

Abstract
Aims
L-type Ca2+ channels (LTCCs) in adult cardiomyocytes are localized to t-tubules where they initiate excitation-contraction coupling. Our recent work has shown that a subpopulation of LTCCs found at the surface sarcolemma in caveolae of adult feline cardiomyocytes can also generate a Ca2+ microdomain that activates nuclear factor of activated T-cells signaling and cardiac hypertrophy, although the relevance of this paradigm to hypertrophy regulation in vivo has not been examined.
Methods and results
Here we generated heart-specific transgenic mice with a putative caveolae-targeted LTCC activator protein that was ineffective in initiating or enhancing cardiac hypertrophy in vivo. We also generated transgenic mice with cardiac-specific overexpression of a putative caveolae-targeted inhibitor of LTCCs, and while this protein inhibited caveolae-localized LTCCs without effects on global Ca2+ handling, it similarly had no effect on cardiac hypertrophy in vivo. Cardiac hypertrophy was elicited by pressure overload for 2 or 12 weeks or with neurohumoral agonist infusion. Caveolae-specific LTCC activator or inhibitor transgenic mice showed no greater change in nuclear factor of activated T-cells activity after 2 weeks of pressure overload stimulation compared with control mice.
Conclusion
Our results indicate that LTCCs in the caveolae microdomain do not affect cardiac function and are not necessary for the regulation of hypertrophic signaling in the adult mouse heart.
Keywords: Calcium, Hypertrophy, Signalling, Cardiac myocytes
1. Introduction
Hypertrophic growth of the heart typically occurs in response to injury or disease as an attempt to preserve cardiac function although prolongation of this state ultimately predisposes the heart to decompensation and failure.1 Cardiac hypertrophy is activated by a variety of signaling pathways including Ca2+-responsive proteins such as Ca2+/calmodulin-dependent protein kinase II (CaMKII), protein kinase C, and calcineurin/nuclear factor of activated T-cells (NFAT).1 The Ca2+ pool that activates these hypertrophic signaling pathways is less well-understood but implicated effectors include the L-type Ca2+ channel (LTCC),2,3 the transient receptor potential (TRP) family of non-selective cation channels and store-operated Ca2+ channels,4–10 Ca2+ release from intracellular stores via inositol triphosphate (IP3) receptors,11 and leaky type 2 ryanodine receptors (RyR2).12 In addition to a proposed role in generating a Ca2+ signalling microdomain in cardiac myocytes,13 the LTCC serves as the primary source of Ca2+ influx for inducing contraction. For this later function, LTCCs are predominantly localized to the t-tubule network in cardiac myocytes where they provide the trigger Ca2+ required for Ca2+-induced Ca2+ release from closely apposed ryanodine receptor 2 channels (RyR2) in the sarcoplasmic reticulum (SR).14 L-type Ca2+ channels can also be localized to caveolae in cardiomyocytes where they have been implicated in the regulation of a Ca2+ microdomain that underlies cardiac hypertrophy.15 Indeed, a genetically-encoded LTCC inhibitor consisting of a truncated Rem GTPase fused to a caveolae targeting sequence was shown to inhibit Ca2+ influx-induced NFAT nuclear translocation in adult feline cardiac myocytes.16
The LTCC is a multiprotein complex composed of a pore-forming α subunit, of which calcium channel (Cav) 1.2 is the primary form expressed in the heart, and accessory subunits including CaVβ proteins.17 These β subunits function as modulators of channel activity and direct channel membrane trafficking via interaction with the I-II intracellular loop of α subunits.17 Ca2+ channel accessory β subunit (CaVβ subunit) are encoded by four separate genes, each of which generates multiple splice variants with differing properties.18 We have previously demonstrated that overexpression of CaVβ2A in the mouse heart by transgenesis resulted in increased LTCC activity, dilated cardiomyopathy, and profound myocardial Ca2+ overload that resulted in opening of the mitochondrial permeability transition pore and myocyte death.19 However, it remains unknown if LTCCs function as hypertrophic mediators in vivo through their partial localization to non-contractile caveolae microdomains as we and others have previously proposed in cultured cardiomyocytes.13,16
Here, we describe the creation of a putative activator of caveolae-localized LTCCs consisting of a CaVβ2A protein in which the native palmitoylation sites20 were replaced with a caveolae targeting signal21 (CBD-β2A). We find that while this putative activator has no effect on global Ca2+ handling, it did potentiate NFAT nuclear localization activated by Ca2+ influx in isolated feline cardiomyocytes. However, cardiac-specific transgenic mice overexpressing CBD-β2A showed no augmentation in the cardiac hypertrophic response. Moreover, inhibition of LTCC specifically in caveolae with a targeted Rem fusion protein (CBD-Rem) showed no reduction in pressure overload or agonist infusion-induced cardiac hypertrophy in vivo. Importantly, these overexpressed modulatory proteins were localized to membrane fractions containing caveolin-3 (Cav3), they could interact with LTCCs, and they did not disrupt global Ca2+ handling in cardiac myocytes isolated from these transgenic mice. These results suggest that caveolae-resident LTCCs do not significantly contribute to a signaling microdomain that underlies hypertrophic remodelling in the mouse heart in vivo.
2. Methods
2.1 Cloning, virus production and transgenic mice
A cDNA encoding mouse CaVβ2AC3S/C4S fused C-terminal to the canonical caveolin binding domain RNVPPIFNDVYWIAF21 (CBD-β2A) was synthesized (Biobasic, Ontario, Canada), cloned into the pShuttle-CMV vector, and adenovirus was produced. CBD-β2A, as well as a cDNA encoding Rem1-265 fused N-terminal to the same canonical caveolin binding domain (CBD), were cloned into the murine α-myosin heavy chain (MHC) promoter expression vector22 and used to inject newly-fertilized oocytes to generate transgenic mice (FVB/N background was used for all mice). To test the inducibility of our transgenic system,22 we administered doxycycline (DOX) chow and observed loss of most CBD-Rem expression in the heart. However, DOX was not administered in subsequent experiments since we observed no developmental effect on the heart with constitutive CBD-Rem or CBD-β2A expression, and the mice were normal up to approximately 4 months of age. NFAT-luciferase (NFAT-luc) transgenic reporter mice were previously described.23 Both sexes of mice were used and no animals were discarded in the statistical analysis. Experiments involving animals were approved by the Institutional Animal Care and Use Committee of either Cincinnati Children’s Hospital or Temple University School of Medicine and were in accordance with the National Institutes of Health Guidelines for the care and use of laboratory animals.
2.2 Cell isolation, culture, and adenoviral transduction
Adult feline left ventricular cardiomyocyte isolation was described previously.24–26 Cats were anesthetized and euthanatized with sodium pentobarbital by intravenous infusion with a dosage of 100 mg/kg. Isolated feline cardiomyocytes were washed three times in serum-free Medium 199 (Sigma-Aldrich, St Louis MO, USA), supplemented with penicillin, streptomycin and gentamycin and plated on laminin-coated glass cover-slips or culture plates. Cardiomyocytes were infected with adenovirus expressing CBD-β2A, CBD-Rem, and/or NFATc3-enhanced green fluorescent protein (eGFP) for 12 h at a multiplicity of infection (MOI) of 100. Culture media was changed once per day, and infection efficiency was determined by NFATc3-eGFP fluorescence intensity 36-48 hours after infection. Adult mouse cardiomyocytes were isolated as previously described.27 Mice were anesthetized and then euthanatized by CO2 inhalation.
2.3 Immunoprecipitation
HEK293 cells were transfected with pEGFP-C1 vector and pShuttle-CMV CBD-β2A or GFP-CaV1.2 and CBD-β2A using Xtremegene-9. Forty-eight hours post-infection, cells were lysed in immunoprecipitation buffer containing 20 mM Tris-HCl (pH 7.5), 250 mM NaCl, 1% Triton X-100, 10 mM MgCl2, 0.5 mM dithiothreitol, and protease inhibitors. Two milligrams of protein was used for immunoprecipitation with eGFP antibody (Novus Biologicals, Littleton, CO, USA) and protein A/G agarose (Santa Cruz Biotechnology, Dallas TX, USA) for 12 h at 4 °C. Immunoprecipitated proteins and approximately 120 µg of supernatant from the input were run on 6% SDS-PAGE, transferred and immunoblotted. Caveolae immunoaffinity isolations were performed as previously described.16
2.4 Sucrose density gradient
As previously described, membrane rafts were fractionated from cultured adult feline ventricular cardiomyocytes.16 Approximately 1–2 × 106 cardiomyocytes were scraped into ice cold, detergent-free tricene buffer (250 mM sucrose, 1 mM EDTA, 20 mM tricene, pH 7.4) and centrifuged at 1400 g for 5 min at 4 °C. Cell pellets were resuspended in 1 mL tricene buffer, Dounce homogenized, and centrifuged at 1400 g for 10 min at 4 °C. Supernatant was collected, mixed with 30% percoll (Sigma) in tricene buffer and subjected to ultracentrifugation for 25 min with a Beckman MLS50 rotor, 77 000 g, 4 °C (Beckman Coulter, Brea, CA USA). The separated plasma membranes (PM) were collected, sonicated with 30 second bursts on ice three times, and mixed with 60% sucrose to a final concentration of 40% sucrose. This mixture was overlaid with a 30–5% step sucrose gradient and subjected to overnight ultracentrifugation (Beckman MLS50 rotor, 87 000 g, 4 °C). Fractions were collected every 0.4 mL from the top sucrose layer and proteins were precipitated using a solution of 0.1% weight/volume deoxycholic acid in 100% weight/volume trichloroacetic acid.
2.5 Western blotting
For experiments in adult feline cardiomyocytes, protein fractions from sucrose density gradient centrifugation were loaded onto SDS-PAGE gels, transferred to nitrocellulose membranes, and immunoblots were performed with the appropriate primary antibody, followed by anti-mouse or anti-rabbit horseradish peroxidase-conjugated secondary antibodies (GE Healthcare, Little Chalfont, Buckinghamshire, UK). Membranes were exposed to Western Lightning ECL chemiluminescence substrate (Perkin Elmer, Waltham, MA, USA), and immunoblots were scanned, digitized, and quantified using Image J software. For transgenic mouse experiments, hearts were surgically removed, frozen in liquid nitrogen and stored at −80 °C. Cardiac ventricles were homogenized in buffer containing 20 mM Tris–HCl (pH 7.5), 250 mM NaCl, 1% Triton X-100, 10 mM MgCl2, 0.5 mM dithiothreitol, and protease inhibitors. Homogenates were centrifuged at 14 000 rpm for 10 min and supernatants were used for blotting. Twenty-five to one hundred micrograms of protein was loaded on SDS-PAGE gels and transferred to PVDF membranes. Immunoblots were performed using the appropriate primary antibody and fluorescent conjugated secondary antibodies (LI-COR, Lincoln, NE, USA) in combination with an Odyssey CLx Infrared Imaging System (LI-COR). Primary antibodies used were: endothelial-nitric oxide synthase (eNOS, BD Biosciences, San Jose, CA, USA), CaV1.2 (Millipore, Billerica, MA, USA), Cav3 (BD Biosciences), eGFP (Novus Biologicals, Littleton, CO, USA), Rem GTPase (Santa Cruz Biotechnology, Dallas TX, USA), CaVβ2 (Santa Cruz Biotechnology, Dallas, TX), and β-tubulin (Developmental Studies Hybridoma Bank, University of Iowa, Iowa City, USA). An additional CaVβ2A antibody was used for some experiments and was a gift from Kevin Campbell, University of Iowa Carver College of Medicine, Iowa City, USA.
2.6 Fractional shortening and intracellular Ca2+ measurements
As previously described,16 adult feline cardiomyocytes were plated on laminin coated glass coverslips, which were then broken and pieces with affixed cardiomyocytes were placed in a heated chamber (35 °C) on the stage of an inverted microscope. For experiments using adult mouse cardiomyocytes, cells in suspension were placed in the chamber and allowed to settle. Cardiomyocytes were then perfused with a normal physiological Tyrode’s solution containing 150 mM NaCl, 5.4 mM KCl, 1.2 mM MgCl2, 10 mM glucose, 2 mM Na-pyruvate, 1 mM CaCl2, and 5 mM HEPES, pH 7.4 . Cardiomyocytes were loaded with 5–10 μM Fluo-4 AM (Molecular Probes) for 15 min to measure [Ca2+]i, paced at 0.5 Hz, and cardiomyocyte fractional shortening (FS) was measured with edge detection. For fluorescence measurements, the F0 (or F unstimulated) was measured as the average fluorescence of the cell 50 ms prior to stimulation. The maximal Fluo-4 fluorescence (F) was measured at peak amplitude, as previously described.28
2.7 Ca2+ current measurement
Adult feline ventricular cardiomyocytes and adult mouse cardiomyocytes were processed for Ca2+ measurements as previously described,16 using a chamber mounted on an inverted microscope (Nikon, Tokyo Japan) initially perfused with Tyrode’s solution at 37 °C. Low resistance (1–4 MΩ) patch pipettes were filled with solution containing 130 mM Cs-aspartate, 10 mM NMDG, 20 mM TEA-Cl, 2.5 mM Tris-ATP, 0.05 mM Tris-GTP, 1 mM MgCl2, 10 mM EGTA, pH 7.2, and used in whole-cell voltage clamp experiments. Adult cardiomyocytes were dialyzed with this solution and superfused with normal physiological salt solution for 10 min before beginning experiments. Cardioyocytes were then placed in Na+ and K+ free bath solution containing 150 mM NMDG, 2 mM CaCl2, 5.4 mM CsCl, 1.2 mM MgCl2, 10 mM glucose, 5 mM HEPES, 2 mM 4-AP 2, pH 7.4. These experiments were performed in Na+ and K+ free (in and out) solutions so that Ca2+ currents were measured with little contamination from overlapping ionic currents. Standard techniques used to measure membrane potential and whole cell currents were described in detail previously.29 Only cardiomyocytes with minimal (<10%) rundown of ICa,L were included in the data sets. Junction potentials were not corrected and were less than 10 mV. The cell capacitance was measured using small hyperpolarizing test steps. Membrane potentials were controlled with an Axopatch 2A (Axon Instruments, Sunnyvale, CA, USA) voltage-clamp amplifier using pClamp8 (Axon Instruments) software and acquired with a Digidata 1322A analogue to digital converter (Axon Instruments).
2.8 Adrenergic receptor agonist experiments
Electrophysiology experiments were performed as described above in the presence of 100 nM isoproterenol (Iso) or 1 µM CGP (a β1-adrenergic receptor antagonist) and 1 μM zinterol (a β2-adrenergic receptor agonist).
2.9 NFAT translocation
Adult feline ventricular cardiomyocytes were infected with adenovirus expressing NFATc3-eGFP or NFATc3-eGFP with CBD-β2A at a MOI of 100 as previously described.16 NFATc3-eGFP translocation to the nucleus was measured before and after pacing at 1 Hz for 15–60 min by confocal microscopy. NFAT translocation was quantified as the nuclear to cytoplasmic fluorescence signal.
2.10 Echocardiography, pressure overload and drug treatment
Echocardiography was performed on mice after isoflurane inhalation for anaesthesia (dosage was to effect) using a Hewlett Packard 5500 instrument with a 15-MHz microprobe and measurements were taken on M-mode in triplicate for each mouse and averaged. Analgesia was not used. Eight to ten week-old mice of the relevant genotypes were subjected to transverse aortic constriction (TAC) or sham surgical procedure as previously described.23 Mice were anesthetized by isoflurane inhalation to effect. Doppler echocardiography was performed on mice subjected to TAC in order to determine pressure gradients across the aortic constriction. For 2 week TAC experiments, NFAT activity assays were performed on heart homogenates as described previously.23 To generate an additional model of cardiac disease, Alzet osmotic minipumps no. 2002 (Durect Corp, Cupertino, CA, USA) were dorsally implanted in anesthetized 8–10 week-old mice, for 2 weeks. For this surgical procedure, mice were anesthetized by isoflurane inhalation to effect. The Alzet pumps were filled with solutions containing angiotensin II (432 μg/kg/d) and phenylephrine (PE) (100 mg/kg/d), or phosphate buffered saline as a control.
2.11 Immunofluorescence
Cardiomyocytes from adult transgenic mice were isolated as previously described.7 Immunofluorescence was performed using primary antibodies against Cav3 (Abcam, Cambridge, UK) and Rem GTPase (Santa Cruz Biotechnology). Samples were imaged using a Nikon A1 confocal microscope in enhanced resolution mode. Image deconvolution was performed using NIS Elements Advanced Research software (Nikon) and analysed using Imaris software (Bitplane, Belfast, UK). For experiments with adult feline ventricular cardiomyocytes, immunofluorescence was performed using primary antibodies against CaV1.2 (Millipore) and Cav3 (BD Biosciences).
2.12 Statistics
Results are presented in all cases as mean ± SEM. Statistical analysis was performed using Prism 7 (Graphpad Software, La Jolla, CA, USA). Patch-clamp data were analysed with Clampfit 10 software (Axon Instruments). For feline cardiomyocyte experiments and Ca2+ handling experiments in isolated transgenic mouse cardiomyocytes, unpaired t-test or ANOVA were used to detect significance as appropriate. For electrophysiology experiments in Figures 2B and5D, two-way ANOVA for repeated measures followed by Sidak’s multiple comparisons test was used to establish whether drug treatment resulted in a significant increase in activity. For TAC experiments, two-way ANOVA followed by Newman-Keuls multiple comparisons test was used. P-values less than 0.05 were considered significant.
Figure 2.

CBD-β2A potentiates Ca2+ influx-dependent NFAT nuclear translocation in adult feline cardiomyocytes. (A) Current-voltage plot of adenoviral-CBD-β2A infected or adenoviral-eGFP control infected feline cardiomyocytes in unstimulated conditions or after application of CGP and zinterol and (B) graph representing peak current density from (A). Two-way ANOVA for repeated measures and Sidak’s multiple comparisons test was used for statistical analysis. *P < 0.05 as indicated. (C–D) Immunofluorescence images of isolated adult feline cardiomyocytes infected with adenoviral-NFATc3-eGFP (control, C) or adenoviral-NFATc3-eGFP and adenoviral-CBD-β2A (D) resting or after pacing for 15 min at 1 Hz. White arrows show regions of NFATc3 nuclear accumulation. Scale bar is 100 µm. (E) Plot of NFAT nuclear to cytoplasmic ratio from (C–D) at different time points of pacing. The number of cardiomyocytes analysed is given within the graph and these were generated from one cat heart disassociation (A–B) or four cat heart disassociations (C–E). Two-way ANOVA with Newman-Keuls post hoc test used for statistical analysis. *P < 0.05 vs. unpaced cardiomyocytes with the same adenoviral infection. #P < 0.05 versus paced, NFATc3-eGFP infected control cardiomyocytes.
Figure 5.
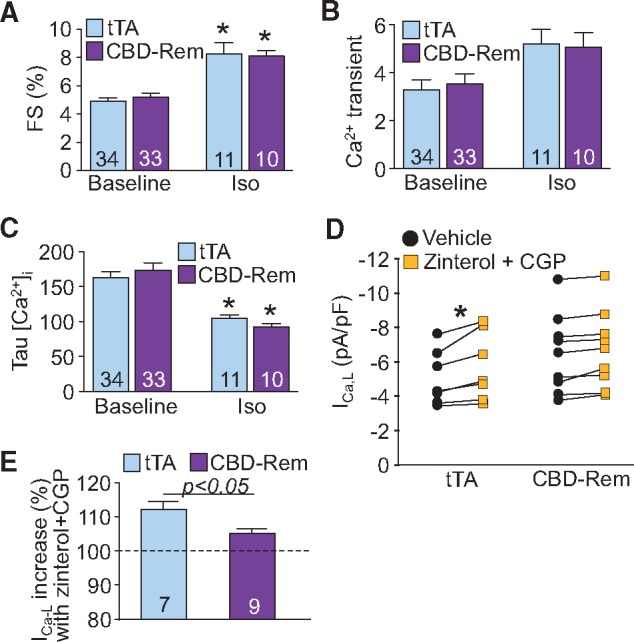
CBD-Rem does not alter global Ca2+ handling or adrenergic responsiveness in the mouse heart. (A) FS of cardiomyocytes isolated from CBD-Rem (high) DTG mice or tTA controls in unstimulated conditions or after treatment with 100 nM Iso. (B) Mean amplitude of Ca2+ transients and (C) rate of Ca2+ transient decay in cardiomyocytes isolated from CBD-Rem (high) DTG mice or tTA controls, paced at 0.5 Hz, in unstimulated conditions or after treatment with Iso. (D) LTCC current density in cardiomyocytes isolated from CBD-Rem (high) DTG mice or tTA controls in unstimulated conditions or after treatment with the β2AR agonist zinterol and β1AR antagonist CGP. There was no significant difference in baseline currents from tTA (control) or CBD-Rem (high) expressing myocytes by Student’s t-test. (E) Percentage increase in current after CGP and zinterol treatment from (D). For each experiment, number of cardiomyocytes analysed is given within the graph, which was generated from at least 3 separate mice for each genotype. Two-way ANOVA with Newman-Keuls multiple comparisons test was used for statistical analysis in panels (A-C). Two-way ANOVA for repeated measures with Sidak’s multiple comparisons test was used for statistical analysis of drug effect in panel (D). Student’s t-test used for statistical analysis in panel (E). *P < 0.05 vs. vehicle control treatment of the same genotype.
3. Results
3.1 Overexpression of CBD-β2A increases NFAT nuclear localization in feline cardiomyocytes
Although LTCCs are predominantly localized to t-tubules in cardiomyocytes (Figure 1A), a subpopulation of channels are localized to the surface sarcolemma where previous work showed that they can be found in caveolae as marked by Cav3, and partially regulated by β2 adrenergic receptors (β2AR).13 Our previous work demonstrated that adenoviral overexpression of a truncated Rem1-265 fused N-terminal to a canonical CBD in feline cardiomyocytes resulted in abrogation of β2AR mediated increases in LTCC activity without affecting global Ca2+ handling.16 Furthermore, viral expression of CBD-Rem inhibited NFAT nuclear translocation after a pacing protocol, suggesting that this caveolae-localized fraction of channels generates a Ca2+ microdomain signal sufficient to activate the calcineurin/NFAT signalling pathway.16 To extend these findings, we employed a similar strategy to localize a putative LTCC activator to caveolae using a CBD fusion to the LTCC β2A subunit, which is known to increase channel conductance and to slow channel inactivation.18 We also mutated the known cysteine palmitoylation sites responsible for general membrane localization20 (CaVβ2AC3S/C4S) so that the CBD would predominate in targeting the β2A subunit (CBD-β2A, Figure 1B) to caveolae. We first confirmed that the CBD-β2A fusion retained its ability to interact with LTCCs via immunoprecipitation in HEK293 cells (Figure 1C). Feline cardiomyocytes were then infected with a recombinant adenovirus expressing this CBD-β2A chimeric protein, which partitioned into the buoyant fractions containing Cav3, eNOS, and a subpopulation of LTCCs upon sucrose density gradient centrifugation (Figure 1D). In a separate experiment, feline cardiomyocytes infected with adenovirus expressing CBD-β2A were fractionated and caveolae were immunoaffinity isolated from PM fractions (lane PM, Figure 1E) using a Cav3 antibody and we identified both CaV1.2 and CBD-β2A as proteins associated with the Cav3 complex by immunoblotting (lane B, Figure 1E). These results suggest that overexpression of CBD-β2A protein produces the desired localization to lipid rafts and that it associates in complex with Cav3 and CaV1.2 in adult feline cardiomyocytes without displacing other caveolae resident proteins.
Figure 1.

CBD-β2A does not affect global Ca2+ handling in feline cardiomyocytes. (A) Immunofluorescence demonstrating localization of CaV1.2 primarily to t-tubules in feline cardiomyocytes. Cav3 is found both at the surface sarcolemma and in t-tubules. Scale bar is 10 µm. (B) Schematic representation of CBD-β2A chimeric fusion cDNA in which β2AC3S/C4S is fused C-terminal to a canonical caveolin binding domain (CBD). (C) Western blot from HEK293 cells transfected with GFP and CBD-β2A or GFP-CaV1.2 and CBD-β2A and immunoprecipitated (IP) with GFP antibody. Western blot of ‘input’ supernatant is included as a control for expression. (D) LTCC, eNOS, β2A, and Cav3 immunoblots from adenoviral-CBD-β2A infected feline cardiomyocyte protein fractions obtained after sucrose density gradient centrifugation. PM and total homogenate (H) fractions are also shown. (E) Western bot of immunoaffinity isolated caveolae from feline cardiomyocytes infected with adenovirus expressing CBD-β2A. H, homogenate; PM, total plasma membrane; B, bound (immunoprecipitated) fraction; U, unbound fraction. (F) Current-voltage plot of CBD-β2A infected or uninfected control feline cardiomyocytes in unstimulated conditions or after application of Iso and (G) graph representing peak current density from (F). Two-way ANOVA followed by Newman-Keuls multiple comparisons test was used for statistical analysis. (H) Fractional shortening (FS) of isolated feline cardiomyocytes infected with CBD-β2A expressing adenovirus or from uninfected control cells. Student’s t-test used for statistical analysis. (I) Mean amplitude of Ca2+ transients from isolated feline cardiomyocytes infected with CBD-β2A expressing adenovirus or from uninfected control cells, paced at 0.5 Hz. Student’s t-test used for statistical analysis. At least two cat hearts were disassociated to generate and analyze all the cardiomyocytes shown in panels A, D–I. *P < 0.05 as indicated.
Functionally, overexpression of CBD-β2A in adult feline cardiomyocytes had no effect on total cellular LTCC current density nor did it alter upregulation of the channel activity upon β-adrenergic stimulation with Iso (Figure 1F–G, see Supplementary material online, Figure S1). Overexpression of CBD-β2A also had no effect on cardiomyocyte contractility (Figure 1H) or the global Ca2+ transient (Figure 1I), suggesting that its effects are likely restricted to caveolin-containing microdomains. These results are in contrast to previous data showing that overexpression of the native β2A subunit dramatically augments global LTCC activity and contractility.19
In our previous study we demonstrated that the CBD-Rem could inhibit β2AR-mediated increases in caveolae-localized LTCCs.16 Here we performed a similar experiment in which feline cardiomyocytes were infected with adenovirus expressing either GFP (control) or CBD-β2A and the response of the myocytes to zinterol (a β2AR agonist) and CGP (a β1AR antagonist) was measured by whole-cell patch clamp electrophysiology (Figure 2A and B, see Supplementary material online, Figure S2A and B). We found that both the control and CBD-β2A expressing cardiomyocytes showed an approximate 10% increase in peak current density (Figure 2B, see Supplementary material online, Figure S2C). However, we detected no obvious augmentation of this response by CBD-β2A infection, although we cannot rule out a more subtle effect of CBD-β2A on the local Ca2+ microdomain at the mouth of the LTCC that would be undetectable at the whole-cell level. We therefore sought to determine whether CBD-β2A could activate caveolae-localized LTCCs to affect hypertrophic signalling pathways, and here employed adult feline cardiomyocytes infected with adenoviruses expressing NFATc3-eGFP and CBD-β2A or NFATc3-eGFP alone as a control. We previously reported that feline cardiomyocytes infected with NFATc3-eGFP showed increased nuclear accumulation of NFAT after pacing at 1 Hz for 1 h.16 Indeed, 15 min after a 1 Hz pacing protocol, NFATc3-eGFP accumulation in the nucleus was clearly observed in the double-infected cardiomyocytes (Figure 2D and E, white arrows) as compared with the single-infected controls (Figure 2C and E). These data demonstrate that CBD-β2A further potentiates NFAT activation by enhancing the activity of caveolae-localized LTCCs, which supports the idea that this Ca2+ microdomain participates in hypertrophic signaling as we had previously observed.16
3.2 Overexpression of CBD-β2A does not enhance cardiac hypertrophy in the mouse heart
To investigate whether caveolae-localized LTCCs play a role in regulating hypertrophy in vivo, we used a cardiac-specific binary transgenic system in which the presence of both a tet-transactivator (tTA) protein under the control of the α-MHC promoter and a CBD-β2A responder transgenic line together produces overexpression of the CBD-β2A protein in the absence of DOX, as described previously.22 Double transgenic (DTG) CBD-β2A mice showed robust protein expression by Western blotting (Figure 3A) but had no baseline cardiac phenotype at 10–12 weeks of age except for a small reduction in FS (Figure 3B–G). Since there was no induction of baseline hypertrophy with CBD-β2A overexpression, CBD-β2A DTG and tTA single transgenic control mice were subjected to pressure overload stimulation via TAC surgery (Figure 3B–G). By 2 weeks post-surgery, both DTG and control mice demonstrated a similar increase in diastolic septum (IVSd, Figure 3B) and posterior wall thickness (LVPWd, Figure 3D), increased ventricle weight normalized to body weight (VW/BW, Figure 3F), and decreased diastolic left ventricle chamber internal diameter (LVIDd, Figure 3C). Fractional shortening (FS, Figure 3E) and lung weight normalized to body weight (LW/BW, Figure 3G) were also not significantly different between genotypes after TAC.
Figure 3.
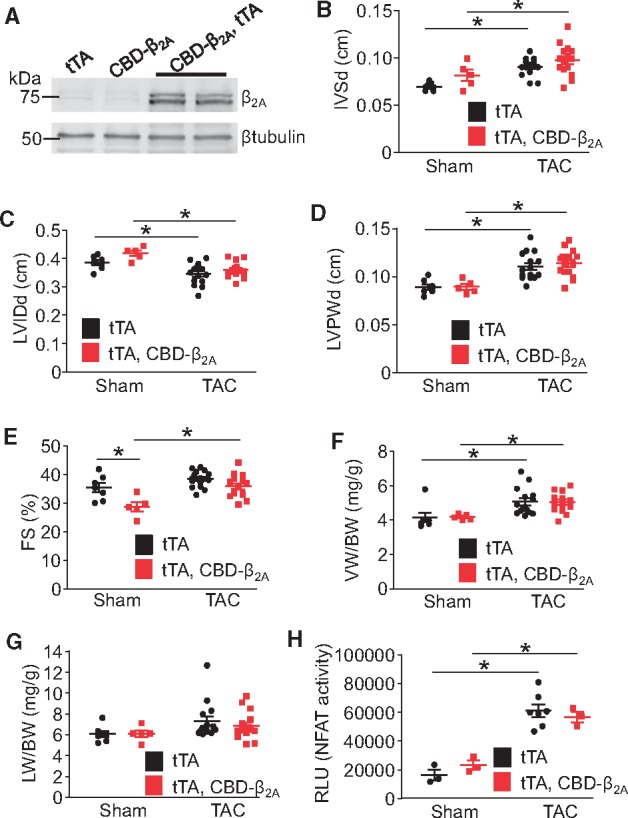
CBD-β2A does not enhance cardiac hypertrophy after 2 weeks of TAC in the mouse. (A) Immunoblot for β2A and β-tubulin from the hearts of CBD-β2A and tTA DTG, CBD-β2A single transgenic, or tTA control mice. (B) Echocardiographic measurements of diastolic intraventricular septum thickness (IVSd), (C) diastolic LVIDds, (D) diastolic left ventricle posterior wall thickness (LVPWd), and (E) FS percentage from CBD-β2A and tTA DTG mice or tTA control mice after 2 weeks of TAC surgery. (F) Gravimetric measurements of ventricle weight normalized to body weight (VW/BW) and (G) lung weight normalized to body weight (LW/BW) from CBD-β2A and tTA DTG mice or tTA control mice after 2 weeks of TAC surgery. (H) NFAT luciferase activity as measured from heart lysates of CBD-β2A, NFAT-luc and tTA expressing triple transgenic mice or NFAT-luc and tTA expressing DTG control mice after 2 weeks of TAC surgery. For each experiment, a two-way ANOVA with Newman-Keuls multiple comparisons test was used for statistical analysis. *P < 0.05 as indicated.
To determine if CBD-β2A overexpression altered calcineurin/NFAT-mediated hypertrophic signalling after TAC, DTG mice were crossed to contain the NFAT luciferase (NFAT-luc) reporter transgene and then subjected to 2 weeks of TAC surgery. While pressure overload induced a significant increase in NFAT activity in the heart compared with sham controls, there was no further increase due to CBD-β2A overexpression (Figure 3H). These results suggest that caveolae-localized LTCCs are not significant inducers of cardiac hypertrophic signalling in the adult mouse heart at baseline or with a disease stimulus.
3.3 Overexpression of CBD-Rem does not inhibit cardiac hypertrophy in the mouse heart
Although CBD-β2A overexpression did not augment pathological hypertrophy induced by pressure overload, it was possible that this domain of LTCC activity was already maximally activated during disease such that addition of CBD-β2A produced no greater effect. For this reason, we again created inducible transgenic mice with cardiac-specific overexpression of a similarly designed caveolae-targeted LTCC modulator, but this time it consisted of a putative inhibitor based on a Rem protein (Rem1-265) fused N-terminal to a canonical CBD. We have previously reported that adenoviral-mediated expression of CBD-Rem in adult feline cardiomyocytes results in localization to low-density caveolin-containing fractions by sucrose density gradient centrifugation, that the overexpressed CBD-Rem interacts with a complex containing Cav3 by immunoprecipitation, that CBD-Rem does not affect global Ca2+ handing but does eliminate β2AR-mediated increases in LTCC activity, and that CBD-Rem can reduce Ca2+-influx mediated NFAT nuclear translocation.16
DTG mice expressing both CBD-Rem and tTA showed high levels of CBD-Rem protein expression (high line) in the absence of DOX (Figure 4A). Transgenic mice containing only the CBD-Rem responder arm of the bi-transgenic system showed a low level of ‘leaky’ expression of the protein and is referred to as the ‘low line’ (Figure 4A). Protein extracts from the hearts of these mice showed that the CBD-Rem chimeric protein partitioned to buoyant fractions containing Cav3 and LTCC when examined by sucrose density gradient centrifugation followed by Western blotting, although some CBD-Rem was also present in the less buoyant fractions (Figure 4B). Nevertheless, cytosolic CBD-Rem was predicted to be innocuous, as Rem requires close coupling to high voltage-activated channels in membrane-associated regions to inhibit their activity, and cytosolic Rem1-265 has no effect on channel function.30 Moreover, some LTCCs were also present in the less buoyant fractions, in keeping with their primary localization to t-tubule structures (Figure 4B). Confocal microscopy followed by image deconvolution similarly revealed that although CBD-Rem was concentrated in the vicinity of the surface sarcolemma, a substantial proportion was cytosolic (Figure 4C). Cav3 was shown to be localized to both the surface sarcolemma and t-tubules as previously shown in feline cardiomyocytes (Figure 1A), although the majority of CBD-Rem and Cav3 co-localization occurred at the surface of the myocytes (Figure 4C).
Figure 4.

CBD-Rem is localized to caveolae-containing subsarcolemmal microdomains. (A) Immunoblot for Rem from the hearts of tTA control, single transgenic CBD-Rem (low expression), DTG CBD-Rem and tTA on DOX, and DTG CBD-Rem and tTA expressing mice without DOX (high expression). The blue arrow shows the migration of the CBD-Rem fusion protein. (B) LTCC, CBD-Rem, and Cav3 immunoblots of protein fractions obtained from CBD-Rem (High) transgenic mouse hearts after sucrose density gradient centrifugation. Plasma membrane (PM) and total homogenate (H) fractions are also shown. (C) Immunofluorescence deconvolution confocal microscopy for Rem (green, left) and Cav3 (red, right) of an adult cardiomyocyte from a DTG mouse (CBD-Rem expression). Merged image shown on bottom. Scale bar is 5 µm.
As previously demonstrated for CBD-Rem expression in feline cardiomyocytes,16 transgenic overexpression of CBD-Rem had no effect on isolated cardiomyocyte contractility (Figure 5A), global Ca2+ handling as measured by amplitude of the Ca2+ transient (Figure 5B), or Ca2+ transient decay time (Figure 5C). Additionally, although transgenic overexpression of CBD-Rem showed a trend towards increased baseline LTCC current density, possibly reflecting long-term compensation of the system because of the CBD-Rem inhibitor, it did not prove to be significant (Figure 5D). CBD-Rem overexpression also had no effect on increased shortening, faster Ca2+ decay, and the trend towards increased Ca2+ transient amplitude elicited by Iso stimulation in isolated cardiomyocytes (Figure 5A–C). However, CBD-Rem was effective at eliminating the effect of β2AR stimulation on LTCCs in isolated mouse cardiomyocytes via treatment with zinterol and CGP, which resulted in a significant drug-mediated augmentation of current density of approximately 10% in adult cardiomyocytes from tTA control mice, but was insignificant in adult cardiomyocytes from transgenic mice expressing CBD-Rem (Figure 5D and E, see Supplementary material online, Figure S3). These data recapitulate CBD-Rem mediated inhibition of caveolae-localized LTCCs we previously observed in feline cardiomyocytes16 and strongly support the conclusion that transgenic overexpression of CBD-Rem regulates the same caveolae-localized LTCC population in mouse cardiomyocytes.
DTG mice overexpressing CBD-Rem in the heart had no baseline cardiac phenotype as measured to 10 weeks (Figure 6A–F). We subjected these DTG mice and their appropriate controls to TAC surgery for 2 weeks to test whether inhibition of caveolae-localized LTCCs could interrupt hypertrophic signaling after pressure overload. In control mice we observed increased posterior wall thickness (LVPWd, Figure 6C), increased intraventricular septum thickness (IVSd, Figure 6A) and increased ventricle weight normalized to body weight (VW/BW, Figure 6E), which was identical to the response observed in DTG mice. CBD-Rem overexpression also had no effect on reducing the activity of the NFAT-luciferase reporter in triple transgenic mice after 2 weeks of TAC (Figure 6G), nor was FS or lung weight normalized to body weight (LW/BW) altered (Figure 6D and F). Finally, CBD-Rem DTG mice showed no reduction in hypertrophy after 2 weeks of infusion with the neurohumoral agonists angiotensin II (AngII) and PE when compared with tTA control mice (Figure 6H). These results show that inhibition of caveolae-localized LTCC does not significantly alter hypertrophic signaling or ventricular growth and remodelling following pathologic stimulation in the mouse heart.
Figure 6.
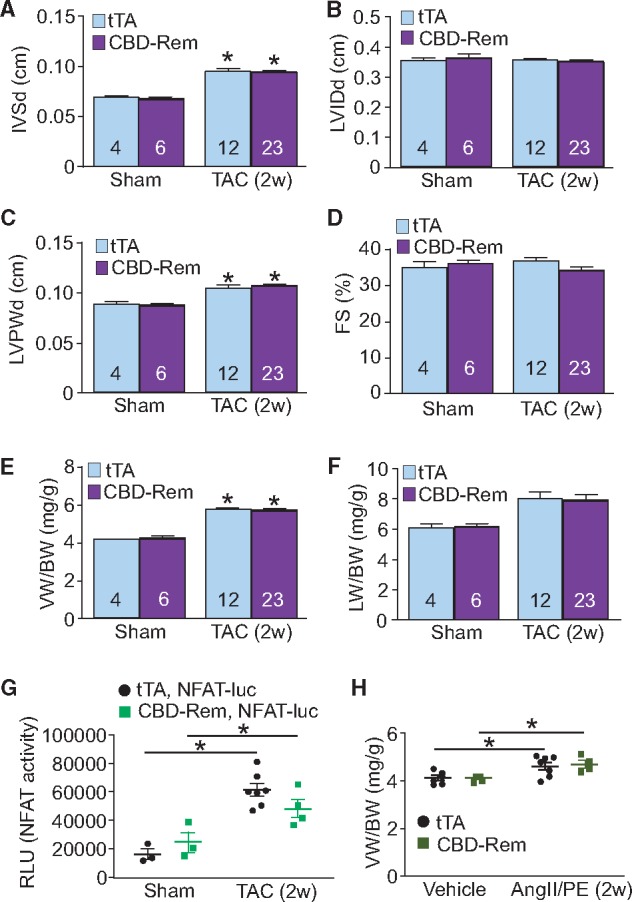
CBD-Rem does not protect against cardiac hypertrophy after 2 weeks of TAC or AngII/PE infusion in the mouse. (A) Echocardiographic measurements of diastolic intraventricular septum thickness (IVSd), (B) diastolic LVIDds, (C) diastolic left ventricle posterior wall thickness (LVPWd), and (D) FS percentage from CBD-Rem (high) DTG or tTA control mice after 2 weeks of TAC surgery. (E) Gravimetric measurements of ventricle weight normalized to body weight (VW/BW) and (F) lung weight normalized to body weight (LW/BW) from CBD-Rem (high) DTG or tTA control mice after 2 weeks of TAC surgery. (G) NFAT luciferase activity as measured from lysates of CBD-Rem, NFAT-luc and tTA expressing triple transgenic mice or NFAT-luc and tTA expressing DTG control mice after 2 weeks of TAC surgery. (H) Gravimetric measurements of VW/BW from CBD-Rem (high) DTG or tTA control mice after 2 weeks of AngII/PE infusion. For each experiment, number of mice analysed is given within the graph. Two-way ANOVA with Newman-Keuls multiple comparisons test was used for statistical analysis. *P < 0.05 vs. sham in panels A–F, and *P < 0.05 as indicated in panels G–H.
While CBD-Rem had no effect on cardiac hypertrophy after 2 weeks of TAC surgery, we were also interested in more long-term effects of pathologic stimulation, such as the transition to heart failure that is typically observed with 10–12 weeks of TAC in the mouse (Figure 7A–F).31 However, as demonstrated with 2 weeks of TAC, after 12 weeks there was still no effect of CBD-Rem overexpression compared to tTA single transgenic controls on posterior wall thickness (LVPWd, Figure 7C), increased intraventricular septum thickness (IVSd, Figure 7A) and increased ventricle weight normalized to body weight (VW/BW, Figure 7E), although there was a very minor but significant reduction in left ventricle chamber internal dimension (LVIDd) in the CBD-Rem transgenic mice (Figure 7B). CBD-Rem mice also showed no change in fractional shortening (FS%, Figure 7D) compared with controls, nor was pulmonary oedema prevalent (Figure 7F). Finally, ventricular weight normalized to body weight was also similarly increased in both experimental and tTA control groups (Figure 7E). Thus, even with protracted hypertrophic stimulation the presence of the CBD-Rem caveolae-specific putative inhibitor had almost no impact on the progression of hypertrophic pathology compared with controls.
Figure 7.
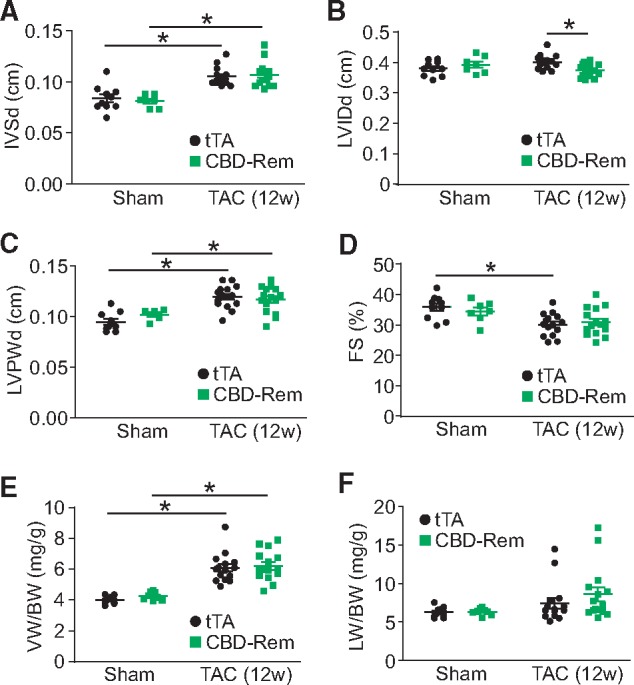
CBD-Rem does not protect against cardiac hypertrophy after 12 weeks of TAC in the mouse. (A) Echocardiographic measurements of diastolic intraventricular septum thickness (IVSd), (B) diastolic LVIDds, (C) diastolic left ventricle posterior wall thickness (LVPWd), and (D) FS percentage from CBD-Rem (high) DTG or tTA control mice after 12 weeks of TAC surgery. (E) Gravimetric measurements of ventricle weight normalized to body weight (VW/BW) and (F) lung weight normalized to body weight (LW/BW) from CBD-Rem (high) DTG or tTA control mice after 12 weeks of TAC surgery. Two-way ANOVA with Newman-Keuls multiple comparisons test was used for statistical analysis. *P < 0.05 as indicated.
4. Discussion
Although LTCCs are the major Ca2+ influx pathway in the heart to initiate contraction, these channels are mostly confined to t-tubules (Figure 1A) where they generate the trigger Ca2+ required for SR Ca2+ release. Hence it was unclear how these channels might also produce a more regulated Ca2+ signal required for activation of proteins such as calcineurin,15 which is known to require a high and sustained Ca2+ signal.32 A solution to this dilemma was proposed 10 years ago when Kamp and colleagues demonstrated that a subpopulation of LTCCs could exist in caveolae signalling microdomains,13 which we confirmed to also be present in adult feline cardiomyocytes and capable of regulating calcineurin/NFAT signaling.16 Previous work from our lab also demonstrated that the protein CIB1 (Ca2+ and integrin–binding protein-1) is involved in anchoring calcineurin to membrane locations for efficient activation and that CIB1 could associate with LTCCs.33
Here we generated transgenic mice with putative activation or inhibition of LTCCs in the caveolae microdomain to more directly examine if this Ca2+ signaling domain also regulates hypertrophy in the adult mouse heart in vivo. Both the Rem GTPase and CaVβ2A are regulated by native membrane localization domains20,30,34 and in the case of Rem, are non-functional if truncated and unable to membrane associate.30,34 Fusion of either a truncated Rem1-265 or a palmitoylation deficient CaVβ2AC3S/C4S to a peptide sequence providing interaction with Cav1 and Cav321 successfully directed both chimeric proteins to caveolae. We previously demonstrated that CBD-Rem could successfully eliminate β2AR-mediated activation of LTCC in feline cardiomyocytes without affecting β1AR-mediated signaling or global Ca2+ handling, and that this inhibition of caveolae-localized LTCCs reduced Ca2+ influx-mediated NFAT nuclear localization suggesting a role for this pathway in hypertrophic signalling.16 Here, we observed that CBD-β2A was properly localized to buoyant fractions containing caveolae in isolated feline cardiomyocytes and that it did not interfere with β1AR signalling (Figure 1F–G). CBD-β2A also increased nuclear translocation of NFATc3-eGFP when feline cardiomyocytes were paced by field stimulation (Figure 2C–E).
Surprisingly, overexpression of CBD-β2A or CBD-Rem showed no effect on altering TAC-induced NFAT activity in the mouse heart, in contrast to the regulation observed in cultured adult feline cardiomyocytes. These results suggest that the mouse heart may not rely on this caveolae-based Ca2+ microdomain of signaling. However, we do not believe that the lack of effect observed in the mouse heart with caveolae-directed CBD-β2A or CBD-Rem was the result of a technical issue such as improper localization. Transgenic expression resulted in high levels of CBD-Rem protein in Cav3 containing fractions that co-localized with Cav3 by immunofluorescence (Figure 4). Additionally, our electrophysiology experiments in transgenic mice expressing CBD-Rem show elimination of LTCC current regulated by β2AR stimulation in the caveolae fraction (Figure 5D–E, see Supplementary material online, Figure S3), as we observed in feline cardiomyocytes.16
Given that CBD-β2A and CBD-Rem were properly localized and active, it seems likely that caveolae-localized LTCCs play no appreciable role in hypertrophic signaling elicited by TAC or neurohumoral agonist stimulation in the mouse heart. These results have implications for the ongoing debate over the role of Cav3 in scaffolding LTCCs in the mouse heart. As discussed above, data from Kamp and colleagues demonstrated that both Cav3 and LTCCs were associated specifically with β2AR but not β1AR in neonatal and adult mouse cardiomyocytes.13 However, a study from McKnight and colleagues suggested that in the adult mouse heart both β1 and β2ARs are associated with Cav3 and LTCCs, and that β1AR signaling was the primary pathway by which LTCCs are phosphorylated.35 In addition, A-kinase anchoring protein 5 (AKAP5) that is associated with Cav3 was required for Iso-induced increases in LTCC current to produce increases in Ca2+ transients and RyR2 phosphorylation at S2808.35 Those results suggest that LTCCs involved with excitation contraction-coupling may also regulate hypertrophic signaling pathways. We find that transgenic expression of CBD-Rem in the mouse heart does not prevent Iso-mediated alterations in Ca2+ handling (Figure 5A–C) and excitation-contraction coupling is unaffected by inhibition of LTCCs associated with Cav3. This would argue that the population of LTCCs associated with Cav3 is not the same population that produces the Ca2+ influx controlling SR Ca2+ release. In conclusion, our transgenic approach showed that, at least in the mouse heart, LTCC-dependent Ca2+ influx within a caveolae signaling microdomain is not a significant regulator of the cardiac hypertrophic response in vivo.
Supplementary material
Supplementary material is available at Cardiovascular Research online.
Conflict of interest: none declared.
Funding
This work was supported by grants from the National Institutes of Health (grant number P01HL108806) (to J.D. Molkentin and S.R. Houser). J.D. Molkentin was also supported by the Howard Hughes Medical Institute. R.N. Correll was supported by a training grant from the National Institutes of Health (F32HL097551).
Supplementary Material
References
- 1. Heineke J, Molkentin JD.. Regulation of cardiac hypertrophy by intracellular signalling pathways. Nat Rev Mol Cell Biol 2006;7:589–600. [DOI] [PubMed] [Google Scholar]
- 2. Chen X, Nakayama H, Zhang X, Ai X, Harris DM, Tang M, Zhang H, Szeto C, Stockbower K, Berretta RM, Eckhart AD, Koch WJ, Molkentin JD, Houser SR.. Calcium influx through Cav1.2 is a proximal signal for pathological cardiomyocyte hypertrophy. J Mol Cell Cardiol 2011;50:460–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Muth JN, Bodi I, Lewis W, Varadi G, Schwartz A.. A Ca(2+)-dependent transgenic model of cardiac hypertrophy: a role for protein kinase Calpha. Circulation 2001;103:140–147. [DOI] [PubMed] [Google Scholar]
- 4. Bush EW, Hood DB, Papst PJ, Chapo JA, Minobe W, Bristow MR, Olson EN, McKinsey TA.. Canonical transient receptor potential channels promote cardiomyocyte hypertrophy through activation of calcineurin signaling. J Biol Chem 2006;281:33487–33496. [DOI] [PubMed] [Google Scholar]
- 5. Kiyonaka S, Kato K, Nishida M, Mio K, Numaga T, Sawaguchi Y, Yoshida T, Wakamori M, Mori E, Numata T, Ishii M, Takemoto H, Ojida A, Watanabe K, Uemura A, Kurose H, Morii T, Kobayashi T, Sato Y, Sato C, Hamachi I, Mori Y.. Selective and direct inhibition of TRPC3 channels underlies biological activities of a pyrazole compound. Proc Natl Acad Sci U S A 2009;106:5400–5405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kuwahara K, Wang Y, McAnally J, Richardson JA, Bassel-Duby R, Hill JA, Olson EN.. TRPC6 fulfills a calcineurin signaling circuit during pathologic cardiac remodeling. J Clin Invest 2006;116:3114–3126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Nakayama H, Wilkin BJ, Bodi I, Molkentin JD.. Calcineurin-dependent cardiomyopathy is activated by TRPC in the adult mouse heart. FASEB J 2006;20:1660–1670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Seth M, Zhang ZS, Mao L, Graham V, Burch J, Stiber J, Tsiokas L, Winn M, Abramowitz J, Rockman HA, Birnbaumer L, Rosenberg P.. TRPC1 channels are critical for hypertrophic signaling in the heart. Circ Res 2009;105:1023–1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wu X, Eder P, Chang B, Molkentin JD.. TRPC channels are necessary mediators of pathologic cardiac hypertrophy. Proc Natl Acad Sci U S A 2010;107:7000–7005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Correll RN, Goonasekera SA, van Berlo JH, Burr AR, Accornero F, Zhang H, Makarewich CA, York AJ, Sargent MA, Chen X, Houser SR, Molkentin JD.. STIM1 elevation in the heart results in aberrant Ca(2)(+) handling and cardiomyopathy. J Mol Cell Cardiol 2015;87:38–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Nakayama H, Bodi I, Maillet M, DeSantiago J, Domeier TL, Mikoshiba K, Lorenz JN, Blatter LA, Bers DM, Molkentin JD.. The IP3 receptor regulates cardiac hypertrophy in response to select stimuli. Circ Res 2010;107:659–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Goonasekera SA, Hammer K, Auger-Messier M, Bodi I, Chen X, Zhang H, Reiken S, Elrod JW, Correll RN, York AJ, Sargent MA, Hofmann F, Moosmang S, Marks AR, Houser SR, Bers DM, Molkentin JD.. Decreased cardiac L-type Ca(2)(+) channel activity induces hypertrophy and heart failure in mice. J Clin Invest 2012;122:280–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Balijepalli RC, Foell JD, Hall DD, Hell JW, Kamp TJ.. Localization of cardiac L-type Ca(2+) channels to a caveolar macromolecular signaling complex is required for beta(2)-adrenergic regulation. Proc Natl Acad Sci U S A 2006;103:7500–7505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bers DM. Calcium cycling and signaling in cardiac myocytes. Annu Rev Physiol 2008;70:23–49. [DOI] [PubMed] [Google Scholar]
- 15. Houser SR, Molkentin JD.. Does contractile Ca2+ control calcineurin-NFAT signaling and pathological hypertrophy in cardiac myocytes? Sci Signal 2008;1:pe31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Makarewich CA, Correll RN, Gao H, Zhang H, Yang B, Berretta RM, Rizzo V, Molkentin JD, Houser SR.. A caveolae-targeted L-type Ca(2)+ channel antagonist inhibits hypertrophic signaling without reducing cardiac contractility. Circ Res 2012;110:669–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Catterall WA. Structure and regulation of voltage-gated Ca2+ channels. Annu Rev Cell Dev Biol 2000;16:521–555. [DOI] [PubMed] [Google Scholar]
- 18. Colecraft HM, Alseikhan B, Takahashi SX, Chaudhuri D, Mittman S, Yegnasubramanian V, Alvania RS, Johns DC, Marban E, Yue DT.. Novel functional properties of Ca(2+) channel beta subunits revealed by their expression in adult rat heart cells. J Physiol 2002;541:435–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Nakayama H, Chen X, Baines CP, Klevitsky R, Zhang X, Zhang H, Jaleel N, Chua BH, Hewett TE, Robbins J, Houser SR, Molkentin JD.. Ca2+- and mitochondrial-dependent cardiomyocyte necrosis as a primary mediator of heart failure. J Clin Invest 2007;117:2431–2444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Chien AJ, Carr KM, Shirokov RE, Rios E, Hosey MM.. Identification of palmitoylation sites within the L-type calcium channel beta2a subunit and effects on channel function. J Biol Chem 1996;271:26465–26468. [DOI] [PubMed] [Google Scholar]
- 21. Couet J, Li S, Okamoto T, Ikezu T, Lisanti MP.. Identification of peptide and protein ligands for the caveolin-scaffolding domain. Implications for the interaction of caveolin with caveolae-associated proteins. J Biol Chem 1997;272:6525–6533. [DOI] [PubMed] [Google Scholar]
- 22. Sanbe A, Gulick J, Hanks MC, Liang Q, Osinska H, Robbins J.. Reengineering inducible cardiac-specific transgenesis with an attenuated myosin heavy chain promoter. Circ Res 2003;92:609–616. [DOI] [PubMed] [Google Scholar]
- 23. Wilkins BJ, Dai YS, Bueno OF, Parsons SA, Xu J, Plank DM, Jones F, Kimball TR, Molkentin JD.. Calcineurin/NFAT coupling participates in pathological, but not physiological, cardiac hypertrophy. Circ Res 2004;94:110–118. [DOI] [PubMed] [Google Scholar]
- 24. Bailey BA, Houser SR.. Sarcoplasmic reticulum-related changes in cytosolic calcium in pressure-overload-induced feline LV hypertrophy. Am J Physiol 1993;265:H2009–H2016. [DOI] [PubMed] [Google Scholar]
- 25. Harris DM, Mills GD, Chen X, Kubo H, Berretta RM, Votaw VS, Santana LF, Houser SR.. Alterations in early action potential repolarization causes localized failure of sarcoplasmic reticulum Ca2+ release. Circ Res 2005;96:543–550. [DOI] [PubMed] [Google Scholar]
- 26. Silver LH, Hemwall EL, Marino TA, Houser SR.. Isolation and morphology of calcium-tolerant feline ventricular myocytes. Am J Physiol 1983;245:H891–H896. [DOI] [PubMed] [Google Scholar]
- 27. Makarewich CA, Zhang H, Davis J, Correll RN, Trappanese DM, Hoffman NE, Troupes CD, Berretta RM, Kubo H, Madesh M, Chen X, Gao E, Molkentin JD, Houser SR.. Transient receptor potential channels contribute to pathological structural and functional remodeling after myocardial infarction. Circ Res 2014;115: 567–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Chen X, Wilson RM, Kubo H, Berretta RM, Harris DM, Zhang X, Jaleel N, MacDonnell SM, Bearzi C, Tillmanns J, Trofimova I, Hosoda T, Mosna F, Cribbs L, Leri A, Kajstura J, Anversa P, Houser SR.. Adolescent feline heart contains a population of small, proliferative ventricular myocytes with immature physiological properties. Circ Res 2007;100:536–544. [DOI] [PubMed] [Google Scholar]
- 29. Piacentino V 3rd, Dipla K, Gaughan JP, Houser SR.. Voltage-dependent Ca2+ release from the SR of feline ventricular myocytes is explained by Ca2+-induced Ca2+ release. J Physiol 2000;523 Pt 3:533–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Finlin BS, Crump SM, Satin J, Andres DA.. Regulation of voltage-gated calcium channel activity by the Rem and Rad GTPases. Proc Natl Acad Sci U S A 2003;100: 14469–14474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Correll RN, Eder P, Burr AR, Despa S, Davis J, Bers DM, Molkentin JD.. Overexpression of the Na+/K+ ATPase alpha2 but not alpha1 isoform attenuates pathological cardiac hypertrophy and remodeling. Circ Res 2014;114:249–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wilkins BJ, Molkentin JD.. Calcium-calcineurin signaling in the regulation of cardiac hypertrophy. Biochem Biophys Res Commun 2004;322:1178–1191. [DOI] [PubMed] [Google Scholar]
- 33. Heineke J, Auger-Messier M, Correll RN, Xu J, Benard MJ, Yuan W, Drexler H, Parise LV, Molkentin JD.. CIB1 is a regulator of pathological cardiac hypertrophy. Nat Med 2010;16:872–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Correll RN, Pang C, Finlin BS, Dailey AM, Satin J, Andres DA.. Plasma membrane targeting is essential for Rem-mediated Ca2+ channel inhibition. J Biol Chem 2007;282:28431–28440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Nichols CB, Rossow CF, Navedo MF, Westenbroek RE, Catterall WA, Santana LF, McKnight GS.. Sympathetic stimulation of adult cardiomyocytes requires association of AKAP5 with a subpopulation of L-type calcium channels. Circ Res 2010;107:747–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.