

Abstract
Aims
To test if a human Hand1 frame shift mutation identified in human samples is causative of hypoplastic left heart syndrome (HLHS).
Methods and results
HLHS is a poorly understood single ventricle congenital heart defect that affects two to three infants in every 10 000 live births. The aetiologies of HLHS are largely unknown. The basic helix–loop–helix transcription factor HAND1 is required for normal heart development. Interrogation of HAND1 sequence from fixed HLHS tissues identified a somatic frame-shift mutation at Alanine 126 (NP_004812.1 p.Ala126Profs13X defined as Hand1A126fs). Hand1A126fs creates a truncated HAND1 protein that predictively functions as dominant negative. To determine if this mutation is causative of HLHS, we engineered a conditional Hand1A126fs mouse allele. Activation of this allele with Nkx2.5Cre results in E14.5 lethality accompanied by cardiac outflow tract and intraventricular septum abnormalities. Using αMHC-Cre or Mef2CAHF-Cre to activate Hand1A126fs results in reduced phenotype and limited viability. Left ventricles of Hand1A126FS mutant mice are not hypoplastic.
Conclusions
Somatically acquired Hand1A126FS mutation is not causative of HLHS. Hand1A126FS mutation does exhibit embryonic lethal cardiac defects that reflect a dominant negative function supporting the critical role of Hand1 in cardiogenesis.
Keywords: Hand1, bHLH, Cardiac development, Transcription, Hypoplastic left heart syndrome
1. Introduction
Hypoplastic left heart syndrome (HLHS) is a poorly understood single ventricle congenital heart defect (CHD) that accounts for 2–3% of all CHDs.1–3 In HLHS, the LV is diminished in size and unable to support the systemic circulation. To account for the insufficiency, the RV is fed with oxygenated blood via an atrial septal defect directly connecting the right and left atriums or ventricular septal defects (VSD) directly connecting the RV and LV. Blood from the RV then exits the ventricle via the pulmonary artery and enters the aorta via a persistent ductus.
Traditional linkage approaches have identified a growing number of mutations that give rise to survivable CHDs.4–8 However, it has proved more difficult to identify CHD-causing genes as a result of early in utero lethality due to catastrophic failure of cardiogenesis, or alternatively, failure of other organ systems required for embryo viability. Such gene mutations cannot be inherited. It is suggested that the acquisition of somatic mutation(s) in such genes, which appear in only a subset of the cells in the developing embryo, could result in greater in utero viability or even in survivable CHDs; however, such a mechanism would likely be very rare.9 HAND1, a bHLH transcription factor, is a prototypical example of a CHD-causing gene whose analysis has been hampered due to early embryonic lethality.10–13 The function of Hand1 is regulated by homo- or hetero-dimerization with other bHLH proteins through its bHLH domain. Hand1 forms dimers that bind E-box (CANNTG) and D-box (CGNNTG) cis-elements.14–16 Deletion of Hand1 or hypomorphic Hand1 expression in mice results in cardiac morphogenic defects, and early embryonic lethality due to placental defects.10,11,13,17,18 Recently, somatic HAND1 mutations have been implicated in the genesis of Tetralogy of Fallot,19 VSDs,20 and HLHS.9,21 These results were obtained from fixed tissues and subsequent studies interrogating unfixed samples were unable to confirm these results.22 Although it is clear that Hand1 plays an important role in cardiac morphogenesis, mutations within the HAND1 coding domains are unlikely to result in viable offspring; therefore, the idea that HAND1 mutations could occur somatically is an intriguing disease model. The controversy regarding somatic mutation as a disease model and the potential caveats of using fixed samples prompted us to address this question directly. We tested a reported Hand1 HLHS mutation (NP_004812.1 p.Ala126Profs13X)21,23 by generating a conditional activation knock-in allele (defined as Hand1A126fs) of this putative Hand1 somatic mutation.
Results show that in tissue culture analysis, the p.Ala126ProfsX13 mutant protein localizes to the nucleus and inhibits both DNA binding and transcriptional activation. Phenotypic and molecular analysis of Hand1SFA126fs mice generated with Nkx2.5Cre24 shows that expression of this dominant negative Hand1 protein disrupts cardiac development. Hand1A126fs/+ mice die by E15.5 and display outflow tract (OFT) abnormalities, thin myocardium and VSDs. Although cardiac Hand1 expression and linage commitment is largely left ventricular,10,25 we observe normal sized LVs in Hand1A126fs/+ mutant mice. Using the αMHC-Cre driver,26 we encounter less severe heart defects and mutant viability. We also employed the anterior heart field (AHF) Mef2cAHF-Cre driver27 and show similar OFT defects to Nkx2.5Cre mutants; however, these mice also survive. Together, these data suggest that although the Hand1 p.Ala126Profs13X mutant protein has altered function, when expressed in mouse cardiomyocytes, it is not causative of HLHS.
2. Methods
2.1 Mouse strains, genotyping
SFHand1A126FSwas generated from ES cells targeted with the constructs described and genotyping by Southern blot10 or with primers H1 5′-CTG CCA TTG GCT CCG GCT AGA GGT-3′ and PGK 5′-GGC TGC TAA AGC GCA TGC TCC AGA CTG-3′ using PCR conditions of 94 °C 1 min, 60 °C 1 min, 72 °C 1 min for 35 cycles. Nkx2.5Cre 24 mice are a Cre-recombinase knock-in allele that express Cre within the cardiac crescent at E7.5 marking myocardium, endocardium.28Hand1 is not expressed in edocardium.25αMHC-Cre transgenic mice express Cre recombinase at E9.5 within the myocardium after which the Cre is dormant until after birth.26Mef2CAHF-Cre is expressed endocardium and myocardium at E7.5.27
No anaesthetic/analgesic agents were used. Euthanasia was performed using CO2 gas in a closed chamber followed by cervical dislocation. All experiments were performed conforming to the NIH guidelines following the Indiana University IACUC animal protocol 10809.
2.2 Cell transfection, luciferase assays, and electrophoretic mobility shift assays
HEK293 CaPO4 transfections and Luciferase assays protocol as described were performed as described.29,30 Ebox reporter, Dbox reporter, Hand1 pIRESNeo, and E12 pIRESNeo expression plasmids are described.14,31 Hand1A126fspIRESNeo was mutagenized using QuickChange Mutagenesis Kit (Agilent Technologies). Data represent six-independent experiments. Error bars denote SE. Asterisk represent significance of P ≤ 0.05 by ANOVA.
2.2.1.Electrophoretic mobility shift assays
EMSAs were carried out as previously described.14 EMSA probe Hand E-box (5′-gga ttc cat tgc atc tgg att cca gag-3′) was used to shift and as specific competitor. Non-specific competitor oligo (5′-gga ttc cat tgGGtc AAg att cca gag-3′).
2.3 Histology
Embryos (E9.5–E18.5) were processed as described.13 Eight viable embryos per genotype were examined for all analyses. Bifurcated heart preparations were generated by cryo-sectioning.
2.4 In situ hybridization
Section and whole mount in situ hybridization (ISH) were performed as described32,33 for probes Hand1, Wnt11, Tbx5, Dkk3, Cited1, Bmp10, Nppa (Anf), Cxcl12, Tbx20, Hey2.
2.5 Lysotracker and EdU immunohistochemistry analysis
Lysotracker and Click-IT EdU Imaging (Life Technologies) was incubated with embryonic hearts as described.26,32
2.6 Quantitative RTPCR (QPCR)
RNA Isolation and quantitative RT–PCR (QRTPCR) was performed as described.32 Error bars denote the maximum and minimum relative level of gene expression set in the QuantStudio 3&5 software. Statistics employed Student’s two-tailed t-test with P ≤ 0.05 significance. N = 6.
3. Results
3.1 Hand1A126FS acts as a dominant negative transcription factor
To confirm that the p.Ala126Profs13X protein (Hand1A126FS) functions as a dominant negative, we employed luciferase analysis, EMSA and cellular localization to test functionality (Figure 1). Hand1A126FS truncates Hand1 within the NH4-terminal portion of the loop domain and is reported to not bind DNA and to repress transcriptional activation of both E- and D-box luciferase reporters in yeast extracts.21 To confirm these findings in a mammalian transcription system, we utilized similar E- and D-box luciferase reporters in HEK293 cells (Figure 1A). Results show that alone neither Hand1, Hand1A126FS (Hand1FS), or E12 transactivate (Figure 1B). When Hand1 is cotransfected with E12 (Hand1 + E12) along with either an E- or D-box reporter significant transactivation (P ≤ 0.05) is observed. Cotransfection of Hand1A126FS and E12 (Hand1FS + E12) results in a decrease of the Hand1-E12 heterodimer transcriptional activity consistent with previous analysis.21 EMSA shows that E12 and (Hand1 + E12) bind to the Ebox probe as homo- (red arrowhead) and heterodimers (black arrowhead), respectively (Figure 1C). Cotranslation of Hand1A126FS with E12 (Hand1A126fs + E12) results in undetectable shifting of the Ebox probe (Figure 1C, asterisks). We engineered a Hand1A126fs–eGFP fusion construct and co-transfected this plasmid with an E12–DsRed fusion plasmid (Figure 1D). Results show Hand1A126fs–eGFP and E12–DsRed nuclear epifluorescence. These data support a model where Hand1A126FS acts as a dominant negative factor.
Figure 1.

Hand1A126FS localizes to the nucleus acting as a dominant negative. (A) Schematic of wild-type HAND1 and HAND1 A126FS and Dbox and Ebox luciferase reporters. (B) Luciferase assays from HEK293 lysates with the indicated plasmids (pIRESNeo), open bars; E12-pIRESNeo (E12), grey bars; Hand1-pIRESNeo (Hand1), black bars; Hand1A126FS-pIRESNeo (Hand1FS), blue bars; Hand1-pIRESNeo + E12-pIRESNeo black diagonal-striped bars; Hand1FS pIRESNeo+ E12-pIRESNeo blue diagonal-striped bars; Hand1-pIRESNeo + Hand1FS-pIRESNeo, waved bars). *,#P ≤ 0.05 by ANOVA compared with pIRESNeo and Hand1FS + E12, respectively. Error bars show standard error. (C) EMSA of in vitro expressed HAND1 (H1), Hand1 A126FS (H1A126fs), and E12 in the indicated combinations. Unprogrammed reticulocyte lysates (UP) show non-specific complexes (ns). E12-programmed lysates reveal homodimer binding (red arrowhead) that is competed with specific competitor (SC) but not non-specific competitor (NS). Hand1-E12 heterodimers (black arrowhead). H1A126fs-E12 heterodimers do not bind DNA (double asterisk). (D) HEK293s co-transfection of Hand1A126fs-eGFP and E12-DsRed. DAPI (blue) marks nuclei. Both Hand1A126fs-eGFP and E12-DsRed co-localize to the nucleus. Transfection and EMSA represent five experiments; only one EMSA is shown. Scale bar in (D), 10 μm.
3.2 Generation and analysis of a conditional Hand1A126FS allele
To test if expression of Hand1A126FS is causative of HLHS, we engineered a conditional-activation allele by introducing the identified p.Ala126Profs13X mutation into the murine Hand1 locus that includes a stop-flox (SF) cassette that bocks the transcription and translation of the mutant Hand1 protein (Figure 2A). The mutation deletes the G in codon 126 (alanine) producing a 13 amino acid frame shift ending in a termination codon. ES targeted clones were identified at a frequency of 59% (Figure 2B). Mice carrying the conditional-activation Hand1SFA126FS/+ allele are viable and fertile. We next generated Nkx2.5Cre; Hand1A126FS/+ and Nkx2.5Cre; Hand1A126FS/fx embryos and show LV myocardium and myocardial cuff expression of Hand1 is detectable from the conditionally activated mutant Hand1 allele (Figure 2C).
Figure 2.

Targeting strategy and expression validation of Hand1A126FS/+ mice. (A) Schematic of the Hand1 locus and targeting construct. Hand1 targeting arms10 were utilized to insert a loxP element-flanked Neomyosin cassette (Flox STOP Neo).32 The 3′ targeting arm includes the A126fs mutation. Upon Cre-recombination and removal of the Stop-Flox cassette, activates the Hand1A126fs allele. (B) Southern blot analysis shows targeting frequency exceeding 50%. (C) E10.5 wholemount Hand1 ISH in control (Hand1SF-A126fs/+), recombined (Nkx2.5Cre/+; Hand1A126fs/+), and compound Hand1 heterozygotes (Nkx2.5Cre/+; Hand1A126fs/fx) single-copy mutants. I2, mandibular component of the first pharyngeal arch; oft, cardiac outflow tract; rv, right ventricle; lv, left ventricle; u, umbilicus. Scale bar 150 μm.
To test if expression of the Hand1A126FS allele models HLHS, we intercrossed Hand1A126FS/+ females to Nkx2.5Cre males and looked for live births. After 59 pups were genotyped and no surviving Nkx2.5Cre; Hand1A126FS/+ were encountered, we initiated embryonic analysis (see Supplementary material online, Table S1). At E14.5, we encountered Nkx2.5Cre; Hand1A126FS/+ embryos at a frequency of 0.08 (n = 20; expected frequency 0.25) and no mutants were recovered beyond E15.5 (n = 46). We then initiated isolating Nkx2.5Cre; Hand1A126FS/+ embryos at E12.5, which we obtained at a frequency of 0.30.
Hand1SFA126FS/+ embryos appear phenotypically normal exhibiting normal OFT structures, forming IVS and trabeculated RV and LV (Figure 3A–E). Endocardial cushions are well developed and positioned correctly above the forming IVS (Figure 3D, black arrow). Two wholemount examples of Nkx2.5Cre; Hand1A126FS/+ show oedema (white arrowheads Figure 3F and K) and elongated OFTs (white bars) indicating that morphological alignment of the forming heart is compromised. Abnormal haemorrhaging is apparent in Nkx2.5Cre; Hand1A126FS/+ mutants (Figure 3K).
Figure 3.

Nkx2.5Cre/+; Hand1A126FS/+ E12.5 mutants present with an elongation of the cardiac OFT. (A) Nkx2.5+/+; Hand1SF-A126FS/+ wholemount control compared with two Nkx2.5Cre/+; Hand1A126FS/+ mutants (F, K). Measure of cardiac OFT (white bars) shows that mutant OFTs extend farther into the forming RV, which appears smaller and shows some signs oedema (white arrowhead). Section analysis of the control shown in (A). (B–E) reveals expected structural development that includes a septated pulmonary trunk (pt) developing IVS and LV. Magnification in (E) shows a well-formed compact zone (black bar). Matched sections from Nkx2.5+/+; Hand1A126FS/+ mutants shown in (F) and (K). (G–O) A poorly formed IVS (asterisk), small RV, and a thinner compact zone (J, O). Size of the LV is indistinguishable between control and Nkx2.5+/+; Hand1A126FS/+ mutant hearts. Data represent an example of each genotype. Ten embryos per genotype were examined. ra, right atria; la, left atria; rv, right ventricle; lv, left ventricle; ivs, intraventricular septum; pt, pulmonary trunk. Scale bars 150 and 500 μm.
Section analysis of Nkx2.5Cre; Hand1A126FS/+ mutants reveals a poorly formed IVS that is positioned to the right of the endocardial cushions (compare Figure 3C and D with H, I and M and N), a thin compact zone (black lines Figure 3E, J, and O), and hypotrabeculation. Given that Hand1 is expressed within the myocardium of the left ventricle and a region of the SHF-derived myocardial cuff, but not the majority of the IVS or endocardium25,29 these phenotypes are consistent with both cell-autonomous and non-cell autonomous defects in cardiogenesis.
3.3 Increased cell death is observed in hearts of Hand1A126FS embryos
To determine the cause of the Nkx2.5Cre; Hand1A126FS/wt mutant heart phenotypes, we looked at cell death using Lysotracker staining at E10.5 and E11.5 (Figure 4). Control Hand1SFA126FS/+ hearts display nearly undetectable levels of cell death as indicated by punctate epifluorescence (Figure 4A and B). Nkx2.5Cre; Hand1A126FS/+ mutant hearts reveal a clearly visible increase in the numbers of dying cells within the cardiac ventricles and OFT (Figure 4C and D, white arrows). Cell death is observed in structures that express Hand1 (LV) and structures that do not express Hand1 (RV) indicating autonomous and non-cell autonomous death. Cell proliferation analysis was performed (Figure 4E–G) and results show that proliferation is indistinguishable between control and Nkx2.5Cre; Hand1A126FS/wt mutant hearts (Figure 4E–G).
Figure 4.

Nkx2.5Cre/+; Hand1A126FS/+ hearts display an increase in both cell autonomous and non-cell autonomous cell death. Control Hand1SF-A126fs/+ (A, B) and Nkx2.5Cre/+; Hand1A126FS/+ mutants (C, D) were assayed for cell death using Lysotracker staining in wholemount at E10.5 and E11.5. Control hearts show little cell death. Increased cell death is observed in OFT, RV, and, LV of Nkx2.5Cre/+; Hand1A126FS/+ mutants (white arrows). (E–H) EdU wholemount proliferation analysis at E10.5 and E11.5. Results show no obvious differences in between control and mutant hearts. Data represent an example of each genotype. Six embryos per genotype were examined. ra, right atria; la, left atria; rv, right ventricle; lv, left ventricle. Scale bars 150 μm.
3.4 Altered gene expression is observed in hearts of Hand1A126FS embryos
We next interrogated gene expression defining cardiogenesis. Wnt11 marks the precardiac mesoderm of the OFT and is essential for SHF development.34,35 E11.5 wholemount ISH of Wnt11 confirms that Nkx2.5Cre; Hand1A126FS/+ mutant OFTs are elongated (Figure 5A and B; white bars). The T-box transcription factor Tbx5 expression is largely restricted to the LV.36 Compared with Hand1SFA126FScontrols, Nkx2.5Cre; Hand1A126FS/+ mutant hearts reveal a broader domain of Tbx5 expression that includes the RV (Figure 5C and D; white arrowhead). Cardiac expression of Dkk3 generally marks the cardiac mesoderm becoming enriched within the IVS.37,38 Comparison of Dkk3 expression in Hand1SFA126FScontrols and Nkx2.5Cre; Hand1A126FS/+ mutant hearts shows a largely comparable expression pattern; however, the level of Dkk3 appears upregulated in Nkx2.5Cre; Hand1A126FS/+ mutants (Figure 5and F, white arrowhead). Cited1 is a trabecular maker and is reported to be downregulated in Hand1 cardiac conditional knockout mice.12 Comparison of control and Nkx2.5Cre; Hand1A126FS/+ mutant hearts reveals no difference in Cited1 expression (Figure 5G and H). LV size of Hand1A126FS/+ mutants is indistinguishable from controls.
Figure 5.
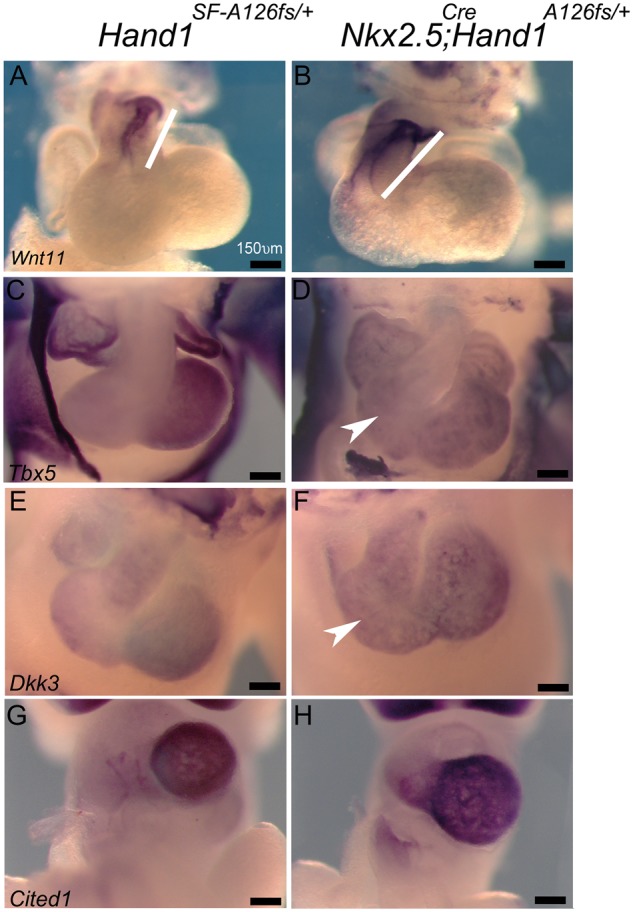
Wholemount ISH of Nkx2.5Cre/+; Hand1A126FS/+ mutant embryos reveals altered cardiac gene expression during heart formation. E11.5 Nkx2.5+/+; Hand1SF-A126FS/+ control (A, C, E, G) and Nkx2.5Cre/+; Hand1A126FS/+mutant (B, D, F, H) embryos hybridized to the indicated riboprobes. Wnt11 marks OFT tissue (white bars; A and B). Tbx5 is an LV chamber maker. Tbx5 expression appears more homogeneous in Nkx2.5Cre/+; Hand1A126FS/+mutants when compared with controls (white arrowhead; C and D). Dkk3 marks the IVS and ventricular chambers. Dkk3 expression appears more robust in Nkx2.5Cre/+; Hand1A126FS/+mutants (white arrowhead; E and F). Cited1 appears unchanged between control and Nkx2.5Cre/+; Hand1A126FS/+mutants (G and H). Data represent an example of each genotype. Eight embryos per genotype were examined. Scale bars 150 μm.
Next we performed section ISH at E10.5 and E12.5 on Hand1SFA126FS controls and Nkx2.5Cre; Hand1A126FS/+ mutants (Figure 6). We looked at the trabecular markers Bmp10,39Nppa (Anf),40 and Cited1 (Figure 6A–L). Results show that expression is largely unaltered. Analysis of Cxcl12 expression, a cardiomyocyte ligand required for coronary formation,41,42 shows Cxcl12 expression is markedly upregulated (Figure 6M–P; arrowheads). Similarly, Dkk3 expression (Figure 6Q–T) shows a marked expansion of expression consistent with E11.5 analysis (Figure 5). Examination of the compact zone markers Tbx2043,44 and Hey245,46 show that Tbx20 expression is upregulated in Nkx2.5Cre; H and 1A126FS/+ mutants (Figure 6U–Xl, black arrowheads) while Hey2 expression is decreased (Figure 6Y–Ab, asterisks).
Figure 6.

Section ISH in E10.5 and E12.5 control and Nkx2.5Cre/+; Hand1A126FS/+ mutants. Bmp10 (A–D) and Anf (E–H) expression marks trabeculae. No significant changes in expression are observed. Cited1 expression (I–L) expression is not altered. Cxcl12 cardiac expression is required for formation of intra-ventricular coronary arteries. At E10.5 (M, N) and E12.5 (O, P), Cxcl12 expression is upregulated (black arrowheads) compared with controls (M and O). Dkk3 (Q–T) shows expanded expression (black arrowheads) in Nkx2.5Cre/+; Hand1A126FS/+mutants (R, T) compared with control (Q, S). The compact zone markers Tbx20 (U–X) and Hey2 (Y–Ab) show altered expression in Nkx2.5Cre/+; Hand1A126FS/+mutants. Tbx20 is upregulated in Nkx2.5Cre/+; Hand1A126FS/+mutants (V, X black arrowheads). Hey2 expression is markedly down in Nkx2.5Cre/+; Hand1A126FS/+mutants (Z, Ab; asterisks) compared with Nkx2.5+/+; Hand1SF-A126FS/+ controls. Data represent an example of each genotype. Eight embryos per genotype were examined. Scale bars 200 μm.
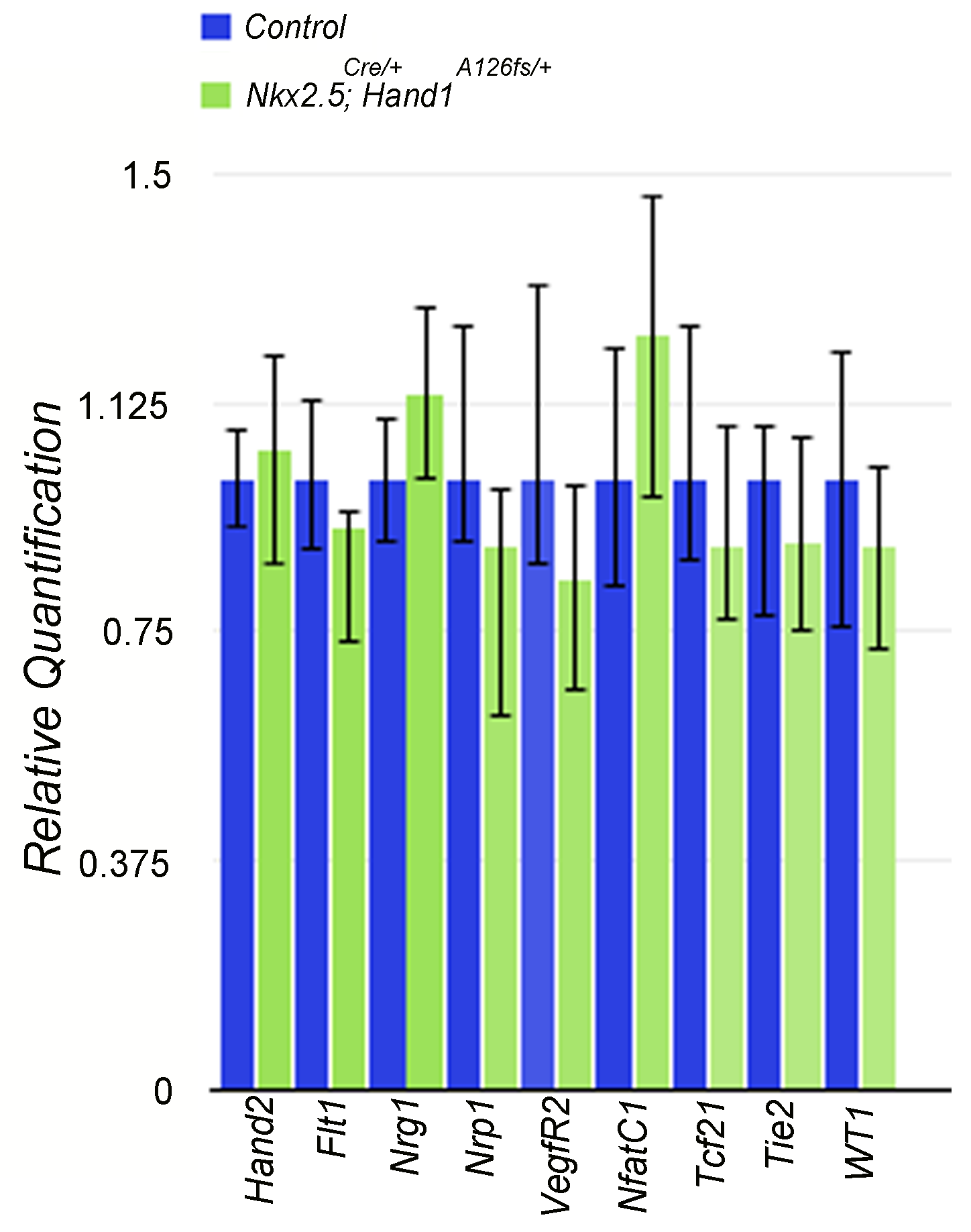
As we observe a thin walled myocardium in the Nkx2.5Cre; Hand1A126FS/+ mutants, we also looked at endocardial and epicardial gene expression which can influence myocardial growth47–49 and production of cardiac myofibroblasts and coronary vasculature.50–53 Expression analysis of the endocardial markers Hand2, Flt1, Tie2, Nrg1, Nrp1, VegfR2, and NfactC1 and epicardial markers WT1 and Tcf21 by QPCR from E10.5 RNA control and mutant hearts revealed no observable changes in expression (see Supplementary material online, Figure S1).
As some Nkx2.5Cre; Hand1A126FS/+ mutants survive to E14.5, we looked at these mutant phenotypes (Figure 7). H&E sections of E14.5 Nkx2.5Cre; Hand1A126FS/+ mutant hearts reveal obvious VSDs both membranous and muscular in nature (Figure 7A and B). Although the LV compact zone is thin when compared with controls (black bars), LV chamber size is clearly proportional to RV size and similar to controls. We observe lower levels of Bmp10 expression (Figure 7C–F), which may reflect either real changes in expression or a near death state of these mutants. Dkk3 maintains its enhanced expression when comparing Nkx2.5Cre; Hand1A126FS/+ mutants to controls suggesting that death is not causative of decreased Bmp10 expression.
Figure 7.
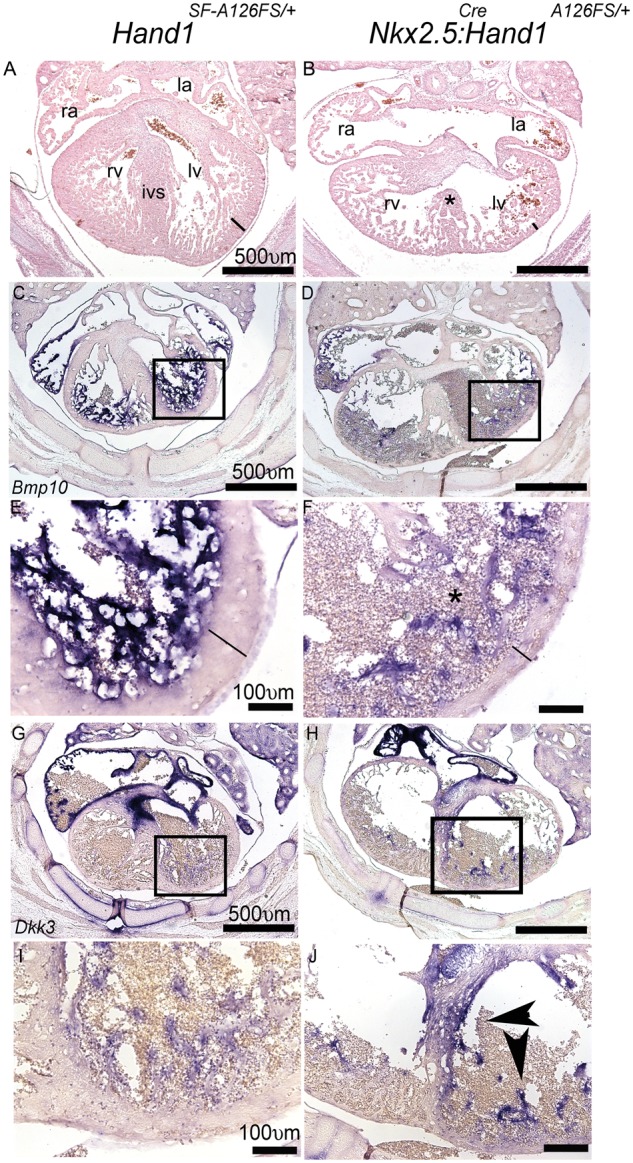
Histological comparison and assessment of gene expression in E14.5 Nkx2.5+/+; Hand1SF-A126FS/+ control and Nkx2.5Cre/+; Hand1A126FS/+mutants. (A) H&E transverse Control hearts show a patent IVS and separate RV and LV with a well-established compact zone (black bar), RA, and LA. (B) Nkx2.5Cre/+; Hand1A126FS/+mutants consistently display VSDs (asterisk) and thin poorly developed compact zone (black bar). LV chamber size is unaffected. (C–F) E14.5 Bmp10 expression reveals a loss trabecular expression. Black boxes in (C, D) define the higher magnification images of (E) and (F). Lines through compact zone measures thickness. (G–J) Dkk3 expression is upregulated in E14.5 Nkx2.5Cre/+; Hand1A126FS/+mutants. Boxes in (G) and (H) define the higher magnification view shown in (I) and (J). Data represent an example of each genotype. Six embryos per genotype were examined. ra, right atria; la, left atria; rv, right ventricle; lv, left ventricle; ivs, intraventricular septum. Scale bars 500 and 100 μm.
3.5 aMHC-cre and Mef2CAHF-cre activation of the Hand1A126FS/+ allele results in improved phenotype and viability
One caveat is that the Nkx2.5Cre knock-in allele could have influence on Hand1 LV expression.54 To help address the Nkx2.5 contribution, we employed the αMHC-Cre transgenic mice that recombines within E9.5 cardiomyocytes.26αMHC-Cretg/+; Hand1A126FS/+ embryos were obtained at expected ratios at E10.5 and E14.5 (see Supplementary material online, Table S2). To our surprise, we identified surviving αMHC-Cretg/+; Hand1A126FS/+ neonatal mice at a frequency of 0.11. We looked at cardiac morphology and expression of Nppa (Anf) at E14.5 (see Supplementary material online, Figure S2A and B). Results show indistinguishable differences in phenotype and in Anf expression between control and αMHC-Cretg/+; Hand1A126FS/+ mutants. We then examined P60 adult hearts for phenotype. Bifurcated αMHC-Cretg/+; Hand1A126FS/+ hearts appear slightly hypertrophied and lack a well define apex (see Supplementary material online, Figure S2D and E). Mutant adults do not exhibit HLHS.
Next, we looked at Mef2CAHF-Cre; Hand1A126FS/+ mutants. Mef2CAHF-Cre27 activates Hand1A126FS within the SHF-derived myocardial cuff. Mef2CAHF-Cre; Hand1A126FS/+ mutants survive to birth at normal frequency (see Supplementary material online, Table S3) exhibiting a normal LV chamber; however, RV size appears reduced at E14.5 and at p60 (see Supplementary material online, Figure S2C and F). We assayed expression of Wnt11, Tbx5, Dkk3, and Cited1 by wholemount ISH in both αMHC- and Mef2CAHF-Cre E10.5 mutants (see Supplementary material online, Figure S3). αMHC-Cre mutants exhibit a more normal OFT (Wnt11 staining white bar) and expression of Tbx5 maintains its LV specific expression (see Supplementary material online, Figure S3A, B, D, E). Expression of Dkk3 appears expanded similar to what is observed in Nkx2.5Cre; Hand1A126FS/+ mutants (white arrowhead in Supplementary material online, Figure S3G and H). No changes in Cited1 patterning are observed in αMHC-cre; Hand1A126FS/+ embryos (see Supplementary material online, Figure S3J and K).
Mef2CAHF-Cre; Hand1A126FS/+ mutants exhibit expanded Wnt11 expression similar to Nkx2.5Cre; Hand1A126FS/+ (see Supplementary material online, Figure S3C). Expression of Tbx5, Dkk3, and Cited1 are unaffected, consistent with Mef2CAHF-Cre expression (see Supplementary material online, Figure S3F, I, L).
4. Discussion
Left-sided cardiac defects have poor clinical outcomes.1–3 In contrast to the increasing mechanistic understanding of congenital defects affecting the OFT,35,55–57 little is understood of the underlying mechanisms causative of HLHS. Hand1 expression and lineage is largely restricted to the LV.10,25 In light of several studies implicating HAND1 in the genesis of Tetralogy of Fallot,19 VSDs,20 and HLHS9,21 in humans along with published evidence demonstrating a high frequency of HAND1 somatic mutations observed from fixed tissues that are not encountered from fresh tissue22—the follow up on these controversial findings in an in vivo system was imperative. The p.Ala126Profs13X Hand1 mutation was identified in hypoplastic hearts from the Leipzig University fixed tissue collection23 and reported to act as a dominant negative in yeast.21 We confirm this result in mammalian expression analysis (Figure 1). Given we observe a loss of DNA binding, we hypothesize that the mutant Hand1 protein dimerizes with both wild-type Hand1, Hand2, and E-proteins when they are coexpressed. Twist-family bHLH factors are well-established to be promiscuous in dimer partner choice and the disruption of phosphoregulation of evolutionarily conserved bHLH residues regulating dimerization is directly associated with human disease such as in Saethre Chotzen Syndrome.14,15,31,58,59 The Hand1 p.Ala126Profs13X localizes to the nucleus and maintains the integrity of the first helix allowing for protein dimerization. All data are consistent with this model.
In this study, we directly tested the HAND1A126fs mutation in mice as causative of HLHS. Our results suggest that when this mutation is activated within cardiomyocytes at E7.5 via Nkx2.5Cre is not causative of HLHS but these mutants are embryonic lethal (Figures 3–7) presenting OFT elongation, thin compact zone, hypotrabeculation, and VSDs. Although Nkx2.5Cre is expressed in the endocardial lineage,28Hand1 is not25 thus the observed phenotypes are driven by Hand1A126FS cardiomyocyte expression. As the Nkx2.5Cre is a knock-in allele genetic interactions could be at play. To account for this, we activated the HAND1A126fs allele using αMHC-Cre26 and Mef2CAHF-Cre27 mice. The αMHC promoter is expressed at E9.5 and is then down regulated until after birth. Activation of the Hand1A126fs allele results in a less-severe phenotype where some mice survive (see Supplementary material online, Table S2) displaying normal looking hearts at E14.5 that become enlarged by P60 (see Supplementary material online, Figure S2). Given the later activation of Hand1A126fs by αMHC-Cre, we cannot distinguish the difference in phenotypes as resulting from genetic interactions with Nkx2.5 or differences in temporal activation or both. Nevertheless, neither Cre driver results in HLHS when crossed to the HAND1SFA126fs allele. Mef2CAHF-Cre generated mutants are born and exhibit elongated OFTs similar to Nkx2.5Cre generated mutants (see Supplementary material online, Figure S3). Wholemount expression shows consistent Wnt11 elongation with no effect on LV gene expression. From this data, we conclude that p.Ala126Profs13X Hand1, even if somatically acquired, is not causative of HLHS. As we considered the high frequency reported in the Leipzig samples (0.77)9,21 for the HAND1A126fs mutation juxtaposed to the finding that no HAND1 mutations were observed from interrogation of non-fixed HLHS patient samples,22 it suggests to us that DNA modifications via tissue fixation is the source of this discrepancy. Given this, we are not confident that HAND1A126fs mutation phenotypes are biologically relevant to human CHDs, although they do reveal some insight into Hand1 role in cardiogenesis. Data from HLHS patients induced pluripotent stem cells show reduced levels of Hand1 and Hand2 in differentiating cardiomyocytes, suggesting that although protein mutations may not be commonly encountered the alteration of transcriptional regulation maybe more frequent explaining the association between Hand1 and CDHs.60 Indeed, changes in HAND1 DNA methylation status is reported in Tetralogy of Fallot patients.61 Additionally, two HAND1 point mutations were reported (p.G73S and p.K152N) in a study from screens of peripheral blood.62 Such simple point mutations are likely far less deleterious than a truncated protein.
The cardiac developmental abnormalities observed in Nkx2.5Cre; Hand1A126FS/+ mouse embryos do present a unique phenotype compared with previously tested Hand1 loss-of and gain-of function mutations. Knockout of Hand110,11 and hypomorphic Hand1 expression13 are embryonic lethal at E9.5 resulting from extraembryonic insufficiencies. Conditional cardiac deletion of Hand1,12 results in neonatal lethality due to a transient decrease in LV size accompanied with endocardial cushion defects. Additionally, in an Mlc2v-Hand1 knock-in model,63 hearts were growth expanded and lacked an IVS. Collectively, these models suggest that cardiomyocyte growth/cell death is affected but the molecular mechanisms affected are still elusive; however, we can conclude that the expression of either wild-type63 or a truncated Hand1 protein (this study) can be more deleterious than the conditional deletion of Hand1.12
In considering the defects observed in Nkx2.5Cre; Hand1A126FS/+ mice, VSDs are a common CHD encountered in human births whereas in mice these defects are often associated with embryonic lethality. VSDs have a wide spectrum of aetiologies and encountering them in mouse mutants is common. Cardiomyocyte cell death is also observed at an abnormal level (Figure 4). This phenotype is not normally encountered in models of CHDs but is common in adult heart disease.64,65 Interestingly, elongated OFT has been reported in Hand1 gain-of-function studies66 supporting a possible active role for the dominant function of Hand1A126FS. Additionally, hypotrabeculation and thin walls are encountered in Nkx2.5Cre; Hand1A126FS/+ hearts. These phenotypes are accompanied by altered trabecular and compact zone gene expression where some genes are down regulated such as Hey2 (Figure 6) and Bmp10 at E14.5 (Figure 7); however, most gene expression changes identified show an increased or expanded expression (CxCl12, Dkk3, Tbx5, Tbx20; Figures 6 and 7). This finding could suggest that Hand1 has repressive transcriptional functions although previous findings do not show such changes in loss-of-function analysis.10–13 Alternatively, expanded expression could result from feedback regulation. Tbx5 for example is restricted to the LV and ChIP-Seq analysis67 suggests that Tbx5 may be a Hand1 regulator. Interestingly, Tbx5 expression is expanded within the RV where Hand1 is not expressed, suggesting a non-cell autonomous mechanism. Tbx5 expansion is only observed within Nkx2.5Cre; Hand1A126FS/+ hearts and not when either the αMHC-Cre or Mef2CAHF-Cre is employed (Figure 5; see Supplementary material online, Figure S3). The Nkx2.5Cre haploinsufficiency combined with the earlier activation of Hand1A126FS/+ expression than αMHC-Cre (E7.5 vs. E9.5) could both influence this phenotype. Mef2CAHF-Cre recombines Hand1 at E7.5 but not within the LV, collectively limiting our ability to distinguish between a temporal and/or genetic mechanism.
Together, these data suggest that although the idea of acquiring a somatic mutation in Hand1 as a mechanism to account for HLHS or any CHD is attractive, the likelihood of this occurring in HAND1 is low. We speculate that it is possible and perhaps likely that mutations within Hand1 enhancer sequences that alter its spatiotemporal expression would be an inheritable HAND1 mutation and identification of human small nucleotide polymorphisms in HAND1 further supports this idea.68
Supplementary material
Supplementary material is available at Cardiovascular Research online.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
Acknowledgements
We would like to thank Danny Carney for technical assistance.
Conflict of interest: none declared.
Funding
Infrastructural support at the Herman B Wells Center for Pediatric Research is in part supported by the generosity of the Riley Children‘s Foundation, Division of Pediatric Cardiology, and the Carrolton Buehl McCulloch Chair of Pediatrics. This work is supported by the National Institutes of Health NIH R01 HL122123-03, HL120920-03 and P01HL134599 (to A.B.F.).
References
- 1. Tchervenkov CI, Jacobs ML, Tahta SA.. Congenital heart surgery nomenclature and database project: hypoplastic left heart syndrome. Ann Thorac Surg 2000;69:S170–S179. [DOI] [PubMed] [Google Scholar]
- 2. Hinton RB, Martin LJ, Tabangin ME, Mazwi ML, Cripe LH, Benson W.. Hypoplastic left heart syndrome is heritable. J Am Coll Cardiol 2007;50:1590–1597. [DOI] [PubMed] [Google Scholar]
- 3. Gordon BM, Rodriguez S, Lee M, Chang RK.. Decreasing number of deaths of infants with hypoplastic left heart syndrome. J Peds 2008;153:354–358. [DOI] [PubMed] [Google Scholar]
- 4. Nemer G, Fadlalah F, Usta J, Nemer M, Dbaibo G, Obeid M, Bitar F.. A novel mutation in the GATA4 gene in patients with Tetralogy of Fallot. Hum Mutat 2006;27:293–294. [DOI] [PubMed] [Google Scholar]
- 5. Liu X-Y, Wang J, Yang YQ, Zhang YY, Chen XZ, Zhang W, Wang XZ, Zheng J-H, Chen YH.. Novel NKX2-5 mutations in patients with familial atrial septal defects. Pediatr Cardiol 2011;32:193–201. [DOI] [PubMed] [Google Scholar]
- 6. Liu X-y, Yang Y-Q, Yang Y, Lin X-P, Chen Y-h.. Novel NKX2-5 mutations identified in patients with congenital ventricular septal defects. Chung Hua I Hsueh Tsa Chih 2009;89:2395–2399. [PubMed] [Google Scholar]
- 7. Chen M-W, Pang Y-S, Guo Y, Liu B-L, Shen J, Song H-D, Liu T-W.. Association between GATA-4 mutations and congenital cardiac septal defects in Han Chinese patients. Chung Hua Hsin Hsueh Kuan Ping Tsa Chih 2009;37:409–412. [PubMed] [Google Scholar]
- 8. Schott JJ, Benson DW, Basson CT, Pease W, Silberbach GM, Moak JP, Maron BJ, Seidman CE, Seidman JG.. Congenital heart disease caused by mutations in the transcription factor NKX2-5. Science 1998;281:108–111. [DOI] [PubMed] [Google Scholar]
- 9. Hickey EJ, Caldarone CA, McCrindle BW.. Left ventricular hypoplasia. J Am Coll Cardiol 2012;59:S43–S54. [DOI] [PubMed] [Google Scholar]
- 10. Firulli AB, McFadden DG, Lin Q, Srivastava D, Olson EN.. Heart and extra-embryonic mesodermal defects in mouse embryos lacking the bHLH transcription factor Hand1. Nat Genet 1998;18:266–270. [DOI] [PubMed] [Google Scholar]
- 11. Riley P, Anson-Cartwright L, Cross JC.. The Hand1 bHLH transcription factor is essential for placentation and cardiac morphogenesis. Nat Genet 1998;18:271–275. [DOI] [PubMed] [Google Scholar]
- 12. McFadden DG, Barbosa AC, Richardson JA, Schneider MD, Srivastava D, Olson EN.. The Hand1 and Hand2 transcription factors regulate expansion of the embryonic cardiac ventricles in a gene dosage-dependent manner. Development 2004;132:189–201. [DOI] [PubMed] [Google Scholar]
- 13. Firulli B, McConville DP, Byers JS, Vincentz JW, Barnes RM, Firulli AB.. Analysis of a Hand1 hypomorphic allele reveals a critical threshold for embryonic viability. Dev Dyn 2010;239:2748–2760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Firulli BA, Redick BA, Conway SJ, Firulli AB.. Mutations within helix I of Twist1 result in distinct limb defects and variation of DNA binding affinities. J Biol Chem 2007;282:27536–27546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Barnes RM, Firulli AB.. A Twist of insight, the role of twist-family bHLH factors in development. Int J Dev Biol 2009;53:909–924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Vincentz JW, Barnes RM, Firulli AB.. Hand factors as regulators of cardiac morphogenesis and implications for congenital heart defects. Birth Defects Res A Clin Mol Teratol 2011;91:485–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Morikawa Y, Cserjesi P.. Extra-embryonic vasculature development is regulated by the transcription factor HAND1. Development 2004;131:2195–2204. [DOI] [PubMed] [Google Scholar]
- 18. Maska EL, Cserjesi P, Hua LL, Garstka ME, Brody HM, Morikawa Y.. A Tlx2-Cre mouse line uncovers essential roles for hand1 in extraembryonic and lateral mesoderm. Genesis 2010;48:479–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wang J, Lu Y, Chen H, Yin M, Yu T, Fu Q.. Investigation of somatic NKX2-5, GATA4 and HAND1 mutations in patients with tetralogy of Fallot. Pathology 2011;43:322–326. [DOI] [PubMed] [Google Scholar]
- 20. Reamon-Buettner SM, Ciribilli Y, Traverso I, Kuhls B, Inga A, Borlak J.. A functional genetic study identifies HAND1 mutations in septation defects of the human heart. Hum Mol Genet 2009;18:3567–3578. [DOI] [PubMed] [Google Scholar]
- 21. Reamon-Buettner SM, Ciribilli Y, Inga A, Borlak J.. A loss-of-function mutation in the binding domain of HAND1 predicts hypoplasia of the human hearts. Hum Mol Genet 2008;17:1397–1405. [DOI] [PubMed] [Google Scholar]
- 22. Esposito G, Butler TL, Blue GM, Cole AD, Sholler GF, Kirk EP, Grossfeld P, Perryman BM, Harvey RP, Winlaw DS.. Somatic mutations in NKX2–5, GATA4, and HAND1 are not a common cause of tetralogy of fallot or hypoplastic left heart. Am J Med Genet A 2011;155:2416–2421. [DOI] [PubMed] [Google Scholar]
- 23. Craatz S, Kunzel E, Spanel-Borowski K.. Classification of a collection of malformed human hearts: practical experience in the use of sequential segmental analysis. Pediatr Cardiol 2002;23:483–490. [DOI] [PubMed] [Google Scholar]
- 24. Moses KA, DeMayo F, Braun RM, Reecy JL, Schwartz RJ.. Embryonic expression of an Nkx2-5/Cre gene using ROSA26 reporter mice. Genesis 2001;31:176–180. [DOI] [PubMed] [Google Scholar]
- 25. Barnes RM, Firulli B, Conway SJ, Vincentz JW, Firulli AB.. Analysis of the Hand1 cell lineage reveals novel contributions to cardiovascular, neural crest, extra-embryonic, and lateral mesoderm derivatives. Dev Dyn 2010;239:3086–3097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Agah R, Frenkel PA, French BA, Michael LH, Overbeek PA, Schneider MD.. Gene recombination in postmitotic cells. Targeted expression of Cre recombinase provokes cardiac-restricted, site-specific rearrangement in adult ventricular muscle in vivo. J Clin Invest 1997;100:169–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Verzi MP, McCulley DJ, De Val S, Dodou E, Black BL.. The right ventricle, outflow tract, and ventricular septum comprise a restricted expression domain within the secondary/anterior heart field. Dev Biol 2005;287:134–145. [DOI] [PubMed] [Google Scholar]
- 28. Zhang Z, Cerrato F, Xu H, Vitelli F, Morishima M, Vincentz J, Furuta Y, Ma L, Martin JF, Baldini A, Lindsay E.. Tbx1 expression in pharyngeal epithelia is necessary for pharyngeal arch artery development. Development 2005;132:5307–5315. [DOI] [PubMed] [Google Scholar]
- 29. Vincentz JW, Casasnovas JJ, Barnes RM, Que J, Clouthier DE, Wang J, Firulli AB.. Exclusion of Dlx5/6 expression from the distal-most mandibular arches enables BMP-mediated specification of the distal cap. Proc Natl Acad Sci USA 2016; 113:7563–7568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Centonze V, Firulli BA, Firulli AB.. Fluorescence Resonance Energy Transfer (FRET) as a method to calculate the dimerization strength of basic helix-loop-helix (bHLH) proteins. Biol Proced Online 2004;6:78–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Firulli B, Howard MJ, McDaid JR, McIlreavey L, Dionne KM, Centonze V, Cserjesi P, Virshup DMa, Firulli AB.. PKA, PKC and the protein phosphatase 2A influence HAND factor function: a mechanisms for tissue specific transcriptional regulation. Mol Cell 2003;12:1225–1237. [DOI] [PubMed] [Google Scholar]
- 32. Firulli BA, Fuchs RK, Vincentz JW, Clouthier DE, Firulli AB.. Hand1 phosphoregulation within the distal arch neural crest is essential for craniofacial morphogenesis. Development 2014;141:3050–3061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Vincentz JW, Barnes RM, Rodgers R, Firulli BA, Conway SJ, Firulli AB.. An absence of Twist1 results in aberrant cardiac neural crest morphogenesis. Dev Biol 2008;320:131–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Eisenberg CA, Eisenberg LM.. WNT11 promotes cardiac tissue formation of early mesoderm. Dev Dyn 1999;216:45–58. [DOI] [PubMed] [Google Scholar]
- 35. Cohen ED, Miller MF, Wang Z, Moon RT, Morrisey EE.. Wnt5a and Wnt11 are essential for second heart field progenitor development. Development 2012;139:1931–1940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Bruneau BG, Logan M, Davis N, Levi T, Tabin CJ, Seidman JG, Seidman CE.. Chamber-specific cardiac expression of Tbx5 and heart defects in Holt-Oram syndrome. Dev Biol 1999;211:100–108. [DOI] [PubMed] [Google Scholar]
- 37. Koshiba-Takeuchi K, Mori AD, Kaynak BL, Cebra-Thomas J, Sukonnik T, Georges RO, Latham S, Beck L, Beck L, Henkelman RM, Black BL, Olson EN, Wade J, Takeuchi JK, Nemer M, Gilbert SF, Bruneau BG.. Reptilian heart development and the molecular basis of cardiac chamber evolution. Nature 2009;461:95–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Mori AD, Zhu Y, Vahora I, Nieman B, Koshiba-Takeuchi K, Davidson L, Pizard A, Seidman JG, Seidman CE, Chen XJ, Henkelman RM, Bruneau BG.. Tbx5-dependent rheostatic control of cardiac gene expression and morphogenesis. Dev Biol 2006;297:566–586. [DOI] [PubMed] [Google Scholar]
- 39. Chen H, Shi S, Acosta L, Li W, Lu J, Bao S, Chen Z, Yang Z, Schneider MD, Chien KR, Conway SJ, Yoder MC, Haneline LS, Franco D, Shou W.. BMP10 is essential for maintaining cardiac growth during murine cardiogenesis. Development 2004;131:2219–2231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Houweling AC, Somi S, Massink MP, Groenen MA, Moorman AF, Christoffels VM.. Comparative analysis of the natriuretic peptide precursor gene cluster in vertebrates reveals loss of ANF and retention of CNP-3 in chicken. Dev Dyn 2005;233:1076–1082. [DOI] [PubMed] [Google Scholar]
- 41. Cavallero S, Shen H, Yi C, Lien CL, Kumar SR, Sucov HM.. CXCL12 signaling Is essential for maturation of the ventricular coronary endothelial plexus and establishment of functional coronary circulation. Dev Cell 2015;33:469–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Harrison MR, Bussmann J, Huang Y, Zhao L, Osorio A, Burns CG, Burns CE, Sucov HM, Siekmann AF, Lien CL.. Chemokine-guided angiogenesis directs coronary vasculature formation in zebrafish. Dev Cell 2015;33:442–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Yamagishi T, Nakajima Y, Nishimatsu S, Nohno T, Ando K, Nakamura H.. Expression of tbx20 RNA during chick heart development. Dev Dyn 2004;230:576–580. [DOI] [PubMed] [Google Scholar]
- 44. Plageman TF Jr, Yutzey KE.. Differential expression and function of Tbx5 and Tbx20 in cardiac development. J Biol Chem 2004;279:19026–19034. [DOI] [PubMed] [Google Scholar]
- 45. Steidl C, Leimeister C, Klamt B, Maier M, Nanda I, Dixon M, Clarke R, Schmid M, Gessler M.. Charterization of the human and mouse HEY1, HEY2, and HEYL genes: Cloning, Mapping, and mutation screening of a new bHLH Gene Family. Genomics 2000;66:195–203. [DOI] [PubMed] [Google Scholar]
- 46. Nakagawa O, Nakagawa M, Richardson JA, Olson EN, Srivastava D.. HRT1, HRT2, and HRT3: a new subclass of bHLH transcription factors marking specific cardiac, somitic and pharyngeal arch segments. Dev Biol 1999;216:72–84. [DOI] [PubMed] [Google Scholar]
- 47. Grego-Bessa J, Luna-Zurita L, del Monte G, Bolós V, Melgar P, Arandilla A, Garratt AN, Zang H, Mukouyama Y-S, Chen H, Shou W, Ballestar E, Esteller M, Rojas A, Pérez-Pomares JM, de la Pompa JL.. Notch signaling is essential for ventricular chamber development. Dev Cell 2007;12:415–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. VanDusen Nathan J, Casanovas J, Vincentz Joshua W, Firulli Beth A, Osterwalder M, Lopez-Rios J, Zeller R, Zhou B, Grego-Bessa J, De La Pompa José L, Shou W, Firulli Anthony B.. Hand2 is an essential regulator for two Notch-dependent functions within the embryonic endocardium. Cell Rep 2014;9:2071–2083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Samsa LA, Givens C, Tzima E, Stainier DY, Qian L, Liu J.. Cardiac contraction activates endocardial Notch signaling to modulate chamber maturation in zebrafish. Development 2015;142:4080–4091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Barnes RM, Firulli BA, VanDusen NJ, Morikawa Y, Conway SJ, Cserjesi P, Vincentz JW, Firulli AB.. Hand2 loss-of-function in Hand1-expressing cells reveals distinct roles in epicardial and coronary vessel development. Circ Res 2011;108:940–949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Weeke-Klimp A, Bax NAM, Bellu AR, Winter EM, Vrolijk J, Plantinga J, Maas S, Brinker M, Mahtab EAF, Gittenberger-de Groot AC, van Luyn MJA, Harmsen MC, Lie-Venema H.. Epicardium-derived cells enhance proliferation, cellular maturation and alignment of cardiomyocytes. J Mol Cell Cardiol 2010;49:606–616. [DOI] [PubMed] [Google Scholar]
- 52. Riley PR, Smart N.. Thymosin beta4 induces epicardium-derived neovascularization in the adult heart. Biochem Soc Trans 2009;37:1218–1220. [DOI] [PubMed] [Google Scholar]
- 53. Winter EM, Gittenberger-de Groot AC.. Epicardium-derived cells in cardiogenesis and cardiac regeneration. Cell Mol Life Sci 2007;64:692–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Lyons I, Parsons LM, Hartley L, Li R, Andrews JE, Robb L, Harvey RP.. Myogenic and morphogenetic defects in the heart tubes of murine embryos lacking the homeo box gene Nkx2-5. Genes Dev 1995;9:1654–1666. [DOI] [PubMed] [Google Scholar]
- 55. Merscher S, Funke B, Epstein JA, Heyer J, Puech A, Lu MM, Xavier RJ, Demay MB, Russell RG, Factor S, Tokooya K, Jore BS, Lopez M, Pandita RK, Lia M, Carrion D, Xu H, Schorle H, Kobler JB, Scambler P, Wynshaw-Boris A, Skoultchi AI, Morrow BE, Kucherlapati R.. TBX1 is responsible for cardiovascular defects in velo-cardio-facial/DiGeorge syndrome. Cell 2001;104:619–629. [DOI] [PubMed] [Google Scholar]
- 56. Vincentz JW, Firulli AB, The Cardiac Neural Crest and Their Role in Development and Disease. Boston: Elsevier; 2014. [Google Scholar]
- 57. Tsuchihashi T, Maeda J, Shin CH, Ivey KN, Black BL, Olson EN, Yamagishi H, Srivastava D.. Hand2 function in second heart field progenitors is essential for cardiogenesis. Dev Biol 2011;351:62–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Firulli BA, Hadzic DB, McDaid JR, Firulli AB.. The basic helix-loop-helix transcription factors dHAND and eHAND exhibit dimerization characteristics that suggest complex regulation of function. J Biol Chem 2000;275:33567–33573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Firulli BA, Krawchuk D, Centonze VE, Vargesson N, Virshup DM, Conway SJ, Cserjesi P, Laufer E, Firulli AB.. Altered Twist1 and Hand2 dimerization is associated with Saethre-Chotzen syndrome and limb abnormalities. Nat Genet 2005;37:373–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Kobayashi J, Yoshida M, Tarui S, Hirata M, Nagai Y, Kasahara S, Naruse K, Ito H, Sano S, Oh H, Hosoda T.. Directed differentiation of patient-specific induced pluripotent stem cells identifies the transcriptional repression and epigenetic modification of NKX2-5, HAND1, and NOTCH1 in hypoplastic left heart syndrome. PloS One 2014;9:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Sheng W, Qian Y, Wang H, Ma X, Zhang P, Diao L, An Q, Chen L, Ma D, Huang G.. DNA methylation status of NKX2-5, GATA4 and HAND1 in patients with tetralogy of fallot. BMC Med Genet 2013;6:46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Cheng Z, Lib L, Li Z, Liu M, Yan J, Wang B, Ma X.. Two novel HAND1 mutations in Chinese patients with ventricular septal defect. Clin Chim Acta 2012;413:675–677. [DOI] [PubMed] [Google Scholar]
- 63. Togi K, Kawamoto T, Yamauchi R, Yoshida Y, Kita T, Tanaka M.. Role of Hand1/eHAND in the dorso-ventral patterning and interventricular septum formation in the embryonic heart. Mol Cell Biol 2004;24:4627–4635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Furtado MB, Nim HT, Boyd SE, Rosenthal NA.. View from the heart: cardiac fibroblasts in development, scarring and regeneration. Development 2016;143:387–397. [DOI] [PubMed] [Google Scholar]
- 65. Bhatt AB, Foster E, Kuehl K, Alpert J, Brabeck S, Crumb S, Davidson WR Jr., Earing MG, Ghoshhajra BB, Karamlou T, Mital S, Ting J, Tseng ZH.. American Heart Association Council on Clinical C Congenital heart disease in the older adult: a scientific statement from the American Heart Association. Circulation 2015;131:1884–1931. [DOI] [PubMed] [Google Scholar]
- 66. Risebro CA, Smart N, Dupays L, Breckenridge R, Mohun TJ, Riley PR.. Hand1 regulates cardiomyocyte proliferation versus differentiation in the developing heart. Development 2006;133:4595–4606. [DOI] [PubMed] [Google Scholar]
- 67. He A, Kong SW, Ma Q, Pu WT.. Co-occupancy by multiple cardiac transcription factors identifies transcriptional enhancers active in heart. Proc Natl Acad Sci USA 2011;108:5632–5637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Sotoodehnia N, Isaacs A, de Bakker PIW, Dörr M, Newton-Cheh C, Nolte IM, van der Harst P, Müller M, Eijgelsheim M, Alonso A, Hicks AA, Padmanabhan S, Hayward C, Smith AV, Polasek O, Giovannone S, Fu J, Magnani JW, Marciante KD, Pfeufer A, Gharib SA, Teumer A, Li M, Bis JC, Rivadeneira F, Aspelund T, Köttgen A, Johnson T, Rice K, Sie MPS, Wang YA, Klopp N, Fuchsberger C, Wild SH, Mateo Leach I, Estrada K, Völker U, Wright AF, Asselbergs FW, Qu J, Chakravarti A, Sinner MF, Kors JA, Petersmann A, Harris TB, Soliman EZ, Munroe PB, Psaty BM, Oostra BA, Cupples LA, Perz S, de Boer RA, Uitterlinden AG, Völzke H, Spector TD, Liu F-Y, Boerwinkle E, Dominiczak AF, Rotter JI, van Herpen G, Levy D, Wichmann H-E, van Gilst WH, Witteman JCM, Kroemer HK, Kao WHL, Heckbert SR, Meitinger T, Hofman A, Campbell H, Folsom AR, van Veldhuisen DJ, Schwienbacher C, O'donnell CJ, Volpato CB, Caulfield MJ, Connell JM, Launer L, Lu X, Franke L, Fehrmann RSN, Te Meerman G, Groen HJM, Weersma RK, van den Berg LH, Wijmenga C, Ophoff RA, Navis G, Rudan I, Snieder H, Wilson JF, Pramstaller PP, Siscovick DS, Wang TJ, Gudnason V, van Duijn CM, Felix SB, Fishman GI, Jamshidi Y, Stricker BHC, Samani NJ, Kääb S, Arking DE.. Common variants in 22 loci are associated with QRS duration and cardiac ventricular conduction. Nat Genet 2010;42:1068–1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.