. The effect of tetracaine on stimulated contractions, sarcoplasmic reticulum Ca2+ content and membrane current in isolated rat ventricular myocytes. J Physiol 1998;507:759–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Baartscheer A, Schumacher CA, Fiolet JW.. Small changes of cytosolic sodium in rat ventricular myocytes measured with SBFI in emission ratio mode. J Mol Cell Cardiol 1997;29:3375–3383. [DOI] [PubMed] [Google Scholar]
- 31. Diarra A, Sheldon C, Church J.. In situ calibration and [H+] sensitivity of the fluorescent Na+ indicator SBFI. Am J Physiol Cell Physiol 2001;280:C1623–1633. [DOI] [PubMed] [Google Scholar]
- 32. Strömer H, de Groot MCH, Horn M, Faul C, Leupold A, Morgan JP, Scholz W, Neubauer S.. Na+/H+ exchange inhibition with HOE642 improves postischemic recovery due to attenuation of Ca2+ overload and prolonged acidosis on reperfusion. Circulation 2000;101:2749–2755. [DOI] [PubMed] [Google Scholar]
- 33. Ch'en FF-T, Dilworth E, Swietach P, Goddard RS, Vaughan-Jones RD.. Temperature dependence of Na+−H+ exchange, Na+− co-transport, intracellular buffering and intracellular pH in guinea-pig ventricular myocytes. J Physiol 2003;552:715–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Raffaello A, De Stefani D, Rizzuto R.. The mitochondrial Ca2+ uniporter. Cell Calcium 2012;52:16–21. [DOI] [PubMed] [Google Scholar]
- 35. Lukyanenko V, Györke I, Györke S.. Regulation of calcium release by calcium inside the sarcoplasmic reticulum in ventricular myocytes. Pflügers Archiv 1996;432:1047–1054. [DOI] [PubMed] [Google Scholar]
- 36. Swietach P, Spitzer KW, Vaughan-Jones RD.. Ca2+-mobility in the sarcoplasmic reticulum of ventricular myocytes is low. Biophys J 2008;95:1412–1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wu X, Bers DM.. Sarcoplasmic reticulum and nuclear envelope are one highly interconnected Ca2+ store throughout cardiac myocyte. Circ Res 2006;99:283–291. [DOI] [PubMed] [Google Scholar]
- 38. Sugai Y, Miura M, Hirose M, Wakayama Y, Endoh H, Nishio T, Watanabe J, ter Keurs HEDJ, Shirato K, Shimokawa H.. Contribution of Na+/Ca2+ exchange current to the formation of delayed afterdepolarizations in intact rat ventricular muscle. J Cardiovasc Pharmacol 2009;53:517–522. [DOI] [PubMed] [Google Scholar]
- 39. Swietach P, Vaughan-Jones RD.. Spatial regulation of intracellular pH in the ventricular myocyte. Ann NY Acad Sci 2005;1047:271–282. [DOI] [PubMed] [Google Scholar]
- 40. Cascio WE, Yan GX, Kléber AG.. Early changes in extracellular potassium in ischemic rabbit myocardium. The role of extracellular carbon dioxide accumulation and diffusion. Circ Res 1992;70:409–422. [DOI] [PubMed] [Google Scholar]
- 41. Coronel R, Wilms-Schopman FJG, Fiolet JWT, Opthof T, Janse MJ.. The relation between extracellular potassium concentration and pH in the border zone during regional ischemia in isolated porcine hearts. J Mol Cell Cardiol 1995;27:2069–2073. [DOI] [PubMed] [Google Scholar]
- 42. Wilensky RL, Tranum-Jensen J, Coronel R, Wilde AA, Fiolet JW, Janse MJ.. The subendocardial border zone during acute ischemia of the rabbit heart: an electrophysiologic, metabolic, and morphologic correlative study. Circulation 1986;74:1137–1146. [DOI] [PubMed] [Google Scholar]
- 43. Kaneko T, Tanaka H, Oyamada M, Kawata S, Takamatsu T.. Three distinct types of Ca2+ waves in langendorff-perfused rat heart revealed by real-time confocal microscopy. Circ Res 2000;86:1093–1099. [DOI] [PubMed] [Google Scholar]
- 44. Li Y, Eisner DA, O’Neill SC.. Do calcium waves propagate between cells and synchronize alternating calcium release in rat ventricular myocytes? J Physiol 2012;590:6353–6361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ruiz-Meana M, Garcia-Dorado D, Hofstaetter B, Piper HM, Soler-Soler J.. Propagation of cardiomyocyte hypercontracture by passage of Na+ through gap junctions. Circ Res 1999;85:280–287. [DOI] [PubMed] [Google Scholar]
- 46. White RL, Doeller JE, Verselis VK, Wittenberg BA.. Gap junctional conductance between pairs of ventricular myocytes is modulated synergistically by H+ and Ca++. J Gen Physiol 1990;95:1061–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Tanaka H, Oyamada M, Tsujii E, Nakajo T, Takamatsu T.. Excitation-dependent intracellular Ca2+ waves at the border zone of the cryo-injured rat heart revealed by real-time confocal microscopy. J Mol Cel Cardiol 2002;34:1501–1512. [DOI] [PubMed] [Google Scholar]
- 48. Tsujii E, Tanaka H, Oyamada M, Fujita K, Hamamoto T, Takamatsu T.. In situ visualization of the intracellular Ca2+ dynamics at the border of the acute myocardial infarct. Mol Cell Biochem 2003;248:135–139. [DOI] [PubMed] [Google Scholar]
- 49. Wasserstrom JA, Shiferaw Y, Chen W, Ramakrishna S, Patel H, Kelly JE, O'Toole MJ, Pappas A, Chirayil N, Bassi N, Akintilo L, Wu M, Arora R, Aistrup GL.. Variability in timing of spontaneous calcium release in the intact rat heart is determined by the time course of sarcoplasmic reticulum calcium load. Circ Res 2010;107:1117–1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Nivala M, Ko CY, Nivala M, Weiss JN, Qu Z.. The emergence of subcellular pacemaker sites for calcium waves and oscillations. J Physiol 2013;591:5305–5320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Fujiwara K, Tanaka H, Mani H, Nakagami T, Takamatsu T.. Burst emergence of intracellular Ca2+ waves evokes arrhythmogenic oscillatory depolarization via the Na+–Ca2+ exchanger: simultaneous confocal recording of membrane potential and intracellular Ca2+ in the heart. Circ Res 2008;103:509–518. [DOI] [PubMed] [Google Scholar]
- 52. Stewart LC, Kelly RA, Atkinson DE, Ingwall JS.. pH heterogeneity in aged hypertensive rat hearts distinguishes reperfused from persistently ischemic myocardium. J Mol Cell Cardiol 1995;27:321–333. [DOI] [PubMed] [Google Scholar]
- 53. Di Diego JM, Antzelevitch C.. Ischemic ventricular arrhythmias: experimental models and their clinical relevance. Heart Rhythm 2011;8:1963–1968. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.